
Abstract
Resolving haplotypes in polyploid genomes using phase information from sequencing reads is an important and challenging problem. We introduce two new mathematical formulations of polyploid haplotype phasing: (1) the min-sum max tree partition problem, which is a more flexible graphical metric compared with the standard minimum error correction (MEC) model in the polyploid setting, and (2) the uniform probabilistic error minimization model, which is a probabilistic analogue of the MEC model. We incorporate both formulations into a long-read based polyploid haplotype phasing method called flopp. We show that flopp compares favorably with state-of-the-art algorithms—up to 30 times faster with 2 times fewer switch errors on 6 × ploidy simulated data. Further, we show using real nanopore data that flopp can quickly reveal reasonable haplotype structures from the autotetraploid Solanum tuberosum (potato).
1. INTRODUCTION
As genomic sequencing technologies continue to improve, we are increasingly able to resolve ever finer genomic details. Case in point, traditional genotyping only determines whether a particular allele is present in a genome (Scheben et al., 2017). However, when organisms are polyploid (and most eukaryotic organisms are), they have multiple copies of each chromosome. We are then additionally interested in the problem of resolving haplotypes, that is, determining the sequence of alleles on each specific chromosome and not just the presence of an allele within the genome. Phasing is the procedure of resolving the haplotypes by linking alleles within a chromosome (Browning and Browning, 2011).
We focus on phasing polyploid organisms by using third-generation sequencing data. Many plants have ploidy greater than two (i.e., have more than two copies of each chromosome), such as tetraploid potatoes (Solanum tuberosum) or hexaploid wheat and cotton. Haplotype phasing has been used to gain insights into evolution (Eriksson et al., 2018), breeding (Qian et al., 2017), and genome-wide association studies (Maldonado et al., 2019), among other applications.
The most common way of determining haplotypes is to use pooled genotyping information from a population to estimate haplotypes (Browning and Browning, 2011). For unrelated individuals, sophisticated statistical methods are used to determine the most likely haplotypes for each individual (Browning and Browning, 2007; Howie et al., 2009; Delaneau et al., 2019) in the population. For related individuals, identity-by-descent information can be used for haplotype phasing (Gao et al., 2009; Motazedi et al., 2018). However, these types of methods do not work on single individuals because they rely on having population data available.
Instead, in this work, we adopt the approach of single individual phasing by sequencing, which is now common in phasing human haplotypes (Choi et al., 2018). We focus on using sequencing information for phasing, which allows us to phase a single individual without population information or prior haplotype knowledge. This is closely related to genome assembly where overlapping reads are stitched together (Nagarajan and Pop, 2013); in our case, nearby heterozygous alleles are stitched together by read information. For the rest of the article, we use the term “phasing” to mean single individual phasing using sequencing information.
1.1. Related work
The first method for phasing polyploid genomes was HapCompass (Aguiar and Istrail, 2012), which uses a graphical approach. Popular methods that followed include HapTree (Berger et al., 2014, 2020), H-PoP (Xie et al., 2016), and SDhaP (Das and Vikalo, 2015). HapTree and H-PoP heuristically maximize a likelihood function and an objective function based on the minimum error correction (MEC) model, respectively whereas SDhaP takes a semi-definite programming approach. HapTree-X (Berger et al., 2020) additionally incorporates long-range expression correlations to allow phasing even of pairs of variants that cannot be covered by a single read, overcoming some of the problems with short-read phasing.
Due to the increased prevalence of long-read data from Oxford Nanopore or PacBio, newer methods taking advantage of the longer-range correlations that are accessible through long-read data have been proposed (Schrinner et al., 2020; Abou Saada et al., 2021). Unfortunately, because the error profiles of long-read technologies differ considerably from Illumina short-reads [e.g., a higher prevalence of indel errors compared to single nucleotide polymorphisms (SNPs)]. Methods tailored to short-reads (Siragusa et al., 2019; Moeinzadeh et al., 2020) may be ineffective, so altogether new paradigms are required.
At a more theoretical level, in the diploid setting, the standard MEC model (Bonizzoni et al., 2016) has proven to be quite powerful. It is known to be APX-Hard and NP-Hard but heuristically solved in practice with relatively good results. Unfortunately, a good MEC score may not imply a good phasing when errors are present (Majidian et al., 2020). This shortcoming is further exacerbated in the polyploid setting, because similar haplotypes may be clustered together since the MEC model does not consider coverage; this phenomenon is known as genome collapsing (Schrinner et al., 2020). Thus, although the MEC model can be applied to the polyploid setting, it may be suboptimal; however, there is yet to be an alternative commonly agreed-upon formulation of the polyploid phasing problem. Indeed, this is reflected in the literature: The mathematical underpinnings of the various polyploid phasing algorithms are very diverse.
1.2. Contributions
In this article, we first address the theoretical shortcomings highlighted earlier by giving two new mathematical formulations of polyploid phasing. We adopt a probabilistic framework that allows us to (1) give a better notion of haplotype similarity between reads and (2) define a new objective function, the uniform probabilistic error minimization (UPEM) score. Further, we introduce the idea of framing the polyploid phasing problem as one of partitioning a graph to minimize the sum of the max spanning trees within each cluster, which, we show, is related to the MEC formulation in a specific case.
We argue that these formulations are better suited for polyploid haplotype phasing using long-reads. In addition to our theoretical justifications, we also implemented a method we call flopp (
The code for flopp is available at https://github.com/bluenote-1577/flopp. flopp utilizes Rust-Bio (Kster, 2016), is written entirely in the rust programming language, and is fully parallelizable. flopp takes as input either binary alignment map + variant call format (BAM + VCF) files, or the same fragment file format used by AltHap (Hashemi et al., 2018) and H-PoP.
2. METHODS
2.1. Definitions
We represent every read as a sequence of variants (rather than as a string of nucleotides, which is commonly used for mapping/assembly tasks). Let R be the set of all reads that align to a chromosome and m be the number of variants in our chromosome. Assuming that tetra-allelic SNPs are allowed, every read ri is considered as an element of the space
We note that flopp by default only uses SNP information, but the user may generate their own fragments, permitting indels and other types of variants to be used even if there are more than four possible alleles. The formalism is the same regardless of the types of variants used or the number of alternative alleles.
For any two reads
d and s stand for different and same, representing the number of different and same variants, respectively, between two reads.
We use k to denote the ploidy. Given a k-ploid organism, a natural approach to phasing is to partition R into k subsets where the cluster membership of a read represents which haplotype the read belongs to. Let
Define the consensus haplotype
In our formalism, we can phrase the MEC model of haplotype phasing as the task of finding a partition
2.2. Problem formulation
2.2.1. min-sum max tree partition model
Let
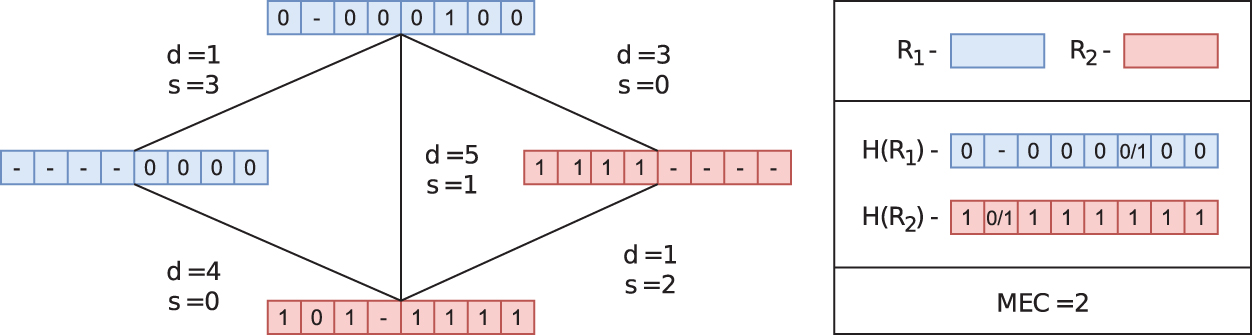
An example of a read-graph (without the weighting w specified) along with corresponding
For a partition of R into disjoint subsets
We formulate the min-sum max tree partition (MSMTP) problem as finding a valid partition
The MSMTP problem falls under a class of problems called graph tree partition problems (Cordone and Maffioli, 2004), most of which are NP-Hard. The following proof shows that MSMTP is NP-Hard.
Proof. Let
Let
Let
If
For the other implication, clearly if a
Therefore, any algorithm that solves MSMTP also decides whether G has a k-coloring, which is NP-Complete for
Intuitively, assuming each
Therefore,
Proof. Take the augmented graph
Now note that
Removing the node
The theorem just cited relies on the assumption that all reads in the set overlap exactly. Although this is obviously not true for the entire genome, flopp takes a local clustering approach where the entire read set R is partitioned into smaller local read sets that overlap significantly.
We verified experimentally that the SMTP score for partitions generated by our method has a strong linear dependence on the MEC score when
2.2.2. UPEM model
The SMTP score has problems with collapsing genomes in the same manner the MEC score does; it does not take into account the assumption that coverage should be uniform between haplotypes. Concretely, if a polyploid organism has two identical haplotypes, the reads from both haplotypes may be clustered together in the MEC model and a noisy read may instead be placed in its own cluster.
Let
The
The UPEM score is a probabilistic version of the MEC model under the hypothesis that the errors and coverage are uniform across haplotypes. The parameter
The UPEM maximization enforces a relatively uniform partition. Further, errors will be distributed among partitions equally due to the non-linearity of the UPEM score; if one cluster is extremely erroneous, the sum of the binomial terms may be higher for a more spread out error even if the overall MEC score is slightly higher. If error and coverage uniformity assumptions are satisfied, clearly these two properties are desirable in an objective function.
2.3. Local graph clustering
We now discuss the algorithms implemented in flopp. A high-level block diagram showing outlining flopp's main processing stages is outlined in Figure 2. Importantly, flopp is a local clustering method, which means that we first attempt to find good partitions for smaller subsets of reads by optimizing the SMTP and UPEM functions and then joining haplotype blocks together afterward. A graphic outlining the local clustering and linking procedure can be found in Figure 3.
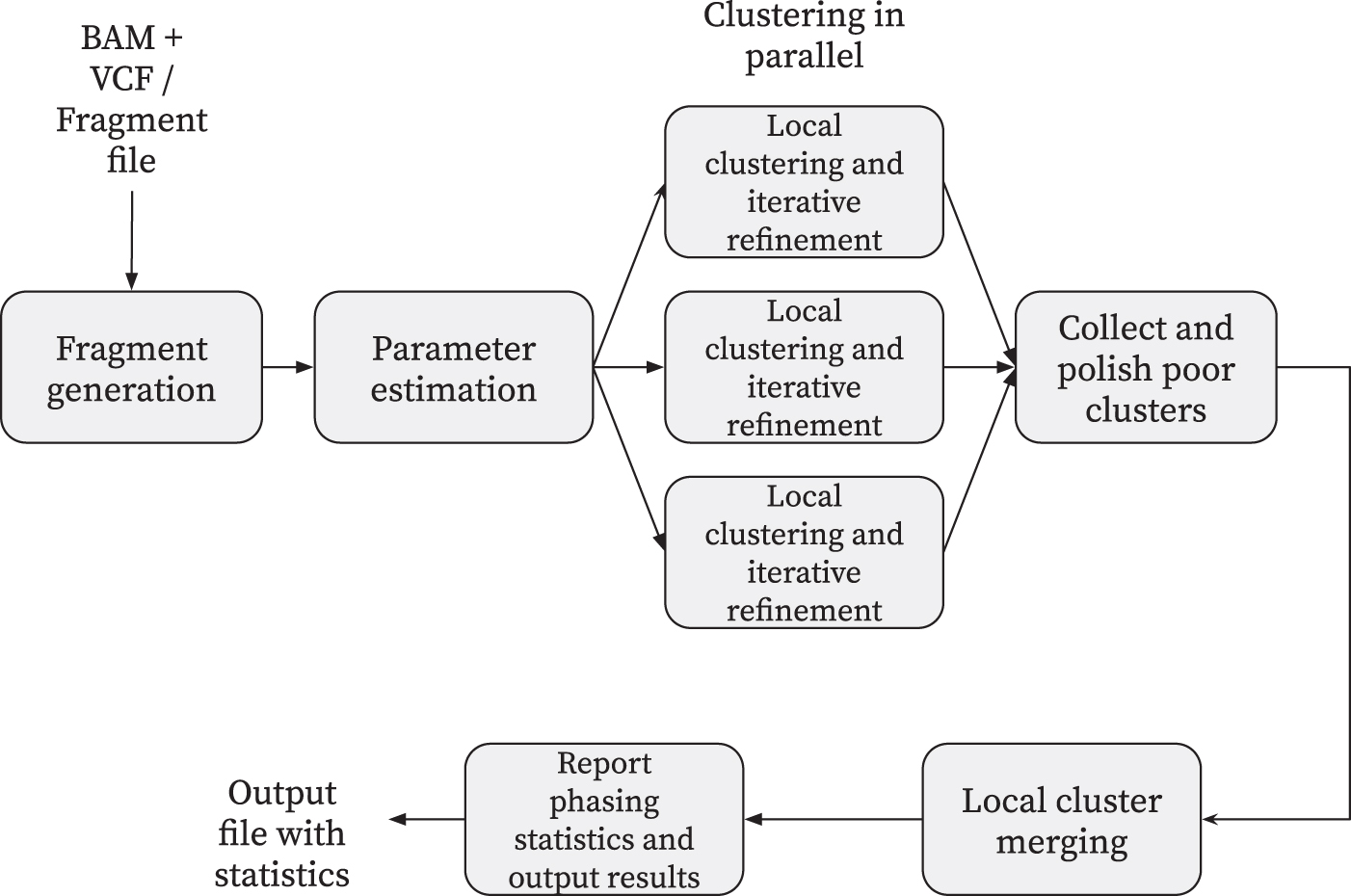
A block diagram outlining flopp's major processing steps. flopp takes BAM+VCF files or a fragment file as input. The fragments are clustered into local haplotype blocks, which are then polished and merged. The final phasing is output to the user as well as various phasing statistics. BAM, binary alignment map; VCF, variant call format.
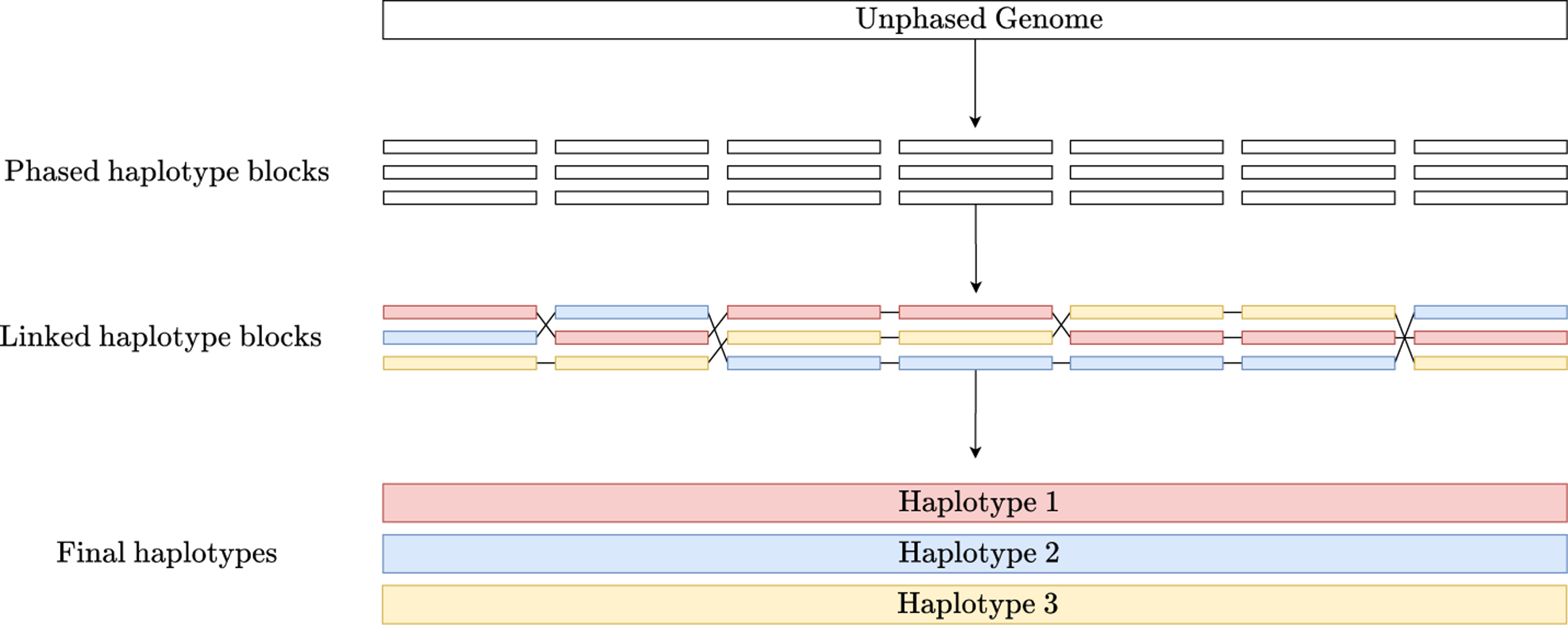
A visual example of the local clustering and linking procedure. flopp breaks up the unphased genome into chunks. The associated reads to each chunk are locally phased and then linked.
2.3.1. Choice of edge weight function
Previous methods that use a read-graph formalism such as SDhaP (Das and Vikalo, 2015), FastHap (Mazrouee and Wang, 2014), WhatsHap Polyphase (WHP) (Schrinner et al., 2020), and nPhase (Abou Saada et al., 2021) all define different weightings between reads. Two previously used weight functions are
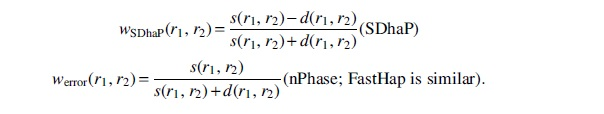
These weightings are quite simple and have issues when the lengths of the reads have high variance, as is the case with long-reads. A more sophisticated approach is to use a probabilistic model. Let
There is a nice interpretation of
In particular, this bound is asymptotically tight up to logarithms, that is,
The proof of the theorem cited earlier is simple; it follows by an application of a Chernoff bound and Sterling's inequality. Arratia and Gordon (1989) show that this approximation is particularly good when
WHP (Schrinner et al., 2020) uses a log ratio of binomial probability densities as their edge weight. Mathematically, the resulting formula is almost the same as Equation 2, except they fix
2.3.2. MSMTP algorithm
We devised a heuristic algorithm for local clustering inspired by the MSMTP problem. From now on, let
For the FindMaxClique method mentioned in Algorithm 1, we use a greedy clique-finding method that takes
The complexity of the local clustering algorithm is
The connection to MSMTP is at line 10. A priori, it is not obvious what metric to use to determine which cluster to put the vertex in. For Kruskal's algorithm, one starts by considering the heaviest edges, so we decided to minimize the maximum edge from the vertex to the cluster so that the heaviest edges are small. Intuitively, a maximum spanning tree is sensitive to a highly erroneous edge, so we prioritize minimizing the worst edge even if on average the vertex connects to the cluster well.
2.3.3. Iterative refinement of local clusters
We refine the output of the local clustering procedure by optimizing the UPEM score using a method similar to the Kernighan-Lin algorithm (Kernighan and Lin, 1970). Pseudocode can be found in Algorithm 2.
In lines 4–14, we check how swapping vertices between partitions affects the overall UPEM score and take a fraction of the best swaps in line 14. We then execute the swaps and check whether the UPEM score has increased. If it has not, we terminate the algorithm; otherwise, we continue until n iterations have passed. We take
2.3.4. Local phasing procedure
Note that Algorithms 1 and 2 work on subsets of reads or subgraphs of the underlying read-graph. Let
The subsets are just all reads that overlap a shifted interval of size b, similar to the work done in Sankararaman et al. (2020). After choosing a suitable b, we run the read-partitioning and iterative refinement on all
It is important to note that computationally, the local clustering procedure is easily parallelizable. The local clustering step has, therefore, a linear speedup in the number of threads.
2.4. Polishing, merging, and parameter estimation
2.4.1. Filling in erroneous blocks
Once we obtain a set of partitions
For genomes with large variations in coverage, we give the user the option to disable this procedure because variation in coverage may affect the UPEM score distribution.
2.4.2. Local cluster merging
Let P represent the final partition of all reads R. We build P given
Then, let
We experimented with more sophisticated merging procedures such as a beam-search approach but did not observe a significantly better phasing on any of our datasets.
2.4.3. Phasing output
Once a final partition P has been found, we build the final phased haplotypes. The output is k sequences in
We constrain the haplotypes using the VCF as follows. For every variant indexed over
For the function
2.4.4. Parameter estimation
We already mentioned in previous sections how we set all parameters for algorithms except for
To estimate
3. RESULTS AND DISCUSSION
3.1. Phasing metrics
There are a plethora of phasing metrics developed for diploid and polyploid phasing (Motazedi et al., 2017). We use three different metrics of accuracy, but argue that each individual metric can be misleading and that all three metrics should be used in unison before drawing conclusions on phasing accuracy.
For a global measure of goodness of phasing, we use the Hamming error rate. Given a set of true haplotypes
where Sk is the set of permutations on k elements, m is the length of each
We define the switch error rate (SWER) similarly to WHP (Schrinner et al., 2020). Let
where
The Hamming error, thoughe easily interpretable, can be unstable. A single switch error can drastically alter the Hamming error rate. The SWER also has issues; for example, if two switches occur consecutively, the phasing is still relatively good but the SWER is worse than if only one switch occurred.
We define a new error rate, called the q-block error rate. For a haplotype 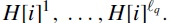 . For a set of haplotypes H, doing this for every haplotype gives a collection of haplotype blocks
. For a set of haplotypes H, doing this for every haplotype gives a collection of haplotype blocks
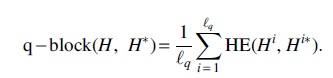
The q-block error rate measures local goodness of assembly and interpolates between the Hamming error rate, when q is the size of the genome, and a metric similar to the switch error when
3.2. Simulation procedure
We used the v4.04 assembly of S. tuberosum produced by the Potato Genome Sequencing Consortium (Hardigan et al., 2016) as a reference. We took the first 3.5 Mb of chromosome 1 and removed all “N”s, leaving about 3.02 Mb and simulated bi-allelic SNPs by using the haplogenerator software (Motazedi et al., 2017), a script made specifically for realistic simulation of variants in polyploid genomes.
We generated SNPs with a mean distance of 45 and 90 bp between SNPs. This is in line with the 42.5 bp average distance between variants, as seen in Uitdewilligen et al. (2013) for S. tuberosum; in that study, they observe that
We used two different software packages for simulating reads. We used PaSS (Zhang et al., 2019) with the provided default error profiles for simulating PacBio RS reads. PaSS has higher error rates than other methods such as PBSIM (Ono et al., 2013), which tends to underestimate error (Zhang et al., 2019). We used NanoSim (Yang et al., 2017a) for simulating nanopore reads by using a default pre-trained error model based on a real human dataset provided by the software.
After generating the haplotypes and simulating the reads from the haplotypes, we obtain a truth VCF from the haplotypes. We map the reads by using minimap2 (Li, 2018) to the reference genome. The scripts for the simulation pipeline can be found at https://github.com/bluenote-1577/flopp_test.
3.3. SMTP versus MEC correlation
We ran flopp on four different simulated datasets and calculated the SMTP score with the weight function
We varied the coverage between 10 × to 20 × for a simulated 4 × ploidy genome. We also varied the length of the local partition blocks by manually changing the parameter b mentioned at the end of Section 2.3 over three different values (20, 50, and 80 SNPs) for each different dataset to investigate how the size of the local clusters affects the SMTP and MEC relationship. The results are shown in Figure 4.
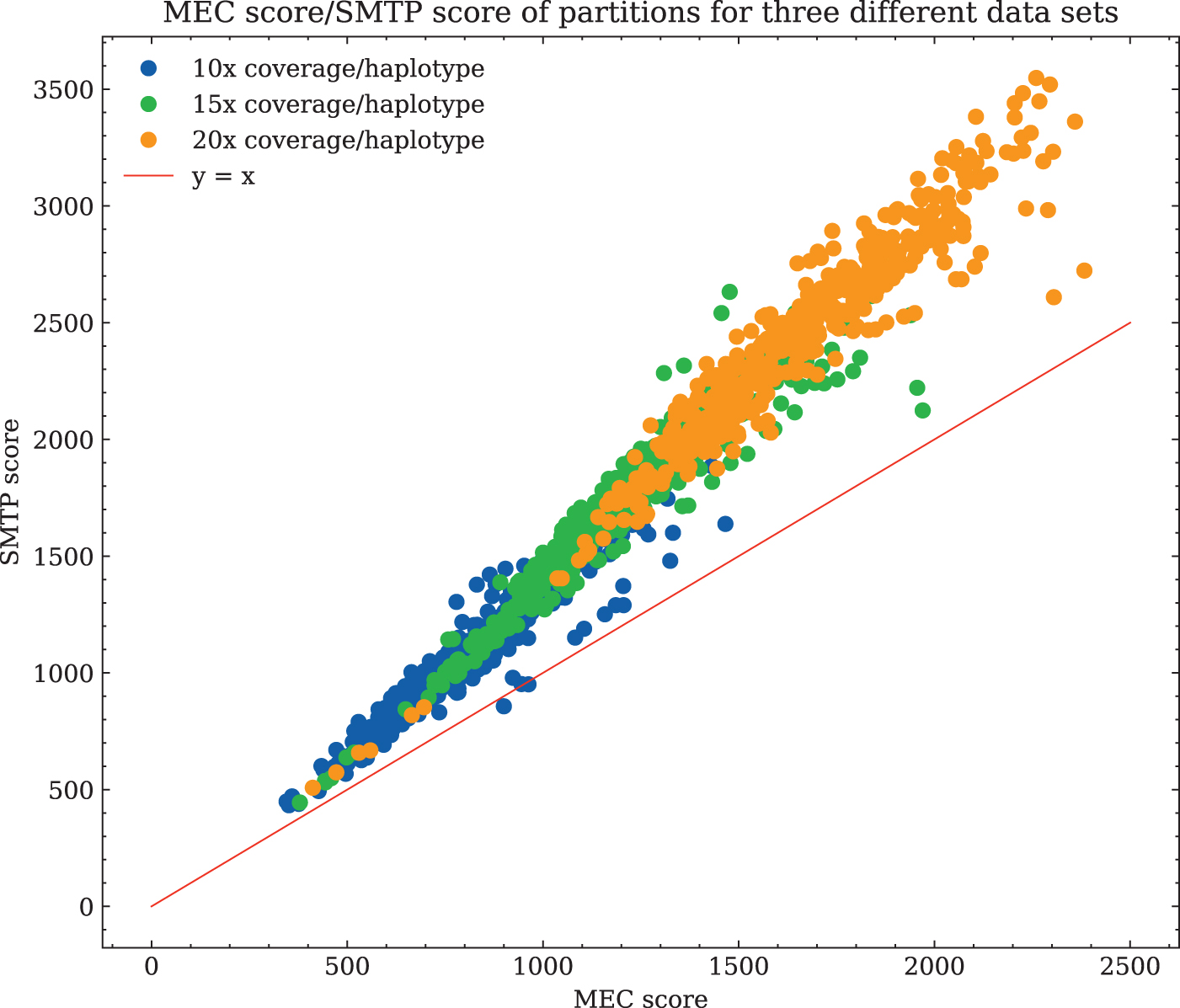
A plot of the SMTP versus MEC score for every local partition (i.e., partition blocks before merging/linking) generated by flopp over three different coverages on 4 × ploidy simulated data, as described in Section 3.2. There is a strong linear relationship between MEC and SMTP scores, and the upper bound for Theorem 2 holds surprisingly well as seen by almost all points being above the line
3.4. Results on simulated data set
We primarily test against H-PoPG (Xie et al., 2016), the genotype constrained version of H-PoP. Other methods such as HapTree and AltHap were tested, but we ran into issues with either computing time or poor accuracy due to the methods not being suited for long-read data. We did not test against nPhase, because the output of nPhase does not have a fixed number of haplotypes. We discuss WHP (Schrinner et al., 2020) at the end of this section.
The switch error and Hamming error rates are shown in Figure 5 for 45 bp average distance between SNPs. For 90 bp, the results are shown in Figure 6. The rest of the analysis in the section pertains to the 45 bp distance case.
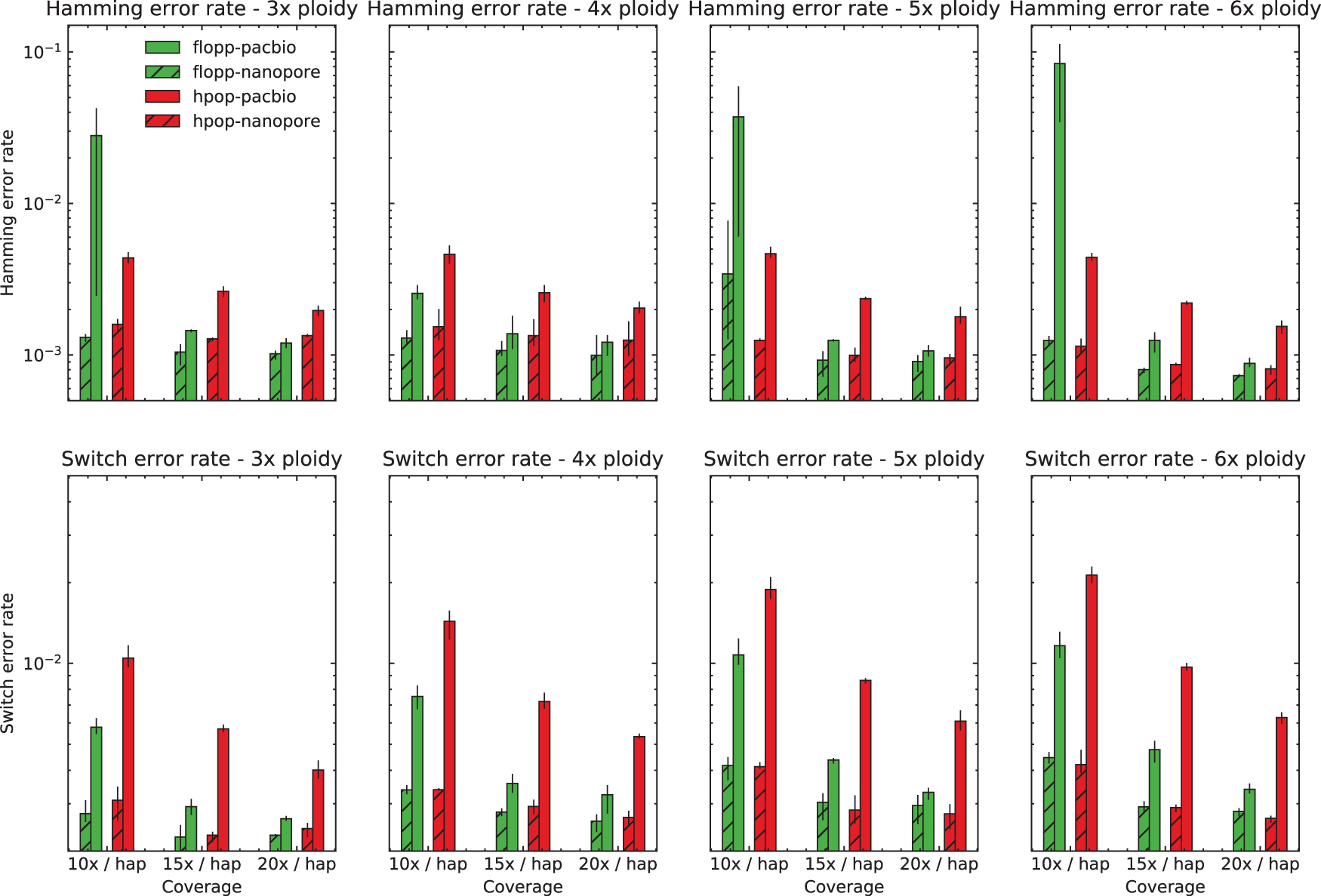
The mean SWER and Hamming error rate from testing on simulated data sets as described in Section 3.2 over a range of ploidies and coverages on two different types of read simulators, with SNPs 45 bp apart on average. The error bars represent the lowest and highest values for the metric over three iterations. The results on the nanopore simulated reads from NanoSim (Yang et al., 2017a) are similar for both methods. flopp achieves much better SWERs on the PacBio simulated reads from PaSS (Zhang et al., 2019), although for low coverage flopp sometimes has a higher Hamming error rate. SNPs, single nucleotide polymorphisms; SWER, switch error rate.
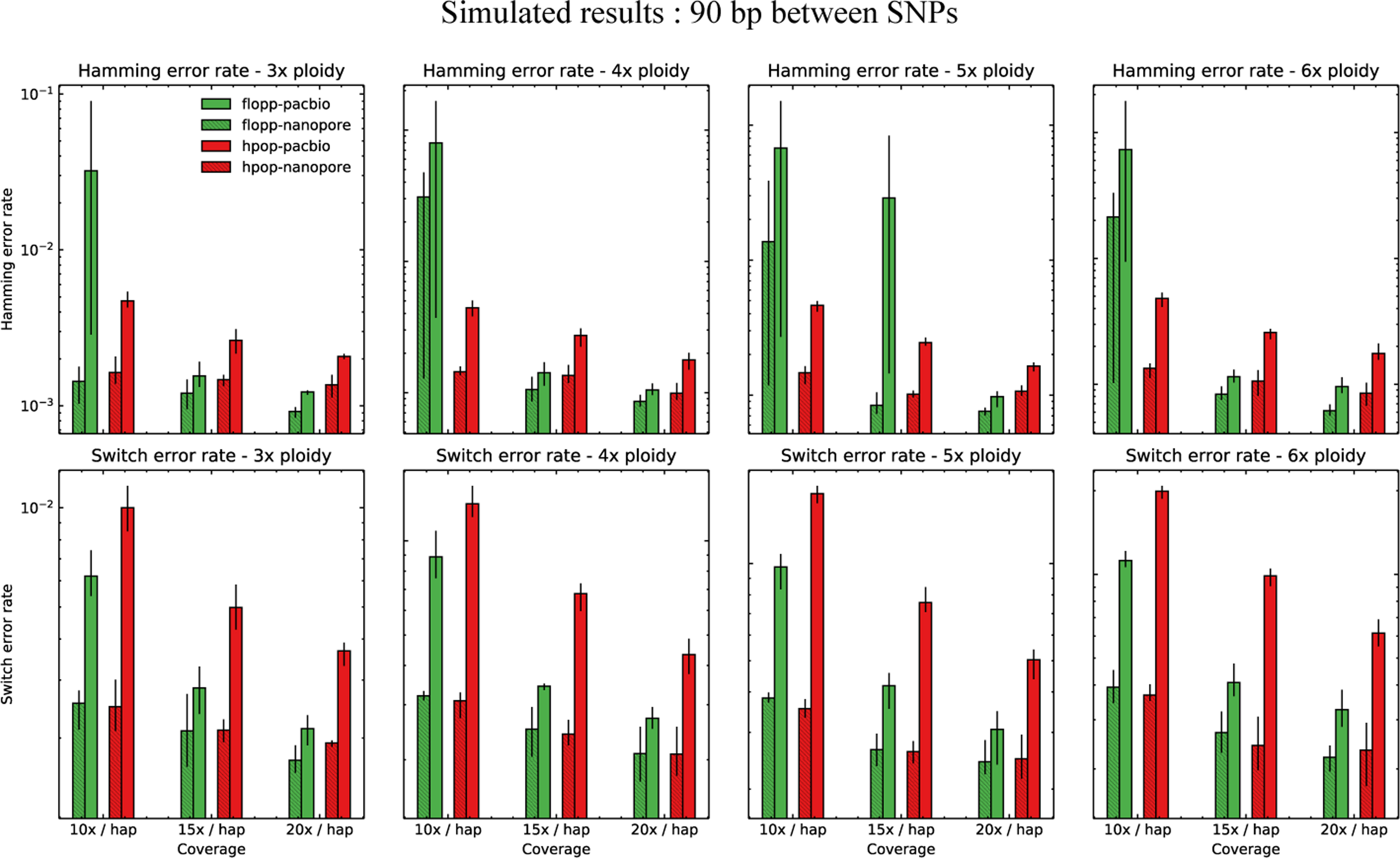
The mean SWER and Hamming error rate from testing on simulated datasets as described in Section 3.2. The same experiment as shown in Figure 5 except 90 bp between SNPs on average.
For each test, we ran the entire pipeline three times; each run at high ploidies takes on the timescale of days to complete. The run times on PacBio reads for H-PoPG, flopp, as well as one instance of WHP on 3 × ploidy data are shown in Figure 7.
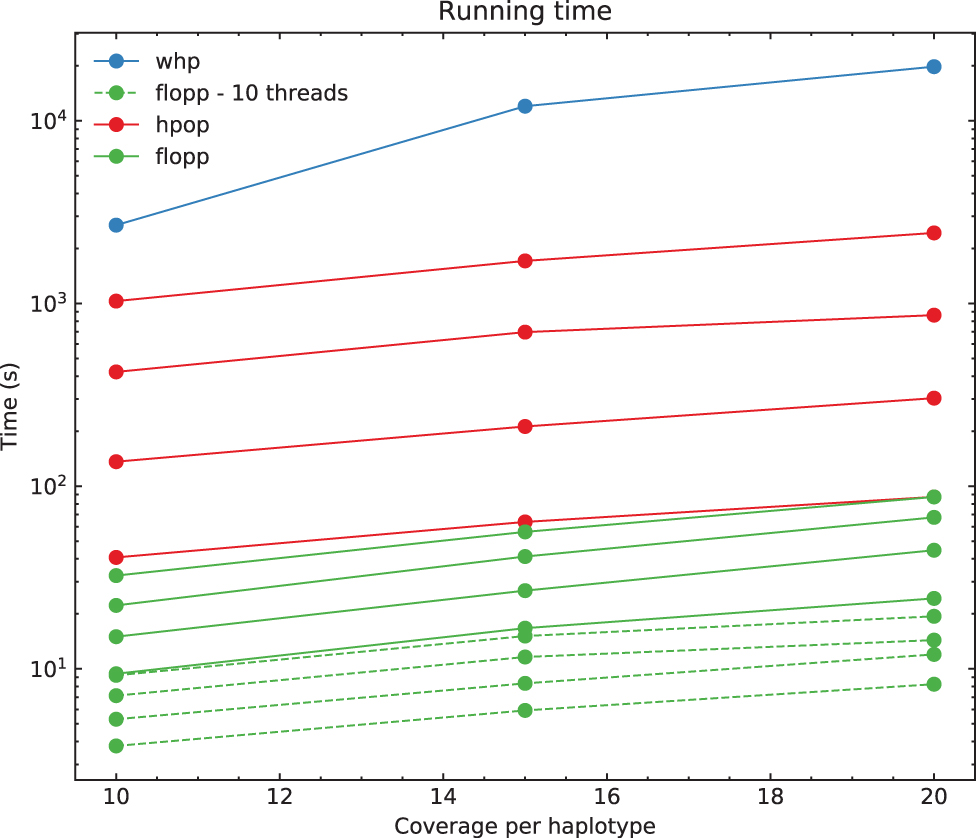
Run times for flopp, H-PoPG, and one instance of WHP. Each line represents a different ploidy, with higher ploidies taking longer. The one instance of WHP was run at 3 × ploidy. H-PoPG; WHP, WhatsHap polyphase.
For the nanopore dataset simulated from NanoSim, the results for H-PoPG and flopp are very similar. The PaSS PacBio simulator outputs reads that are more erroneous, and we can see that flopp generally performs better than H-PoPG across ploidies except for the Hamming error rate when the coverage is relatively low; interestingly, the SWER is still lower in this case. flopp's SWER is consistently 1.5–2 times lower than H-PoPG for the simulated PacBio reads. On the low coverage datasets, H-PoPG's global optimization strategy leads to a better global phasing than flopp's local methodology.
Note that in these tests we phase a contig of length 3.02 Mb, whereas most genes of interest are smaller. In Figure 8, we plot the mean q-block error rates of the 5 × and 6 × ploidy phasings at 10 × coverage; these runs have higher Hamming error rates for flopp than H-PoPG. For blocks of up to around
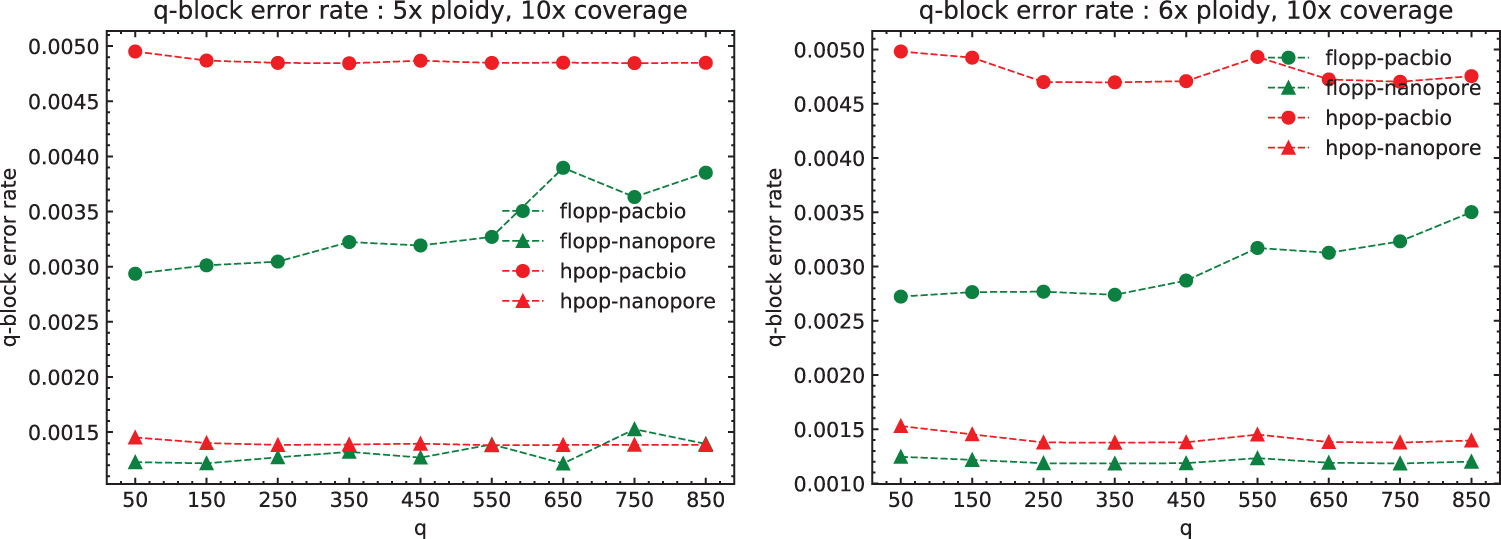
The mean q-block error rates for the 5 × and 6 × ploidy datasets at 10 × coverage per haplotype over three iterations. Each variant is spaced on average 45 bp apart, so each block is of size
Computationally, Figure 7 shows that flopp is at least 3 times faster than H-PoPG and up to 30 times faster than H-PoPG for 6 × ploidy on a single thread. The local clustering step sees a linear speedup from multi-threading. For most datasets, we get a 3–4 times speedup with 10 threads.
We tried testing against WHP (Schrinner et al., 2020), but we found that the accuracy was relatively poor across our datasets and it took a long time to run; see Figure 7. Using the default block-cut sensitivity value, the N50 did not exceed 20 for the 67,000 variants on the simulated contig. We found that WHP takes a conservative approach as
3.5. Phasing tetraploid potato using real nanopore data
We ran flopp on real nanopore data sequenced from an autotetraploid potato (S. tuberosum) specimen generated from Schrinner et al. (2020). We used the DM v6.1 S. tuberosum assembly from Pham et al. (2020) as our reference, and we called SNPs using freebayes (Garrison and Marth, 2012) from short-reads that were also obtained from the same specimen. We aligned all reads with minimap2. We ran flopp on chromosome 2 in the assembly, which was 46 Mb long and had an SNP heterozygosity of 2.0%, that is, 50 bp average distance between SNPs.
flopp only took 1050 seconds to complete on 10 threads. The WHP took 11,492 seconds on the same dataset, and H-PoPG took 8321 seconds. We show two examples of flopp's phasings on two genes in Figures 9 and 10, the first an example of a successful phasing and the second an example of the difficulties of phasing collapsed haplotypes.
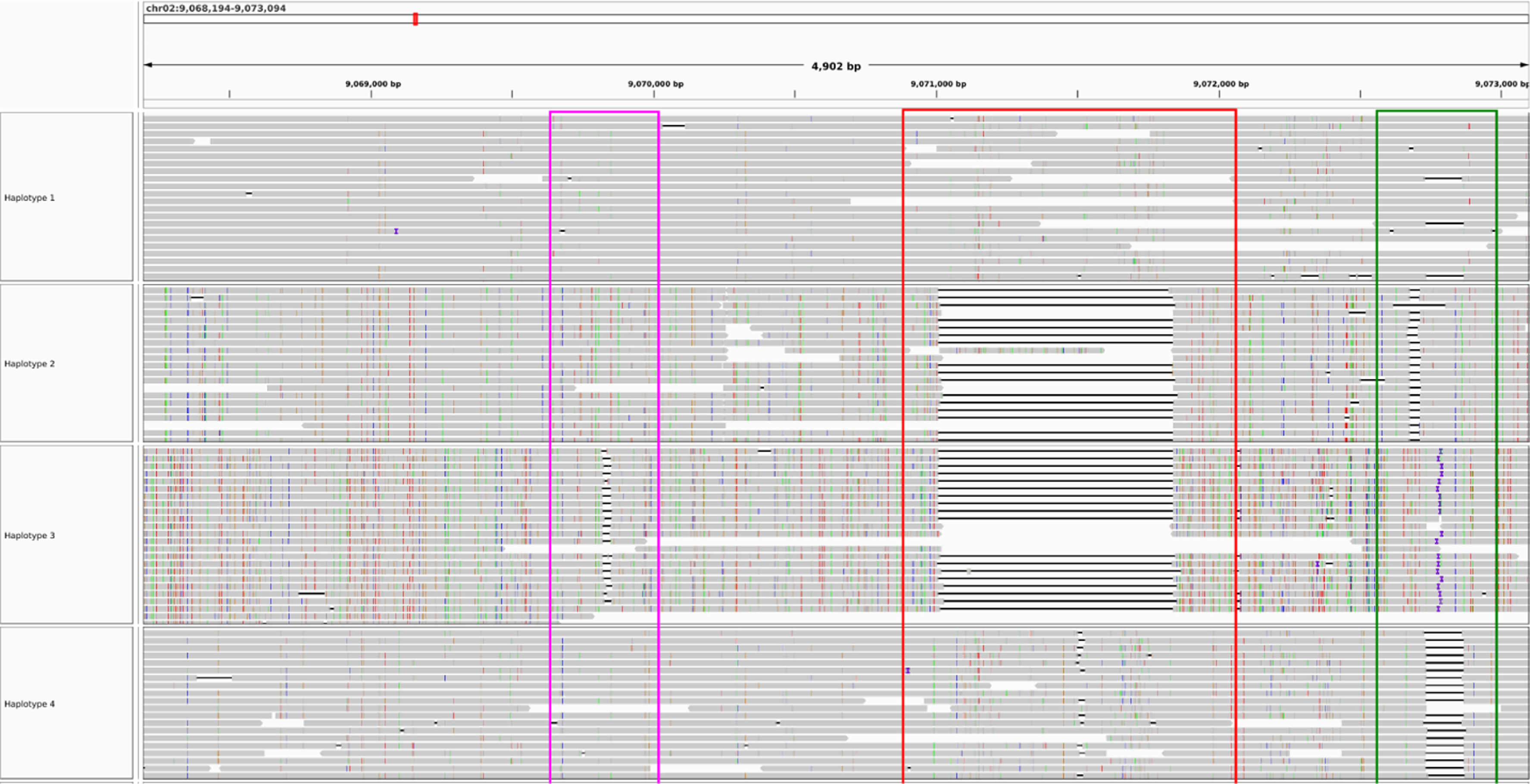
flopp's phasings on a 4902 bp gene. The colored boxes outline structural variations. The clusterings of the structural variations are concordant, implying that the partitioning of reads works reasonably well.
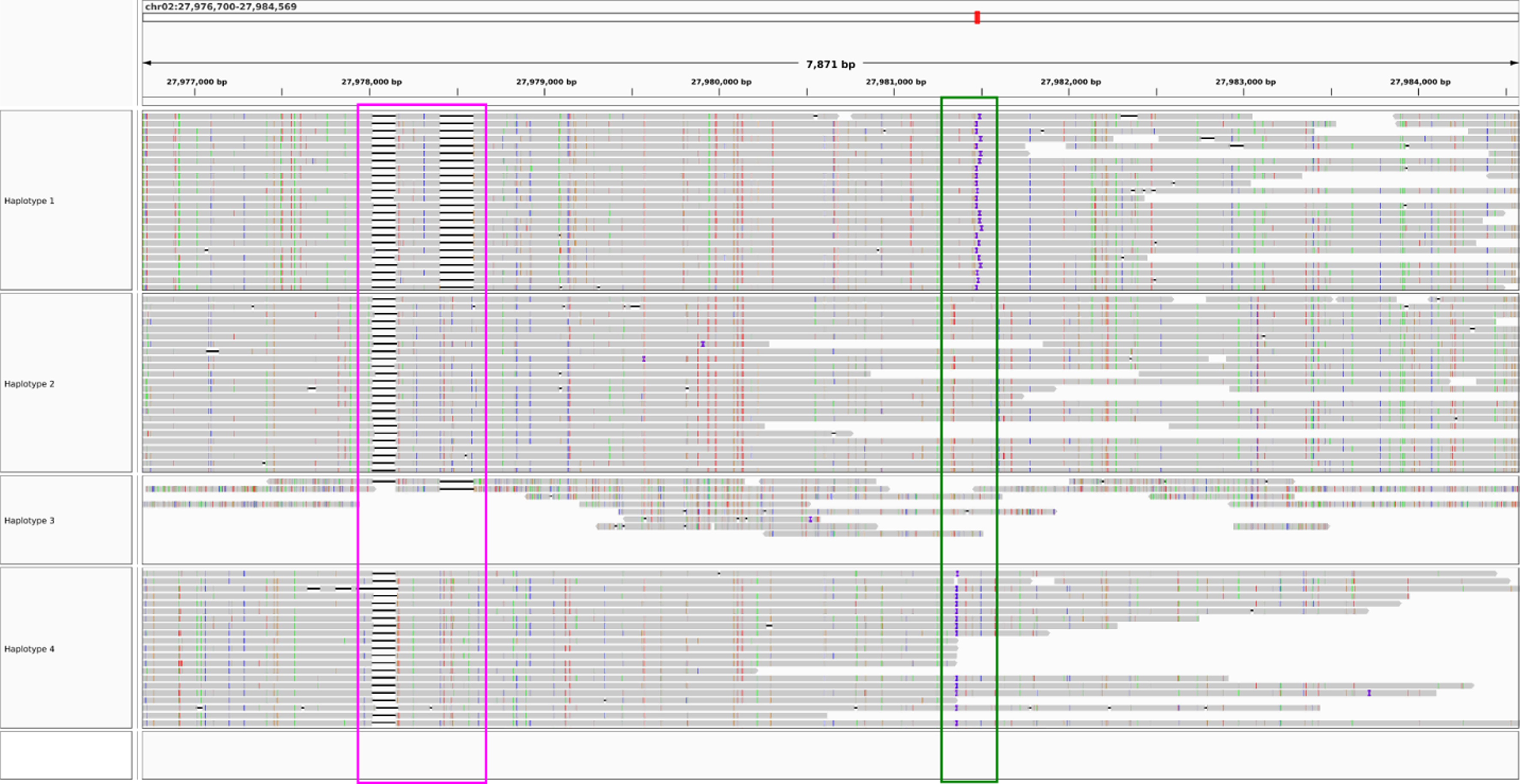
flopp's phasings on a 7871 bp gene. The colored boxes outline structural variations. The phasings for haplotypes 1, 2, and 4 look reasonable, whereas haplotype 3 has very low coverage and it appears that most reads are noisier supplementary alignments. On further inspection, haplotype 2 has twice the coverage of haplotypes 1 and 4, indicating haplotype collapsing and suggesting that haplotype 3 should look similar to haplotype 2.
In Figure 9, the clustered structural variants between haplotypes show that our phasings are quite good. Note that haplotypes 1 and 4 differed for only 13 out of 180 alleles. Thus, we are able to separate haplotypes that look relatively similar. However, in Figure 10, three of the haplotypes look perfectly phased, whereas haplotype 3 has very low coverage and mostly consists of noisy supplementary alignments. It appears that the UPEM uniformity constraint was not strong enough to deter collapsing in this case because the reads in haplotype 3 were so noisy. We found that haplotype 2 had twice the coverage of haplotypes 1 and 4, suggesting that haplotype 3 should look like a copy of haplotype 2.
Although collapsing is an issue for flopp, we have demonstrated that we can still obtain very sensible local haplotypes, even in the case of collapse. We believe that clever post-processing of flopp's output by looking at coverage can recover correct haplotypes in many instances of collapsing. We envision implementing such a post-processing step in future versions of flopp.
4. CONCLUSION
In this article, we presented two new formulations of polyploid phasing, the MSMTP problem and the UPEM model. The SMTP score is a flexible graphical interpretation of haplotype phasing that is related to the MEC score when using a specific weighting on the read-graph, whereas the UPEM score is a superior version of the MEC score when uniformity assumptions are satisfied. Using our probabilistic formulation, we give a new notion of distance between read fragments based on the Kullback-Leibler divergence, which has a rigorous interpretation as a log p-value of a one-sided binomial test.
We implemented a fast, local phasing procedure by using these new formulations and showed that our software, flopp, is faster and more accurate on high coverage data while always having extremely accurate local phasings across a range of error profiles, coverages, and ploidies.
Footnotes
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported by a faculty startup fund from the University of Toronto at Scarborough Department of Computer and Mathematical Sciences.