
Abstract
A key step in most viral infections is the binding of a viral protein to a host receptor, leading to the virus entering the host cell. Disrupting this protein-protein interaction is an effective strategy for preventing infection and subsequent disease. Building on recent advances in computational tools for structural biology, we introduce Virus Inhibition via Peptide Engineering and Receptor Mimicry (VIPER), a novel approach for the automatic derivation and optimization of biomimetic decoy peptides that mimic binding sites of human proteins. VIPER leverages structural data from human-pathogen protein complexes, yielding peptides that can competitively inhibit viral entry by mimicking the natural receptor. We computationally validated VIPER using molecular dynamics simulations and showcased its applicability on three clinically relevant viruses, highlighting its potential to accelerate therapeutic development. With a focus on reproducibility and extensibility, VIPER can facilitate the rapid development of antiviral inhibitors by automating the design and optimization of biomimetic compounds.
INTRODUCTION
Recently, significant progress has been made in designing antiviral peptides, with major breakthroughs occurring during the Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic (Basit et al., 2021; Chatterjee et al., 2020; Chitsike et al., 2021; Curreli et al., 2020; Huang et al., 2020; Jaiswal et al., 2022; Karoyan et al., 2021; Larue et al., 2021; Odolczyk et al., 2021; Panda et al., 2021; Rajpoot et al., 2021; Renzi et al., 2023). The key idea behind these novel inhibitors is to mimic the binding partner that the viral surface protein (VSP) uses to dock to the human receptor. These biomimetic peptides serve as “decoys” that bind directly to the VSP and prevent it from attaching to and entering human cells. While most work has focused on basic research and preclinical efforts, where antiviral peptides so far have shown promise (Jackman, 2022), a biomimetic antiviral peptide inhibitor, Enfuvirtide, has already been approved for clinical use by the FDA (Matthews et al., 2004).
Improvements in in silico approaches (Grisoni et al., 2018; Müller et al., 2018; Plisson et al., 2020; Yoshida et al., 2018; Zakharova et al., 2022) have contributed to the progress seen in peptide engineering in the past few years (Fosgerau and Hoffmann, 2015; Mustafa et al., 2018; Naeimi et al., 2022; Pountos et al., 2016), by reducing the time and cost needed for wet lab experiments. Recently, Google’s DeepMind released AlphaProteo, a machine-learning model designed to generate proteins that bind to target proteins (Zambaldi et al., 2024). However, the model appears to focus on generating larger proteins (>50 amino acids) rather than small peptides. Moreover, AlphaProteo generates these proteins de novo, without mimicking existing human proteins, which could potentially affect the immunogenicity of the resulting binding proteins. Therefore, to the best of our knowledge, there is no dedicated computational method that facilitates the design of biomimetic antiviral entry inhibitor peptides. To fill this gap, we developed Virus Inhibition via Peptide Engineering and Receptor Mimicry (VIPER), a computational approach to automatically design and optimize decoy peptides using the structural data of the human and pathogen proteins in a complex. VIPER is released as an extensible open-source Python program, with emphasis on reproducibility and customizability, and an extensive manual documenting the software is provided in the GitHub repository.
METHODS
VIPER is a computational approach that automatically derives potential entry inhibitor peptides from the structural data of a viral protein bound to the human receptor. Its modular design allows for customizations and extensions of the core components. The workflow can be divided into four components: (1) input, (2) complex analysis, (3) residue selection, and (4) iterative improvement. An overview of the VIPER control flow is given in Figure 1.
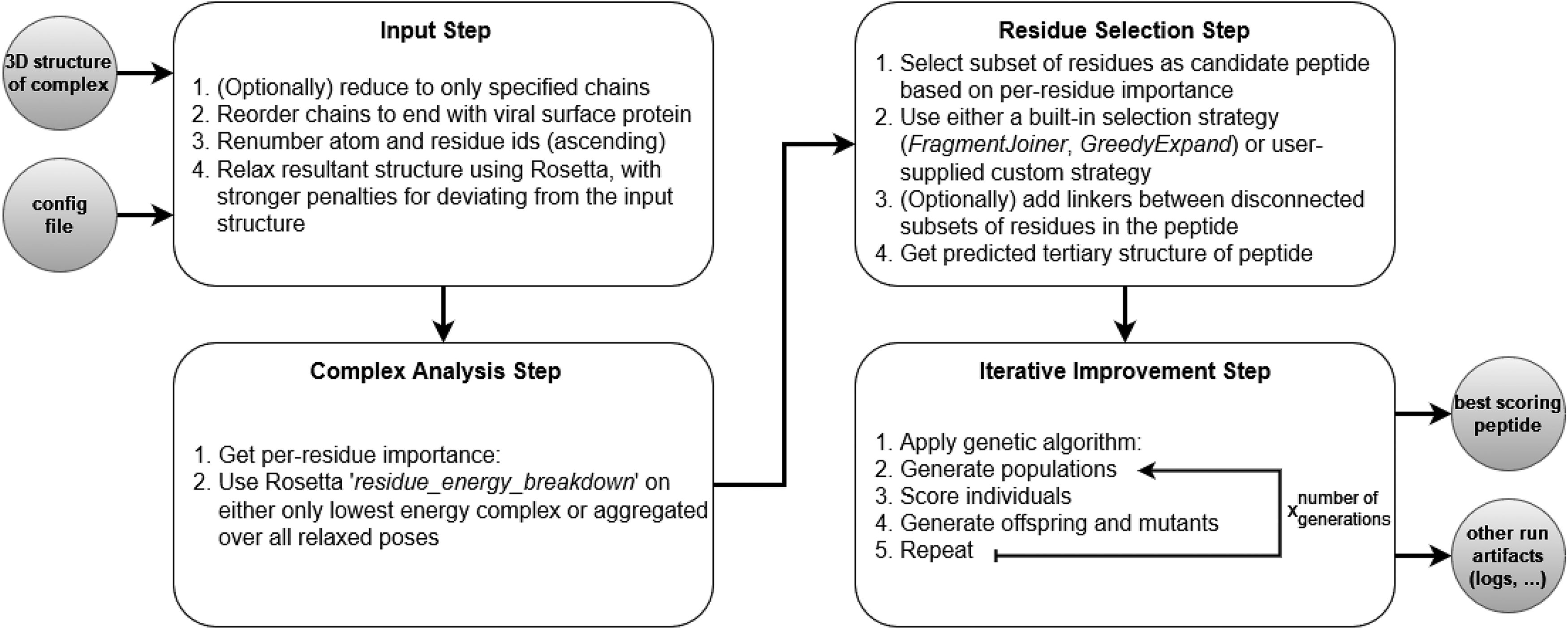
An overview of VIPER’s control flow. VIPER, Virus Inhibition via Peptide Engineering and Receptor Mimicry.
The only necessary input files for VIPER are: (1) a Protein Data Bank (PDB) structure of a VSP bound to a receptor and (2) a configuration file that specifies all settings and options. VIPER makes extensive use of the RosettaCommons software suite (Leaver-Fay et al., 2011) to relax the structures for downstream analysis, with the goal of producing structures that are likely to be more energetically favorable (Conway et al., 2014; Khatib et al., 2011; Nivón et al., 2013; Tyka et al., 2011). All relaxations are performed using the standard relax application, except for additional restraints to the heavy atom positions to avoid deviating too much from the crystal structure. However, these restraints are dropped during the iterative improvement step of the peptide, as there is no experimentally solved structure for the peptide-VSP complex. Further details and all default parameters can be found in Supplementary Data (Supplementary Fig. S1 and Fig. S2).
The residue_energy_breakdown1 (part of the RosettaCommons suite) is used to identify residues strongly involved in binding between the receptor and VSP, as measured by the internal RosettaCommons score function (Alford et al., 2017). Running this application generates a score file that specifies the different types of energy terms such as electrostatic or van der Waals at a per-residue level. This information can then be used to rank residues based on their contribution to the total binding energy score.
Residue selection step
After relaxing the structures and computing the per-residue binding energy, the next step involves identifying a subset of candidate residues (implemented in VIPER in the FragmentJoiner module). FragmentJoiner performs a linear scan through the residues on the receptor chain(s) and starts a fragment when it encounters a residue with a negative (i.e., favorable) energy. This fragment is then extended until a residue is found that does not interact favorably with the VSP, denoted by a positive interaction energy value. At this point, a configurable lookahead window is scanned for favorably interacting residues. If such a residue is found and its inclusion outweighs the penalty of incorporating the residues within the gap, the fragment is extended. If no such residue is found, the fragment is terminated. This procedure is illustrated in Figure 2.
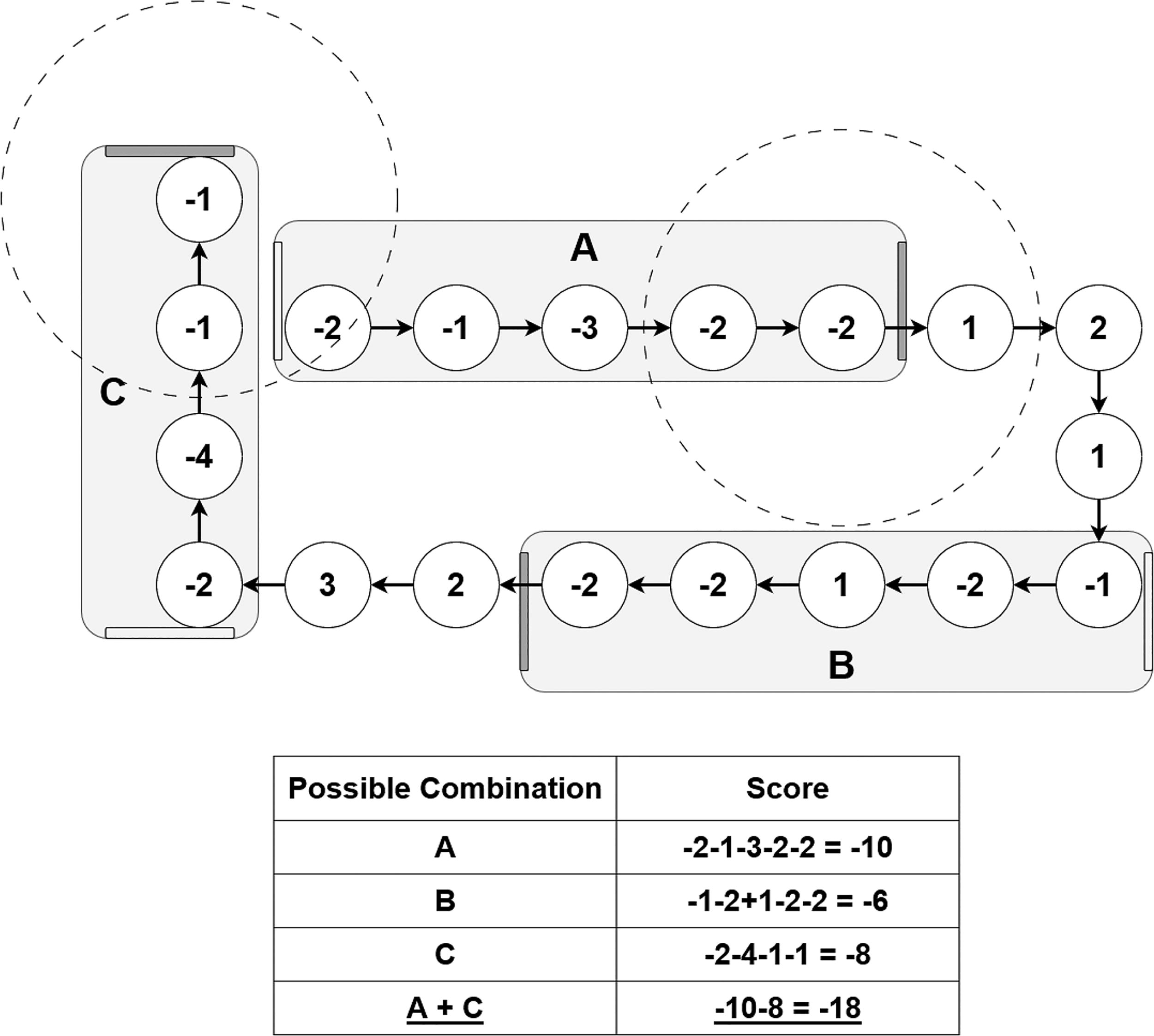
A schematic view of the FragmentJoiner logic. Fragments are identified via a gap-tolerant forward scan of the residues and then combined to maximize the total energy. Only termini of different type (N/C, shown as gray and white bars) are eligible to be combined.
Once all fragments are identified, FragmentJoiner attempts to find the best combination of fragments, that is, the combination with the lowest energy, while staying within the user-defined peptide length limit. Of note, the algorithm can also join connected subsequences of fragments, such that the full-length budget may be used instead of an “all-or-nothing” approach. FragmentJoiner selects the next best-interacting fragment and only adds residues from this fragment up to the residue count limit, starting from the terminus closest to the current set of candidate residues. Therefore, since FragmentJoiner tries to add all residues in a fragment, the only fragments that may be joined only in part are the ones that end up at either end of the final combination of fragments.
Furthermore, FragmentJoiner only considers combinations of fragments where the N-terminus of one fragment is no further away from the C-terminus of the other fragment than a user-defined distance. This distance is equal to the approximate length of the configurable amino acid sequence used as a linker between fragments, whose length can be specified in the configuration options. This constraint ensures that only fragments which could potentially maintain their original relative positions in the receptor protein are joined in the final peptide. For example, two fragments that are 80 Å apart in the receptor would not be joined if the linker is set to be only 10 Å long.
The steps outlined above generate a single candidate peptide derived from segments of the native human receptor sequence. However, introducing single amino acid substitutions at select spots in the sequence might result in a peptide with a stronger binding affinity for the VSP. Furthermore, conformational peptide stability is another important metric that should be factored in when ranking the different peptides generated by VIPER.
Therefore, to further improve the constructs provided by VIPER’s FragmentJoiner, we implemented a genetic algorithm (GA) optimization approach. The fitness function is a combination of the Rosetta energy function (Alford et al., 2017) and a modified spatial side chain interaction index (sSCII), which provides an estimate of the conformational stability of a peptide. The sSCII is used to derive a configurable percentage-based modifier for the Rosetta energy function, ranging from
The individuals are generated through the GA operators: (1) crossover of the amino acid sequence of two parents and (2) mutation (i.e., an amino acid substitution). The first generation starts with the amino acid sequence generated through the previously described residue selection and randomly mutated derivatives of this “seeding” sequence. The individual peptides all have their tertiary structure predicted and subsequently relaxed.
All specific settings are explained in detail among the other VIPER settings in the manual in the GitHub repository.2 The default run configuration provided in the repository was used for all case studies, only updating the chain identifiers (“vsp_chain,” “partner_chain”) according to the corresponding PDB files.
Side chain interaction index
The sSCII is an extension of the side chain interaction index developed by Gehenn et al. (Gehenn et al., 2004, 2006) that we modified to take conformational information into account. The original SCII works by averaging an index value computed for every amino acid in the peptide. This index value is computed by summing up all contact propensities that are larger than one and where the associated partner amino acid is present in the peptide and dividing this sum by the sum of all contact propensities larger than one (Gehenn et al., 2004, 2006). The contact propensity is a measure introduced by Singh and Thornton (1992) to quantify how often and how well amino acid side chains pack against each other compared with the expected frequencies derived from a representative experimental dataset. A contact propensity larger than one indicates a more frequently observed interaction, meaning a favorable interaction. The original formulation is given in Equation 1.
Here
In the original SCII (see Equation 1), an index value is calculated for each amino acid in the sequence, which is then averaged. This per-amino acid value incorporates all inter-residue interactions, regardless of the distance between them or even the order of amino acids in the sequence. Therefore, the SCII for a sequence and a permutation of the same sequence are identical. Since protein sequences have been optimized by evolution, we reasoned that the structural information as well as the order of the amino acids in the sequence is important to the tertiary structure and the function of the protein. Hence, we modified the original sSCII to incorporate structural information by only considering residues within a certain distance of the residue in question during the per-residue index calculation step of the SCII. The rationale behind this choice is that only residues that are close to each other can reasonably have their side chains interact. We discuss how we determined this distance and how we validated our modified sSCII in section 2.4.
VIPER can perform secondary structure analysis of the unbound and bound forms of candidate peptides using Dictionary of Secondary Structure in Proteins (DSSP). VIPER saves the DSSP data, logs differences in secondary structure between bound and unbound states, and generates a json file detailing residues with mismatched secondary structure assignments. It also calculates and logs the percentage of residues affected. This feature is particularly valuable for identifying candidates that may incur an entropic penalty when transitioning from a disordered to an ordered state upon binding. Please consult the manual for additional details.
Computational validation
Spatial side chain interaction index
The sSCII was validated by gathering peptides between 7 and 25 residues in length from UniProt (The UniProt Consortium, 2023) and StraPep (Wang et al., 2018) datasets, randomly scrambling their sequence, predicting the tertiary structure for the newly generated peptide, and comparing the sSCII values derived from the scrambled sequences against those of the original sequences.
Only peptides consisting of a single chain and containing canonical amino acids were used, resulting in 1309 peptides. The original structures of these peptides were collected from the PDB (Burley et al., 2023) based on the PDB accession code listed in the databases, whereas the structure of the scrambled peptides was predicted using OmegaFold (Wu et al., 2022), which can run locally and return a prediction within seconds. The sSCII was then calculated for the original and the scrambled peptide, at 7, 10, 12, 15, and 25 Å radii. To identify the optimal threshold to discern “unstable” from “stable” peptides, the
As it would be more detrimental if a potentially stable candidate would be excluded than if a potentially unstable candidate included, a β of 2 was chosen to assign more importance to the recall of the classifier.
VIPER was run on SARS-CoV-2 (PDB code: 6m0j), HIV (PDB code: 6meo), and HSV (PDB code: 1jma). The run configuration can be viewed in the GitHub repository. In short, the maximum length was set to 18 residues and the resultant peptide was improved through five generations of 10 individuals using the built-in GA optimization.
To evaluate the generated peptides and whether the GA improved on the original solution, 100-nanosecond long molecular dynamics (MD) simulations of the peptide-VSP complex were performed using GROMACS 2023.4 (Abraham et al., 2015, 2024; Páll et al., 2015) with the OPLS/AA forcefield (Jorgensen et al., 1996) (as implemented in GROMACS) and the binding free energy calculated using the gmx_mmpbsa package v.1.6.3 (Valdés-Tresanco et al., 2021) and the Poisson-Boltzmann model, with default parameters. This procedure was applied to the original peptide before any GA modifications, to the best peptide as reported by the GA, and to an intermediary solution produced by the GA optimization, if available.
Implementation
VIPER was developed with Python 3.8 and incorporates several other tools as external dependencies, most notably the RosettaCommon software suite (Leaver-Fay et al., 2011) and PEPstrMOD (Kaur et al., 2007; Singh et al., 2015) for the prediction of the tertiary structure of peptides. VIPER also makes use of the Biopython package (Cock et al., 2009), among others which are listed in the code repository.
Furthermore, VIPER includes extensive logging to ensure reproducibility and to make the automated decisions transparent and traceable. It provides a supported mechanism for customizing its behavior aside from configuration options, where the user can write their own logic in a special source file in which function hooks are already provided, sparing the user from manually finding and modifying the specific part in the code base where they would like their code injected. Additionally, the GA implementation can evolve multiple populations in parallel and uses the Python built-in multiprocessing module to speed up execution.
RESULTS
Spatial side chain interaction index
To computationally validate the sSCII peptide stability metric and to identify the optimal radius for the sSCII calculations, we obtained a total of 1309 unique peptide sequences, identified through the procedure outlined in Section 2. We then scrambled the sequences and compared the ability of the sSCII metric to discriminate between the native sequences and the scrambled ones, building different ROC curves for increasing values of the radius used in the sSCII calculation (see Section 2).
A plot of the ROC curves for the sSCII with a radius of 7, 10, 12, 15, or 25 Å is shown in Figure 3. It can be seen that the 7 Å radius has an area under the curve of 0.87, the highest of all tested configurations. Therefore, this configuration was selected for further use. As the sSCII returns a value between 0 and 1, it is necessary to define a threshold above which peptides are considered stable. The threshold that maximizes the
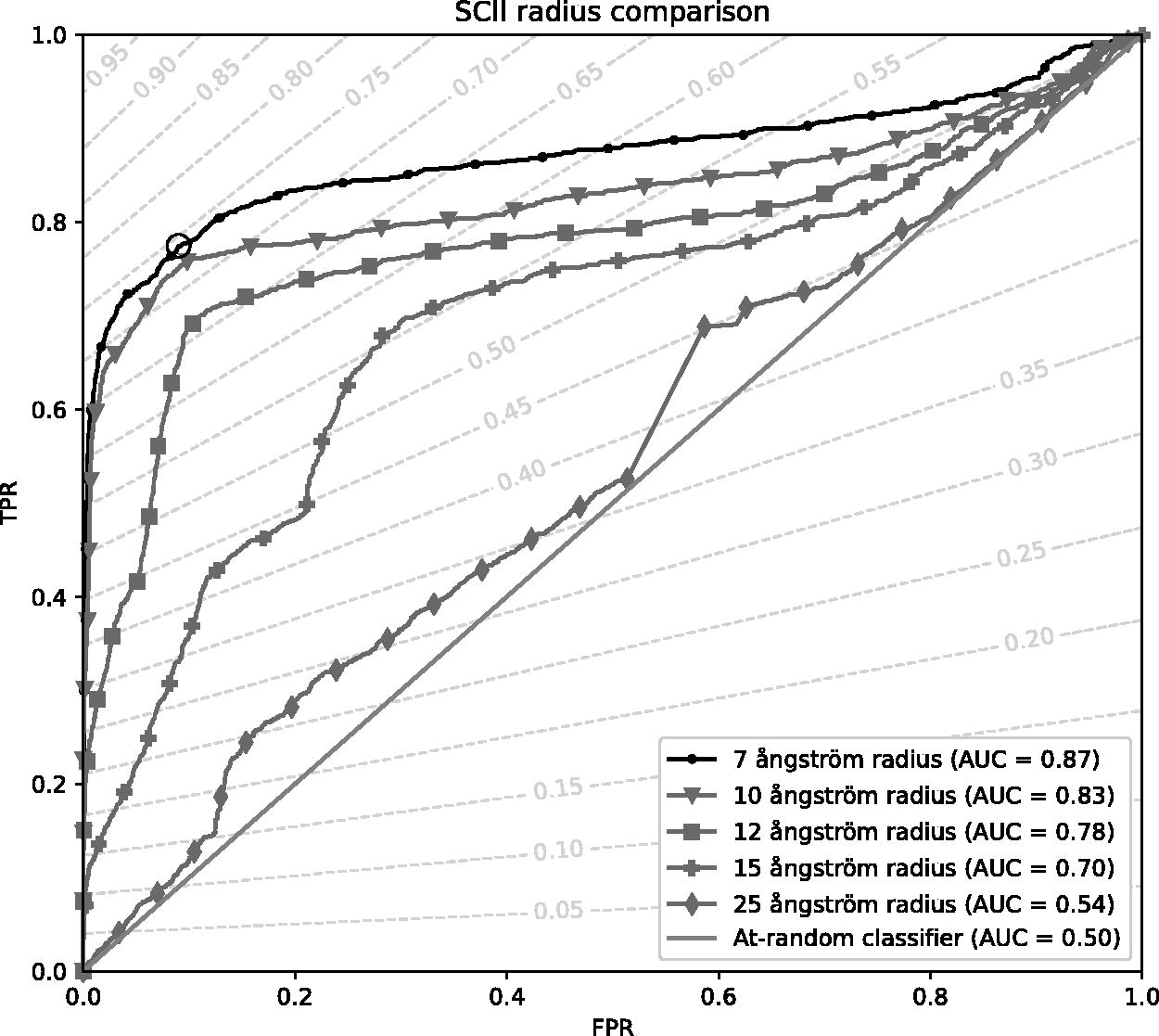
A plot of the receiver operator characteristic curves for different radii and the corresponding area under the curve (AUC). An at-random classifier is shown as an unmarked line. As a guide, dashed lines on which every point has the same
To showcase the applicability of VIPER to clinically relevant viral infections, we ran three case studies for SARS-CoV-2, Human Immunodeficiency Virus (HIV), and Herpes Simplex Virus (HSV), respectively, using VIPER with standard parameters. The final, optimized putative peptide inhibitors for SARS-CoV-2 and HSV are shown in Figure 4, while results for HIV are shown in Supplementary Data (Supplementary Figure S3). As shown in Figure 4, the peptides approximate the conformation of the receptor at the main binding interface of the proteins.
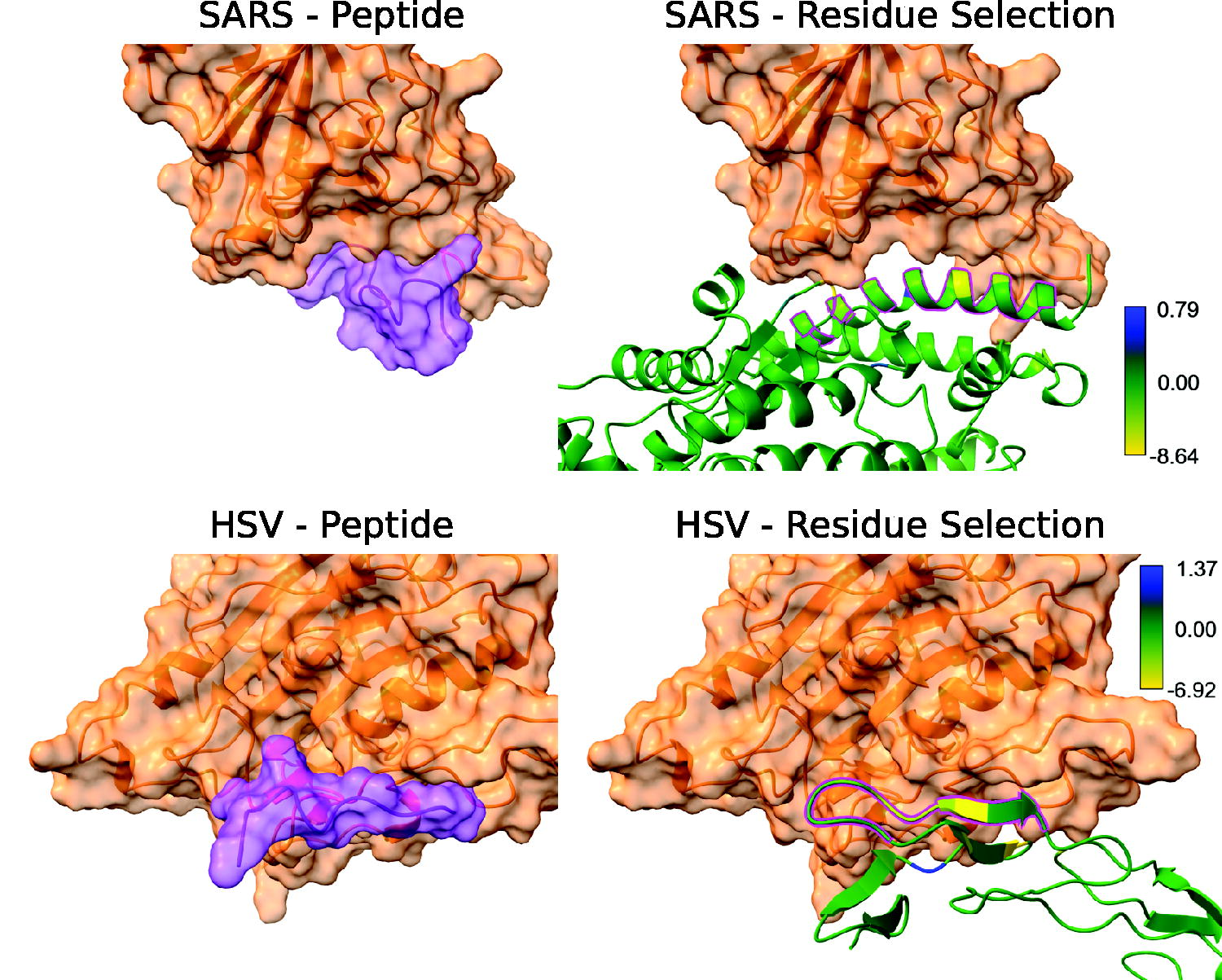
Visualizations for SARS-CoV-2 and HSV. The left-hand side shows the peptide bound to the viral surface protein, whereas the right-hand side shows the per-residue analysis of the receptor bound to the viral surface protein. The residues selected by VIPER to be part of the initial peptide are shown with a magenta outline.
The “SARS-Residue Selection” panel in Figure 4 shows that VIPER with its default configuration mainly selects an
HSV
The surface protein of HSV, a protein called gD, can utilize several different cellular receptors, namely herpesvirus entry mediator (HVEM), nectin-1, or 3-O-sulfated heparan sulfate (Hilterbrand et al., 2021). As a proof of concept, we chose HVEM as the target for VIPER. The “HSV” panels in Figure 4 show that the binding between gD and HVEM is mainly mediated by a long, linear interface between a β-strand in HVEM and a less structurally defined coil region in HSV gD. VIPER selects most of this section, as it is the largest one where multiple close residues interact strongly with the VSP. It is the only selected section though, as the sections are too far from each other to be joined by the standard linker. The initial amino acid sequence generated by VIPER was CGELTGTVCEP, which became CGELTGTVC
Genetic algorithm
To computationally test whether the GA optimization resulted in increased peptide binding affinities for the viral proteins, we performed binding energy calculations with gmx_mmpbsa on MD simulation data (see Section 2 for more details), in addition to the GA score (Rosetta scoring function) proxy. In all tested scenarios, the GA resulted in peptides with higher binding affinity (i.e., lower Rosetta energy units) than the original peptide. Overall, the improvements in the score were sizable, consistent, and well aligned with the results obtained from MD simulation data (Fig. 5). While all peptides initially identified by VIPER had a total negative (i.e., favorable) energy, the GA optimization with default parameters identified at least one peptide for each case study with lower energy, demonstrating the benefit of using the optimization step. Remarkably, values obtained from the GA score (which uses the Rosetta score function) were in very good agreement with those obtained from the MD simulations (Fig. 5).
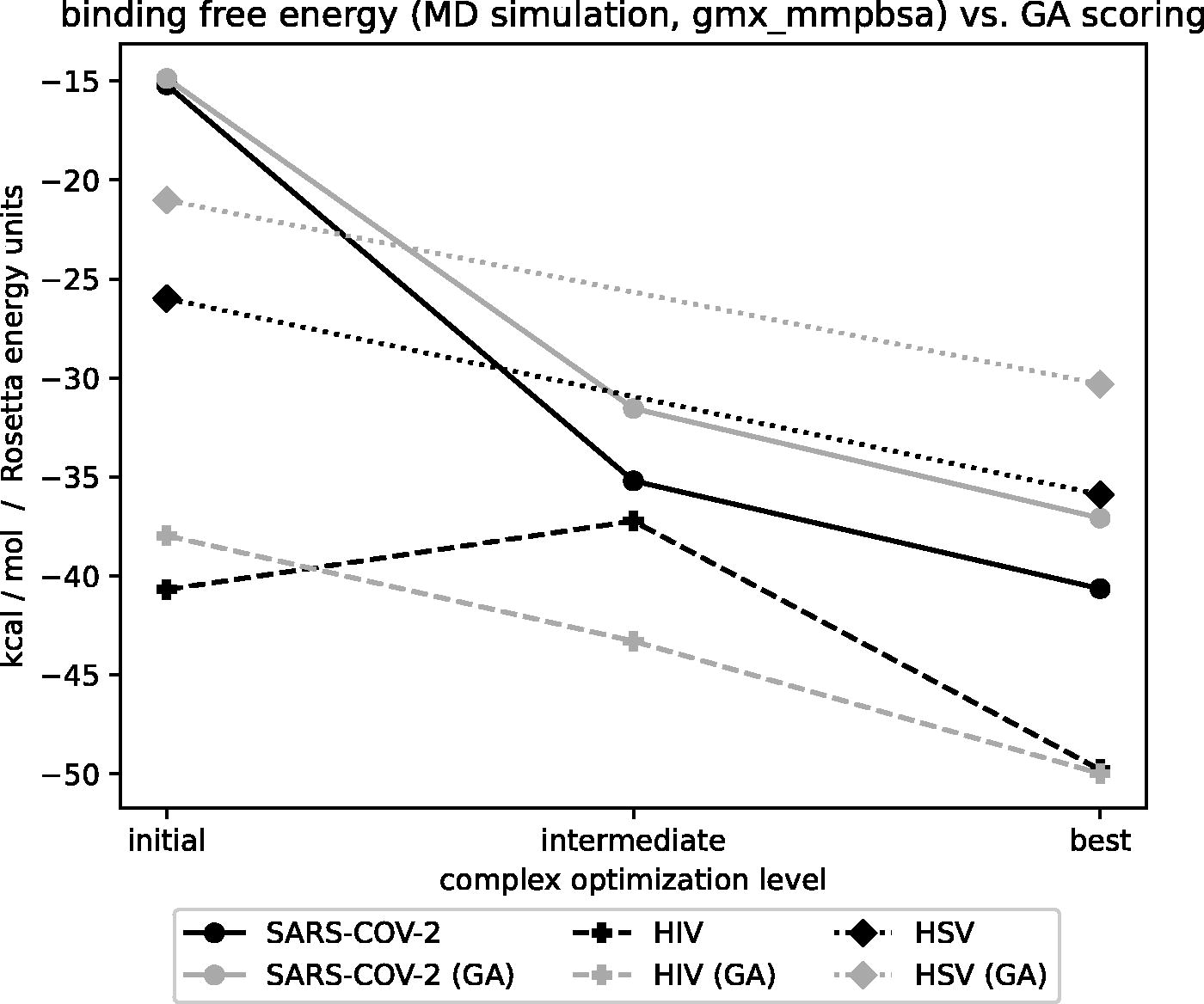
Binding free energy calculation results compared with the GA score for the initial, intermediate (if available), and final peptide. The curves in black refer to the gmx_mmpbsa calculations (kcal/mol), whereas the gray curves refer to the GA score (Rosetta energy units). GA, genetic algorithm.
The actual values for the simulations and projected values (GA scores) are listed in Supplementary Data (Supplementary Table S1).
In this work, we introduce our tool VIPER to design biomimetic viral entry inhibitors using complexes of viral proteins bound to human receptors. VIPER is fully automated and can optionally improve the initial construct by introducing mutations via GA optimization. As the biological activity of the peptide is closely linked to its structure, we sought to include and improve upon a metric to determine the conformational stability of a peptide. We reasoned that improving not just the interaction energy terms but also the conformational stability of a peptide might lead to more desirable candidates. Therefore, we also extended and computationally validated an existing conformational stability measure for peptides (Gehenn et al., 2004, 2006), for use in the GA fitness function. We then showcased VIPER’s use on three model viruses.
While this tool provides users with a structured framework for deriving decoy peptides, there are several limitations that we wish to highlight. Most notably, the computational validation was performed entirely in silico. However, in a previous work (Renzi et al., 2023), we used a similar strategy to the automated workflow in VIPER to design an inhibitory peptide for SARS-CoV-2 that was experimentally validated and shown to actually inhibit the viral entry into human cells, demonstrating the feasibility of the approach.
Another caveat is that the structure of the peptide is predicted independently of the structure of the VSP, which likely neglects any possible interactions that might influence the distribution of conformations adopted by the peptide. Additionally, as an external web server is used for the prediction of the tertiary structure, there is some degree of non-determinism in this tool, although VIPER sets all random seeds for all local tools. This could be an area for future work, especially as computational structure prediction methods have recently made large advances, as can be seen with the AlphaFold family of models (Abramson et al., 2024; Jumper et al., 2021). Another limitation is the fact that the input structure only represents a single point in time, whereas protein-protein interactions can be highly dynamic. Moving from singular peptides to peptide ensembles may alleviate this issue and is therefore a promising avenue for future research.
As the AlphaFold family of models (Abramson et al., 2024; Jumper et al., 2021) supports the structure prediction of multimer proteins, a potential application of these and other forthcoming models is to predict the structure of VSP-receptor complexes if no experimentally solved structure is available and to use these models to generate multiple conformations of the complex to more closely model the dynamic nature of protein-protein interactions.
Deep learning may also help improve the accuracy of VIPER in other ways. Currently, the interaction between proteins is scored using the Rosetta score function (Alford et al., 2017), which relies on physics- and knowledge-based score terms. These, however, do not explicitly model some non-canonical interactions like
Another area for future work is incorporating advanced structural and post-translation modifications like cyclization or acetylation of the peptide, which may improve the pharmaceutical properties of the peptide (Ebrahimi and Samanta, 2023; Wang, 2012; Wang et al., 2022).
Finally, VIPER is agnostic to the host and virus and works purely on the basis of the receptor protein and VSP. This makes VIPER applicable in a wide variety of scenarios, including the development of new antivirals for agriculture, where viruses can cause substantial damage to crops (Tatineni and Hein, 2023), or in animal husbandry (Cui et al., 2023).
Footnotes
ACKNOWLEDGMENTS
The authors thank Austin Seamann and all members of the Ghersi lab for helpful discussions and suggestions.
AUTHORS’ CONTRIBUTIONS
D.G.: Conceptualization, methodology, writing. A.S.K.: Methodology, software, writing. All authors have read and approved the article.
CODE AND DATA AVAILABILITY
All code is available at https://github.com/A-Klingenberg/VIPER/tree/main with an extensive handbook documenting all configuration options and internals of the software tool. The tool is released under the MIT license and also indexed and archived on Zenodo at ![]() for archival stability.
for archival stability.
AUTHOR DISCLOSURE STATEMENT
The authors have no conflicts of interest to declare.
FUNDING INFORMATION
The authors received no financial support for this research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.