
Abstract
Information theory-based measures of variable dependency (previously published) have been implemented into a software package, MIST. The design of the software and its potential uses are described, and a demonstration is presented in the discovery of modifier alleles of the ApoE gene in affecting Alzheimer's disease (AD) by analyzing the UK Biobank dataset. The modifier genes uncovered overlap strongly with genes found to be associated with AD. Others include many known to influence AD. We discuss a range of uses of the dependency calculations using MIST that can uncover additional genetic effects in similar complex datasets, like higher degrees of interaction and phenotypic pleiotropy.
INTRODUCTION
Biological systems generate intrinsically complex datasets in which a large set of variables and attributes interact by storing and transmitting information among one another. This information depends on how each variable interacts with and is related to the other variables of the system. This can be summarized by determining how the variables depend on one another. Characterization of the joint probability distribution of the variables is at the heart of describing their mathematical dependency. We have devised measures of dependency of subsets of variables based on information theory (Galas et al, 2021; Galas et al, 2014).
MIST provides a
It is important to emphasize again that since it is based on Information Theory measures, which quantify the dependencies in the data without making any modeling assumptions, MIST does not answer the question of how the variables are dependent on one another. It does not recover the underlying function governing a dependence. On the other hand, MIST establishes the existence of a dependence among a set of multiple (two, three, or more) variables. Given the dependence identified by MIST, we can choose to find the function underlying the dependence using a model as simple as logistic regression or more complex, like Bayesian networks. Note also that identifying the subsets of dependent variables is like extracting the structure from the data, which may also be seen as a compression of the data.
In this article, we briefly review the previously published mathematical basis of the dependencies (Galas et al, 2021; Galas et al, 2014), describe the design of the software, and then demonstrate MIST's utility by analyzing genetic and clinical data from the UK Biobank (UKBB) database. Specifically, we begin by searching for pairwise functional relationships between single nucleotide polymorphisms (SNPs) and the Alzheimer's disease (AD) phenotype. We then study new genetic dependencies by finding SNPs that modify a well-known genetic variant (APOE4) that affects AD. We also provide a detailed performance analysis of MIST on included test data. The aim of this article is to showcase MIST software that can make the data analysis of large datasets deeper and produce richer results. Although we chose to use genetic data of AD as an example, the same ideas can be applied to other types of data.
METHODS
Information measures
Given a set of N variables and a set of M samples representing information on each of the variables, how do we find functional relationships between the variables? MIST tackles the problem by using a divide-and-conquer approach: while considering the set of all variable tuples as the search space, MIST divides that search space among parallel threads, and then conquers it by computing dependency measures for each tuple.
Symmetric Delta (Galas et al, 2020; Galas et al, 2014) is the measure used in a MIST search. The Symmetric Delta is a novel symmetric measure of functional dependence (it is symmetric under exchange of variables) constructed from joint entropies. Joint entropies between variables (using the Shannon entropy (Shannon and Weaver, 1949) defined as the expectation of the logarithm of each element of the joint probability distribution:
Dependence between two variables X and Y can be directly measured with mutual information
where
A general dependence among three variables, X, Y, and Z, can be measured with symmetric delta.
Given interaction information, differential interaction information
Here
The critical property of this delta measure is that it is zero whenever
Software design
In practice, the bulk of the computational runtime is spent on counting to obtain joint probability distributions, and so we can get a significant speed-up by reducing the number of entropy calculations. First, we avoid doing unnecessary work by carefully defining the search space. For example, it may be that we are only interested in particular combinations of variables, rather than the set of all possible tuples. MIST constructs a TupleSpace from user-defined Variable Groups and Group Tuples that define the sorts of variable tuples to include in the search (Fig. 1). In the AD study below, we reduce one search space size from the order of
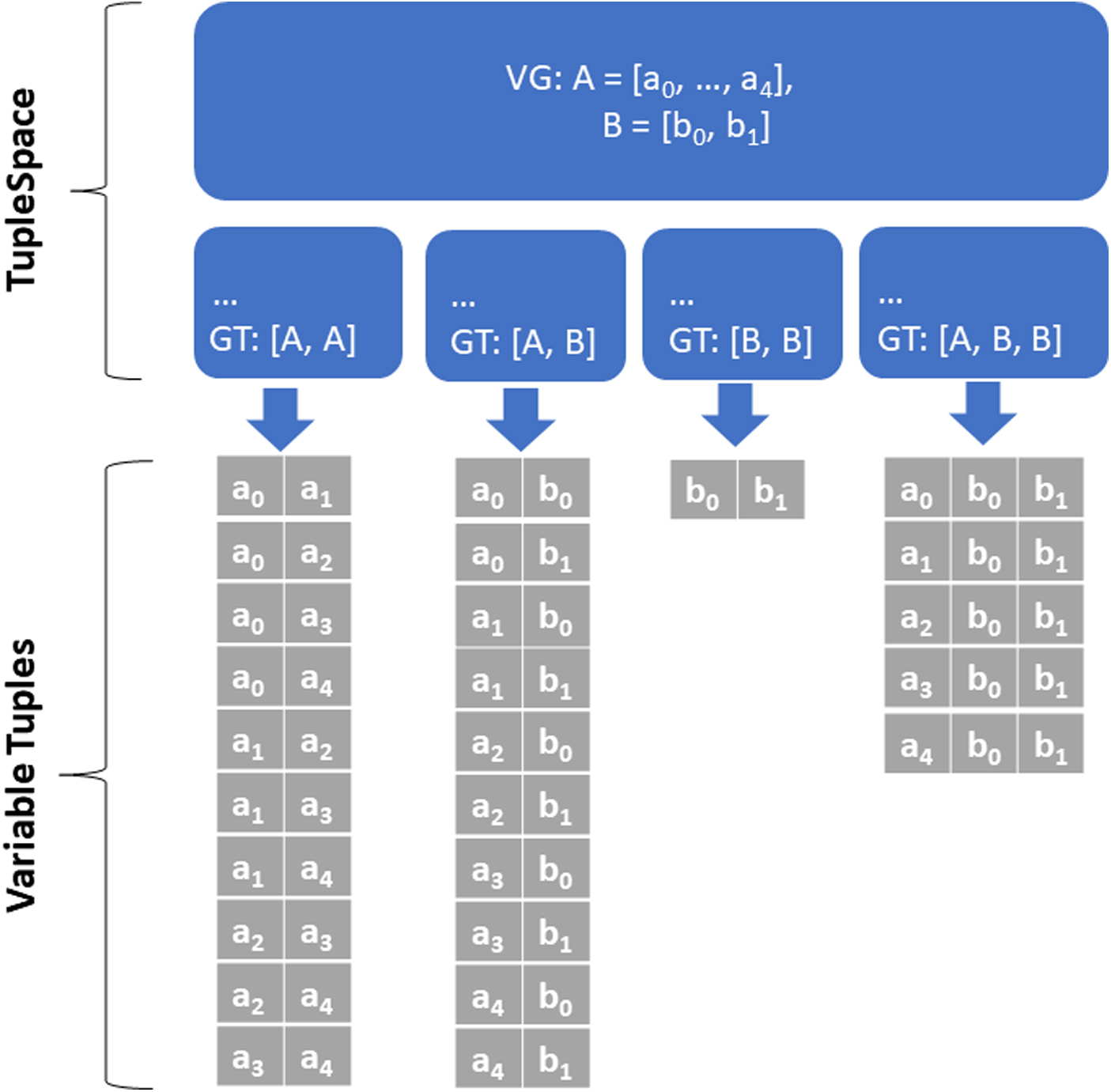
A TupleSpace is composed of VG and GT. The GT act as templates defining the combination of variables drawn from the VG to form Variable Tuples. For example, the Variable at position 0 in the Variable Tuple will be chosen from the group at position 0 in the GT. In this figure, VG A and B are used in four TupleSpaces, each with a different GT. The generated Variable Tuples are displayed underneath each TupleSpace. Since each VG is a disjoint ordered set and each Variable Tuple is an ordered set, Variable Tuples can be determined by simply iterating through the VG. GT, Group Tuples; VG, Variable Groups.
Next, MIST parallelizes the search by dividing the TupleSpace among computation threads. It uses a traversal algorithm to divide the search space into equally sized, contiguous working sets. By treating the search space as an ordered list of tuples, each thread can skip to the starting position in its working set. Thus, MIST achieves scalable parallelization without interprocess communication. As a result, threads can run on separate systems without coordination software like MPI. Generally, increased thread counts reduce runtime, but with diminishing returns as the nonparallel portion of the runtime, plus any threading overhead, take on a larger portion of the runtime (Fig. 2).
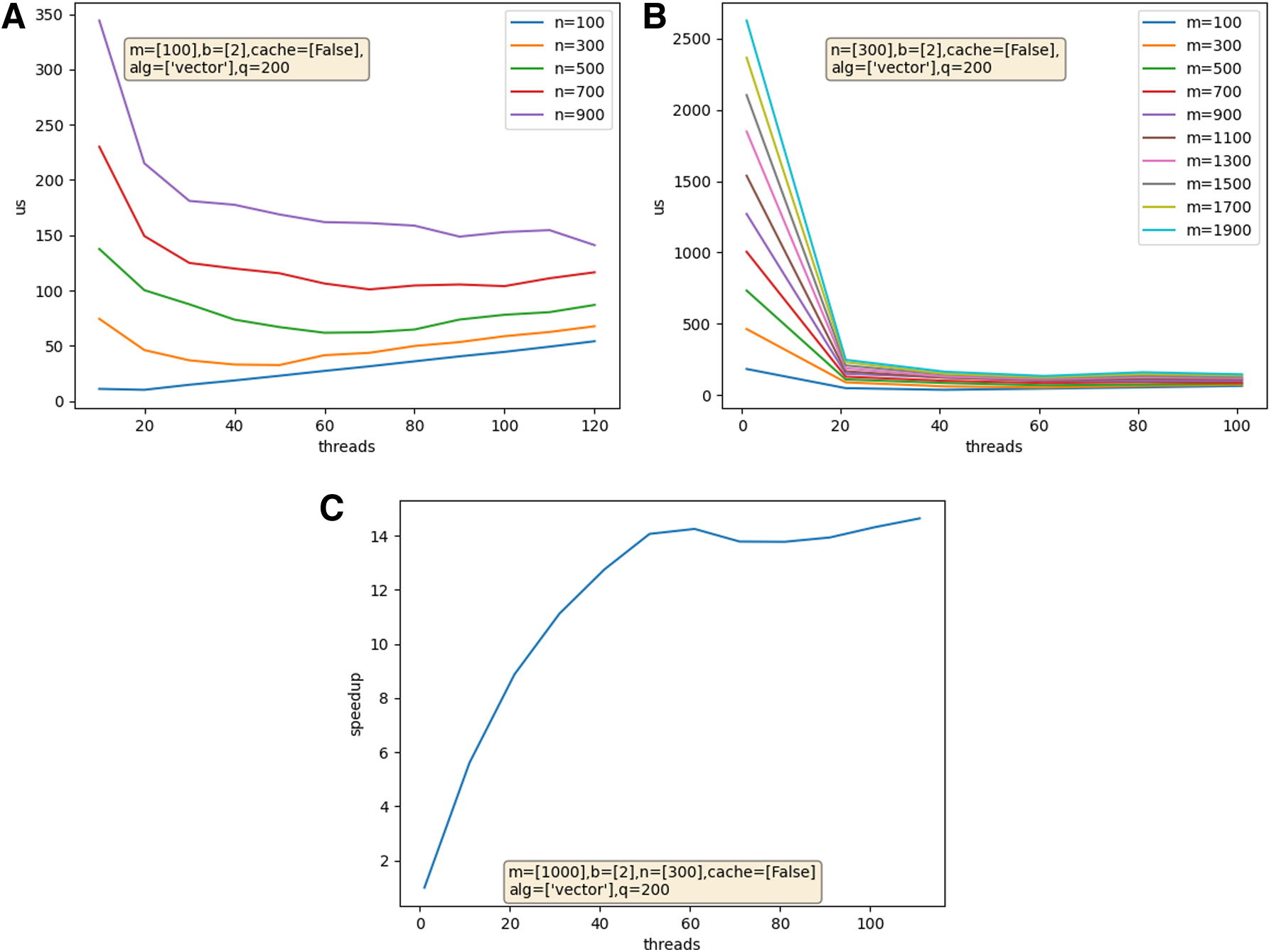
MIST runtime as thread count increases over various N (top, left) and M (top, right) values. The speedup over single core performance (bottom) shows the diminishing returns of thread counts >60. Timings were taken on a server equipped with 4 Intel Xeon Gold 6148 CPUS (total 80 core, 160 threads) and 250GiB RAM. Each measurement was repeated q = 200 times and the mean is used.
The number of entropy calculations is also reduced by reusing subcalculations in Symmetric Delta. MIST does this by keeping an in-memory cache of joint entropies to be reused in later computations. For example, the Symmetric delta of variables A, B, and C involve the joint entropies H(A,B), H(A,C), and H(B,C). If any of these entropies were previously computed, they are retrieved from the cache.
Finally, we can reduce the computation time of each entropy calculation by exploiting the computer architecture. In MIST's probability distribution “bitset” algorithm, each variable is recast as B bitsets of length M, where M is the number of samples and B is the number of bins (number of unique discrete values) of a variable. Each bitset bi corresponds to one value xi, where each bit is 1 when that sample has value xi and 0 otherwise. Then, each element in the probability distribution is the count of 1's in the bitswise AND combination of bitsets:
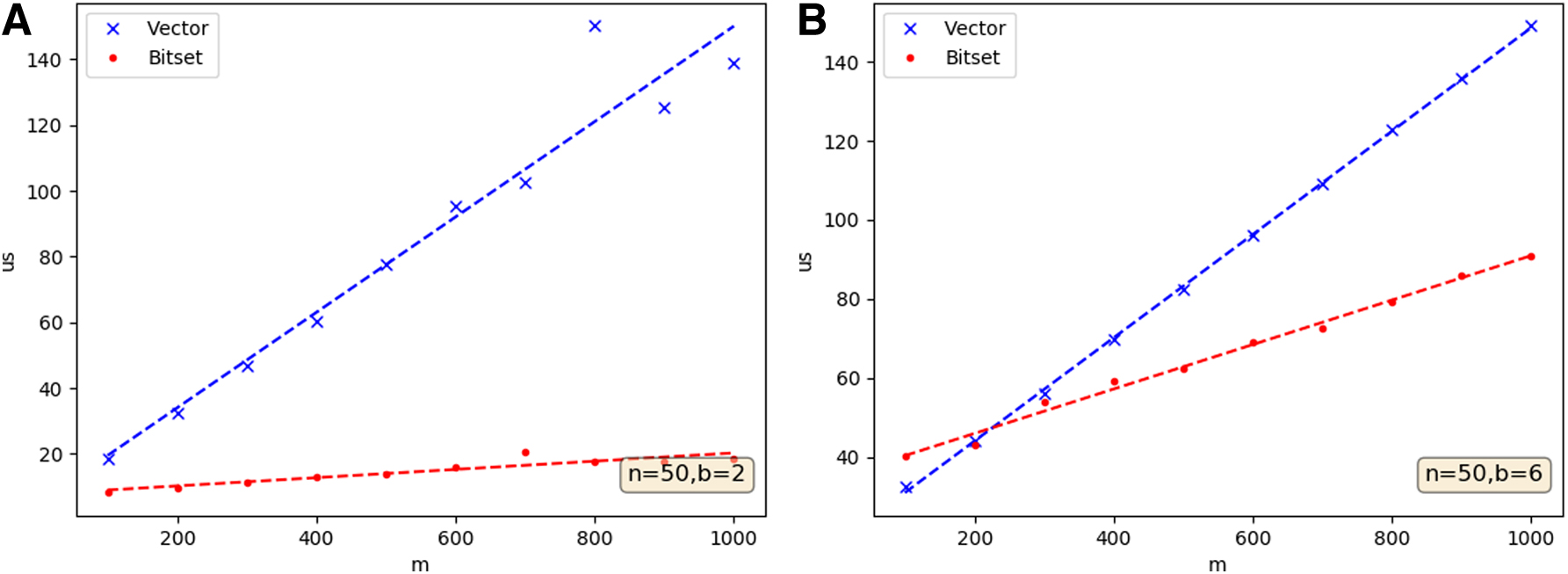
Keeping the number of tuples constant, the runtime is linear in M with a coefficient Bd, where d is the number of variables in a tuple, assuming all variables have the same number of bins. Thus, the bitset algorithm is excellent at speeding up searches over variables with few bins and many samples.
We illustrated MIST on generated test data and real AD diagnosis data from the UKBB.
Test data
The MIST GitHub repository includes a script to create sample data and run three small experiments. The test data contains 90 variables and is specially constructed to hold functional dependencies. These functional dependencies were detected by MIST: a strong f (X) = 2*X dependency between one third of the variables, a weaker dependency for the same function among another third of variables, and a three-way f(X,Y) = XOR(X,Y) among the remaining third. The expected results are packaged with the test data. See (Galas et al, 2021; Galas et al, 2014; Sakhanenko and Galas, 2015) for more examples of the MIST procedure.
Scripts to produce the performance measurements in Figures 1–3 are also included in the repository.
APOE modifiers in AD
We now present an example of how the measures of variable dependence can be used to establish relationships among variables in a dataset. The power of the measures is rather general and can be used in several ways, some of which we describe in a little more abstract, general way. In this study, we demonstrate a specific example by determining a set of modifier alleles of a known allele that predisposes to AD using the data of the UKBB. Note that the AD example is just that, an example of MIST application, and although it produced some biologically very interesting results, it was not our intention to be exhaustive in the analysis of AD so as not to detract from showcasing the MIST software. We piggyback on thorough and vast mathematical theory behind MIST that includes comparisons to other measures of variable relationships (Reshef et al, 2011) or the effects of data features, such as the case–control structure or the minor allele frequency, on the information measures (Ignac et al, 2014). Here is a brief description of the strategy: Suppose we know a pairwise connection of variables like the apoE4 SNP and the AD phenotype.
We can then use the three-way symmetric delta measure (we will call it D3 here) to find any variables that are codependent with these two. This is done by calculating D3 for the three variables (phenotype AD, SNP apoE4, and
From the UKBB, we retrieved 2778 individuals that were diagnosed with AD by selecting all individuals with a recorded first AD diagnosis occurrence (data field 131036: date ICD-10 code G30 reported). The AD phenotype variable is coded 1 for these individuals. For the control group, we selected random individuals with the same age and sex distribution as the AD set. The control group is coded 0 in the AD phenotype variable. Altogether there are 5556 individuals. Although in this example we chose to keep the cases and controls balanced, we could choose a different strategy depending on the problem, for example, we could increase the number of matched controls in an attempt to increase the association power. We have thoroughly reviewed the effects of the case–control structure of the data on the ability to detect a dependency in our previous publications (Ignac et al, 2014). We want to emphasize again that the choice of the case–control structure is problem dependent and is up to the research; MIST provides the vehicle to solve the problem.
There are 784,256 SNPs in the UKBB for the AD and control sets. Each SNP has M values and is coded 0, 1, or 2 for major, minor, and heterozygous alleles. The code-1 is given for subjects that do not have that SNP information in UKBB. Missing values create noise in the data, so we pruned away SNPs with more than 1000 missing values, leaving 767,107 SNPs. Then the data are composed of N = 767,107 + 1 variables, each with M = 5556 measurements.
We start our calculation by computing the pairwise Mutual Information between AD and every SNP. This yields genetic association results somewhat similar to GWAS. For the SNPs with high Mutual Information, we calculate a permutation-based p-value. Specifically, for each SNP we calculated Mutual Information with AD after a permutation such that the linkage between SNPs would be preserved, but the dependence with AD would be randomized. We repeated this process 10,000 times and estimated a p-value. More details on permutation testing can be found in our earlier publications (Ignac et al, 2014; Uechi et al, 2020). Although, we capped this process at 10,000 permutations to save resources, it can be increased to get a better estimate of a p-value (assuming we have enough data). Note that this p-value is not adjusted for multiple hypothesis testing, which is suitable for the exploratory analysis shown in our example. While we know that the APOE alleles should be high scorers, in this study, we carry out the full calculation to demonstrate the full use of MIST.
Table 1 shows SNPs from each chromosome with the strongest association with AD using the Mutual information. Note that 16 of these SNPs have a p-value of <0.0001. Table 2 shows the location and nearest genes for the strongest SNPs, and provides literature indicating the connections to AD.
Single Nucleotide Polymorphisms with the Highest Mutual Information with Alzheimer's Disease for Each Chromosome
Single Nucleotide Polymorphisms with the Highest Mutual Information with Alzheimer's Disease for Each Chromosome
Ordered by p-value. SNPs with p-value 0.0001 are ordered by mutual information value.
Single Nucleotide Polymorphisms with p-Value 0.0001, Their Nearest Genes and Their Relevance to Alzheimer's Disease
AD, Alzheimer's disease; SNPs, single nucleotide polymorphisms.
We use a TupleSpace object to limit the search from all possible variables to only tuples containing one SNP and the AD phenotype variable. This reduces the number of tuples to compute from the default 2.9 × 1011 to 7.6 × 105. The API calls to create this tuple space are:
ts = mist.TupleSpace()
g1 = ts.addVariableGroup(“snps”, snps)
g2 = ts.addVariableGroup(“ad”, ad)
ts.addVariableGroupTuple([g1,g2])
We then calculate a 3-way measure Delta (AD, APOE, SNP), where APOE corresponds to the SNP rs429358, the risk variant for AD. The 3-way measure is calculated for each SNP. We selected 200 SNPs with the largest Delta values and calculated unadjusted permutation-based p-values. Note that we permuted variables AD and APOE to test the hypothesis that we cannot achieve large Delta values with random pairwise dependencies among AD, APOE, and SNP. For more information about different strategies of permutation-based p-value estimation see Ignac et al, 2014. Table 3 shows SNPs with the strongest 3-way dependence with AD and APOE. Note that four of these modifier SNPs have a p-value of <0.0001. Note that these SNPs modify the dependency between the APOE SNP and the AD phenotype, so these are modifier alleles. Table 4 and Figure 4 show the location and nearest genes for the strongest SNPs, and notes the literature indicating the connections to AD.
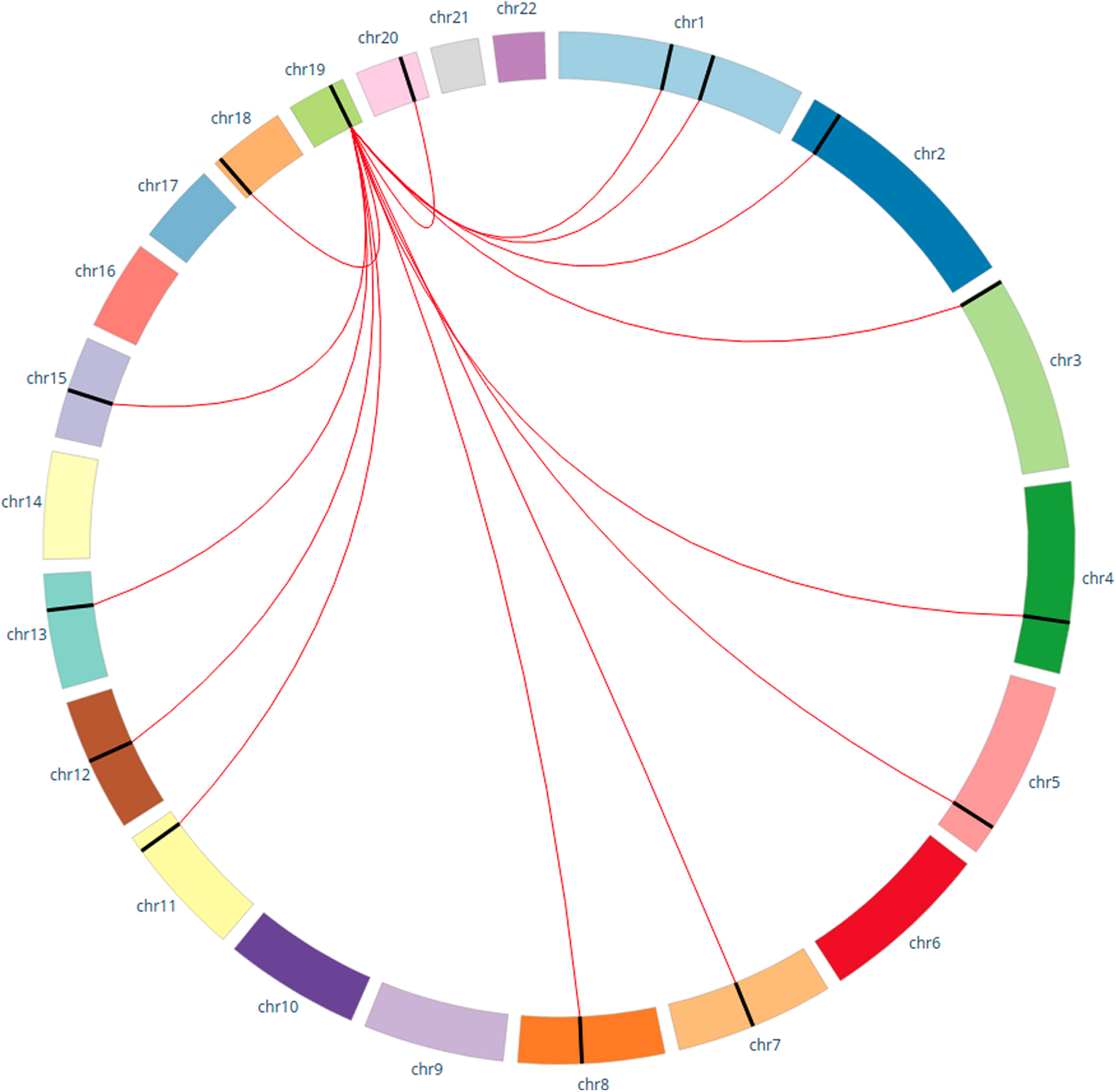
Modifier Single Nucleotide Polymorphisms with the Highest Delta3 Computed with Alzheimer's Disease and APOE and Lowest p-Value
Ordered by p-value. SNPs with p-value of 0.0001 are ordered by D3 values.
Delta3 (Modifier) Single Nucleotide Polymorphisms with p-Value 0.0001, Their Nearest Genes and Their Relevance to Alzheimer's Disease
Similar to the pairwise case, we use a TupleSpace to limit the search to 3-tuples containing one SNP variable, the APOE variable, and the AD variable. The API calls to create this tuple space are:
ts = mist.TupleSpace()
g1 = ts.addVariableGroup(“snps”, snps)
g2 = ts.addVariableGroup(“ad”, ad)
g3 = ts.addVariableGroup(“apoe”, apoe)
ts.addVariableGroupTuple([g1,g2,g3])
It is important to point out that there are many methods that aim to detect genetic interactions. For example, one could use standard logistic regression with an interaction term to model the modifier effect of an SNP on APOE. In this article, the dependencies were detected by MIST without any modeling assumptions, which allows us to detect not only all the linear effects, but also nonlinear ones that are not captured well by the standard methods. Moreover, MIST allows us to go beyond pairwise effects and detect dependencies among multiple variables (see more in the discussion section). There is a large body of research comparing the information theory-based measures with the standard methods, such as those based on correlation (Ignac et al, 2014; Reshef et al, 2011), and is outside the scope of our article.
The MIST software package provides tools to calculate measures of multivariable dependence based on the symmetric delta information theory measures (Galas et al, 2021; Galas et al, 2014). These can be used in many different ways to discover codependency links between variables in complex datasets. The power of the measures is very general. These measures can also be used to formulate some of the key relationships of quantitative genetics (Galas et al, 2021). We have demonstrated the power of MIST in a specific example by determining a set of modifier alleles of a known allele that predisposes to AD in the large dataset of the UKBB (Sudlow et al, 2015). Starting with a pairwise connection of variables like the causative apoE4 SNP and the AD phenotype. We then used the three-way measure (D3) to find any genetic SNP variables that are codependent with these two simply by calculating D3 for the three variables (phenotype AD, SNP apoE4, and any other SNP). These triplets were ranked by their D3 scores and the new SNPs included in the high scoring triplets with apoE4 SNP and AD are identified as codependent with them. These are classed as modifier genes/alleles as shown in Tables 3 and 4 and are known in the literature to represent additional AD-affecting genes as shown in the references.
Some of these identified genes represent interesting factors whose mechanistic, biological roles are yet to be determined. There are a number of other analyses besides the modifier allele discovery that can be performed with MIST. In Figure 5, we diagrammatically summarize the range of simple, direct analyses. To discover modifier alleles, we need to start with a genetic variant for which we wish to find modifiers. In our example we chose the known APOE allele, however, in principle any allele with a phenotypic effect can be used if we have a dataset that is appropriate. In Figure 5, Panel A, we depict the calculations schematically using the APOE4 example (find Allele APOE4, find high scoring triplets with APOE4, AD phenotype, and a modifier-called {Xi}). Going further we could, as shown in Panel B, search for
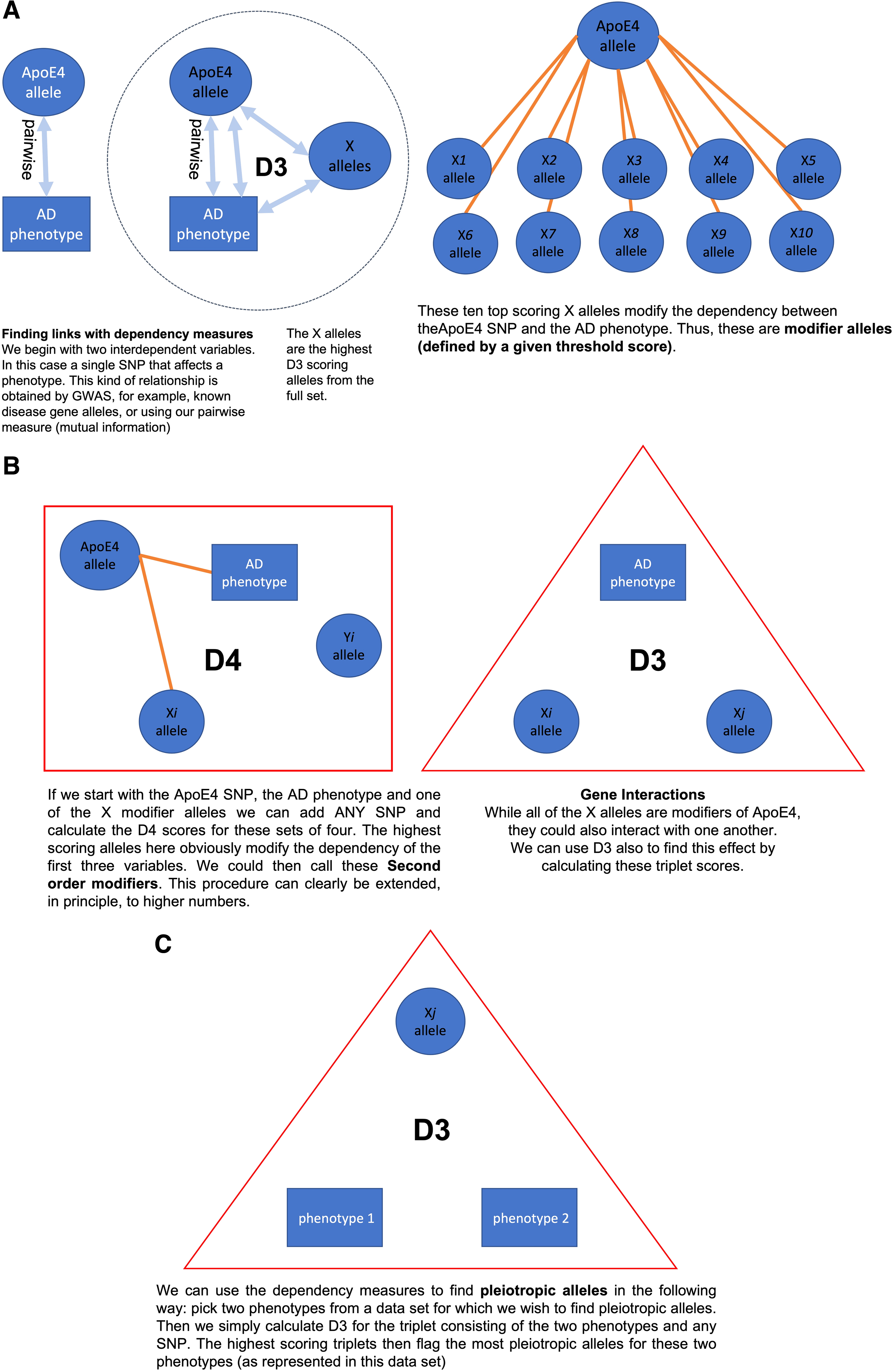
Schematic diagrams of the use of the dependency measures in several analyses of genetic data.
These high-scoring SNPs, {Yi}, we can term second-order modifiers. We might also look for interactions between any two SNPs from any of the above analyses, which is done by applying D3 calculations. Another biologically interesting analysis would be to start with two different phenotypes and search for a third variable, a SNP that induces a good D3 score (Fig. 5, Panel C). Such SNPs are pleiotropic for the two phenotypes. A little thought about possible multivariable dependencies can yield a number of biologically interesting genetic results. These analyses are certainly not limited to genetics. For example, datasets with phenotypes and biochemical or medical data can lead to interesting sets of biomarkers.
MIST is a general tool for analysis. The model-free search up to four dimensions allows for many kinds of analyses. MIST is designed to take on large problems; its parallel algorithm makes it cluster ready, and targeted search spaces help to filter noise from the results.
We utilized generated test data, and real AD diagnosis data from the UKBB. The MIST GitHub repository includes a script to create sample data and run three small experiments. The UKBB AD data comprised individuals with a recorded first AD diagnosis occurrence (data field 131036: date ICD-10 code G30 reported), and a control group of randomly selected individuals with the same age and sex distribution as the AD set. The UKBB data are subject to the legal limitations of a Material Transfer Agreement and may not be shared publicly. To contact UKBB (https://www.ukbiobank.ac.uk/) directly regarding data access, visit https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/contact-us and select “Enquire about access” to contact the Access Management Team.
The MIST source code is available under an MIT license on GitHub by the name mist. The main library is C++ and links to Boost libraries, such as the Boost::Python library to extend to the python language. MIST also includes a CLI program that can run simple searches and exemplifies the library API. Alternatively, MIST can be run from python using the libmist package available on PyPi or through a Docker image that may be built with the Dockerfile included with the source.
Future directions
The software can be improved in various ways. First, we may add more dependency measures to study relationships in other ways. The module software design allows for easy implementation of other measures to compute for each tuple. We are also interested in allowing the user to define their own measures, rather than having to implement them in the library.
The quality of MIST search results may be improved by including more statistical information along with raw signal strength. For example, provide the option to compute statistical measures, such as p-value, during the search rather than after the fact. This would help pare down results based on statistical significance rather than signal strength, which is not always the appropriate filter.
MIST can be further optimized by taking advantage of different computational platforms, such as GPU computing. We expect that the calculation of probability distributions could be translated into CUDA library operations. Optimizations in this module could yield a large benefit in runtime.
Footnotes
ACKNOWLEDGMENT
AUTHORs' CONTRIBUTIONS
A.B.: software (lead) and writing—review and editing (equal). N.A.S.: formal analysis (equal) and writing—review and editing (equal). J.K.-G.: formal analysis (equal) and writing—review and editing (equal). D.J.G.: conceptualization (lead); writing—original draft (lead); writing—review and editing (equal); funding acquisition (lead); and supervision (lead).
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was partially funded by NIH Award no. 1U54DA049098-01, The Bill and Melinda Gates Foundation (Award no. OPP1126103), and the Pacific Northwest Research Institute.