
Abstract
The computation of genomic distances has been a very active field of computational comparative genomics over the past 25 years. Substantial results include the polynomial-time computability of the inversion distance by Hannenhalli and Pevzner in 1995 and the introduction of the double cut and join distance by Yancopoulos et al. in 2005. Both results, however, rely on the assumption that the genomes under comparison contain the same set of unique markers (syntenic genomic regions, sometimes also referred to as genes). In 2015, Shao et al. relax this condition by allowing for duplicate markers in the analysis. This generalized version of the genomic distance problem is NP-hard, and they give an integer linear programming (ILP) solution that is efficient enough to be applied to real-world datasets. A restriction of their approach is that it can be applied only to balanced genomes that have equal numbers of duplicates of any marker. Therefore, it still needs a delicate preprocessing of the input data in which excessive copies of unbalanced markers have to be removed. In this article, we present an algorithm solving the genomic distance problem for natural genomes, in which any marker may occur an arbitrary number of times. Our method is based on a new graph data structure, the multi-relational diagram, that allows an elegant extension of the ILP by Shao et al. to count runs of markers that are under- or over-represented in one genome with respect to the other and need to be inserted or deleted, respectively. With this extension, previous restrictions on the genome configurations are lifted, for the first time enabling an uncompromising rearrangement analysis. Any marker sequence can directly be used for the distance calculation. The evaluation of our approach shows that it can be used to analyze genomes with up to a few 10,000 markers, which we demonstrate on simulated and real data.
1. Introduction
The study of genome rearrangements has a long tradition in comparative genomics. A central question is how many (and what kind of) mutations have occurred between the genomic sequences of two individual genomes. To avoid disturbances due to minor local effects, often the basic units in such comparisons are syntenic regions identified between the genomes under study, much larger than the individual DNA bases. We refer to such regions as genomic markers, or simply markers, although often one also finds the term “genes.”
Following the initial statement as an edit distance problem (Sankoff, 1992), a comprehensive trail of literature has addressed the problem of computing the number of rearrangements between two genomes. In a seminal article in 1995, Hannenhalli and Pevzner (1999) introduced the first polynomial time algorithm for the computation of the inversion distance of transforming one chromosome into another one by means of segmental inversions. Later, the same authors generalized their results to the HP model (Hannenhalli and Pevzner, 1995), which is capable of handling multi-chromosomal genomes and accounts for additional genome rearrangements. Another breakthrough was the introduction of the double cut and join (DCJ) model (Yancopoulos et al., 2005; Bergeron et al., 2006), which is able to capture many genome rearrangements and whose genomic distance is computable in linear time. The model is based on a simple operation in which the genome sequence is cut twice between two consecutive markers and re-assembled by joining the resulting four loose cut-ends in a different combination.
A prerequisite for applying the DCJ model in practice is that their genomic marker sets must be identical and that any marker occurs exactly once in each genome. This severely limits its applicability in practice. Linear time extensions of the DCJ model allow markers to occur exclusively in one of the two genomes, computing a genomic DCJ-insertion/deletion (indel) distance that minimizes the sum of DCJ and indel events (Braga et al., 2011; Compeau, 2013). Still, markers are required to be singleton, that is, no duplicates can occur. When duplicates are allowed, the problem is more intricate and all approaches proposed so far are NP-hard (see for instance, Sankoff, 1999; Bryant, 2000; Bulteau and Jiang, 2013; Angibaud et al., 2009; Martinez et al., 2015; Shao et al., 2015; Yin et al., 2016). From the practical side, more recently, Shao et al. (2015) presented an integer linear programming (ILP) formulation for computing the DCJ distance in the presence of duplicates, but restricted to balanced genomes, where both genomes have equal numbers of duplicates. Yin et al. (2016) then developed a branch and bound approach to compute the DCJ-indel distance of quasi-balanced genomes, which have not only an equal number of duplicated common markers but also markers that occur exclusively in one of the two genomes. An ILP that computes the DCJ-indel distance of unbalanced genomes was later presented by Lyubetsky et al. (2017), but their approach does not seem to be applicable to real data sets; see Section 6.1.2 for details.
In this article, we present the first feasible exact algorithm for solving the NP-hard problem of computing the distance under a general genome model where any marker may occur an arbitrary number of times in any of the two genomes, called natural genomes. Specifically, we adopt the maximal matches model where only markers appearing more often in one genome than in the other can be deleted or inserted. Our ILP formulation is based on the one from Shao et al. (2015), but with an efficient extension that allows to count runs of markers that are under- or over-represented in one genome with respect to the other, so that the pre-existing model of minimizing the distance allowing DCJ and indel operations (Braga et al., 2011) can be adapted to our problem. With this extension, once we have the genome markers, no other restriction on the genome configurations is imposed.
The evaluation of our approach shows that it can be used to analyze genomes with up to a few 10,000 markers, provided the number of duplicates is not too large. The complete source code of our ILP implementation and the simulation software used for generating the benchmarking data in Section 6.2 are available from https://gitlab.ub.uni-bielefeld.de/gi/ding.
This article is an extended version of earlier work that was presented at RECOMB 2020 (Bohnenkämper et al., 2020).
2. Preliminaries
A genome is a set of chromosomes, and each chromosome can be linear or circular. Each marker in a chromosome is an oriented DNA fragment. The representation of a marker m in a chromosome can be the symbol m itself, if it is read in direct orientation, or the symbol
Given two genomes A and B, let
2.1. The DCJ-indel model
A genome can be transformed or sorted into another genome with the following types of mutations:
A DCJ is the operation that cuts a genome at two different positions (possibly in two different chromosomes), creating four open ends, and joins these open ends in a different way. This can represent many different rearrangements, such as inversions, translocations, fusions, and fissions. For example, a DCJ can cut linear chromosome 1 2 ¯4 ¯3 5 6 before and after ¯4 ¯3, creating the segments Since the genomes can have distinct multiplicity of markers, we also need to consider insertions and deletions of segments of contiguous markers (Yancopoulos and Friedberg, 2009; Braga et al., 2011; Compeau, 2013). We refer to insertions and deletions collectively as indels. For example, the deletion of segment
In this article, we are interested in computing the DCJ-indel distance between two genomes A and B, that is denoted by
Singular genomes: The genomes contain no duplicate markers, that is, each common marker is singular in each genome. (The exclusive markers are not restricted to be singular, because it is mathematically trivial to transform them into singular markers when they occur in multiple copies.) Formally, we have that, for each
Balanced genomes: The genomes contain no exclusive markers, but can have duplicates, and the number of duplicates in each genome is the same. Formally, we have
Quasi-balanced genomes: The genomes contain exclusive markers and can have duplicates, but still the number of duplicates in each genome is the same. Formally, we have
Natural genomes: These genomes can have exclusive markers and duplicates, with no restrictions on the number of copies. Since these are generalizations of balanced genomes, computing the distance for this set of instances is also NP-hard. In the present work, we present an efficient ILP formulation for computing the distance in this case.
3. DCJ-indel Distance of Singular Genomes
First, we recall the problem when common duplicates do not occur, that is, when we have singular genomes. We will summarize the linear time approach to compute the DCJ-indel distance in this case that was presented in Braga et al. (2011), already adapted to the notation required for presenting the new results of this article.
3.1. Relational diagram
For computing a genomic distance, it is useful to represent the relation between two genomes in some graph structure (Hannenhalli and Pevzner, 1995; Bergeron et al., 2006; Friedberg et al., 2008; Braga et al., 2011). Here, we adopt a variation of this structure, defined as follows. For each marker m, denote its two extremities by
If the marker extremities
All vertices have degree one or two, therefore
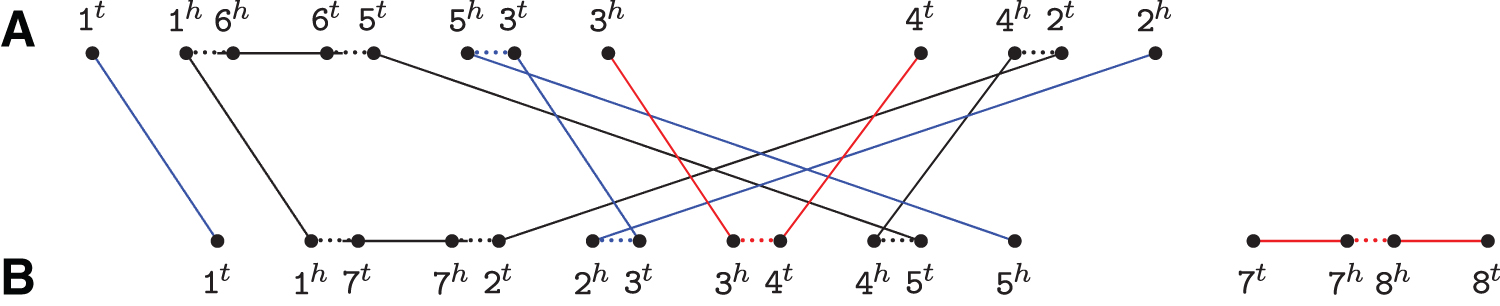
For genomes A={1 ¯6 5 3, 4 2} and B={1 7 2 3 4 5, 7 ¯8}, the relational diagram contains one cycle, two AB-paths (represented in blue), one AA-path, and one BB-path (both represented in red). Short dotted horizontal edges are adjacency edges, long horizontal edges are indel edges, and top–down edges are extremity edges.
The numbers of telomeres and of AB-paths in
3.2. Runs and indel-potential
The approach that uses DCJ operations to group exclusive markers for minimizing indels depends on the following concepts.
Given two genomes A and B and a component C of
While sorting components separately with optimal DCJs only, runs can be merged (when two runs become a single one), and also accumulated together (when all its indel edges alternate with adjacency edges only and the run can be inserted or deleted at once) (Braga et al., 2011). The indel-potential of a component C, denoted by
Figure 2 shows a BB-path with 4 runs, and how its indel-potential can be achieved.
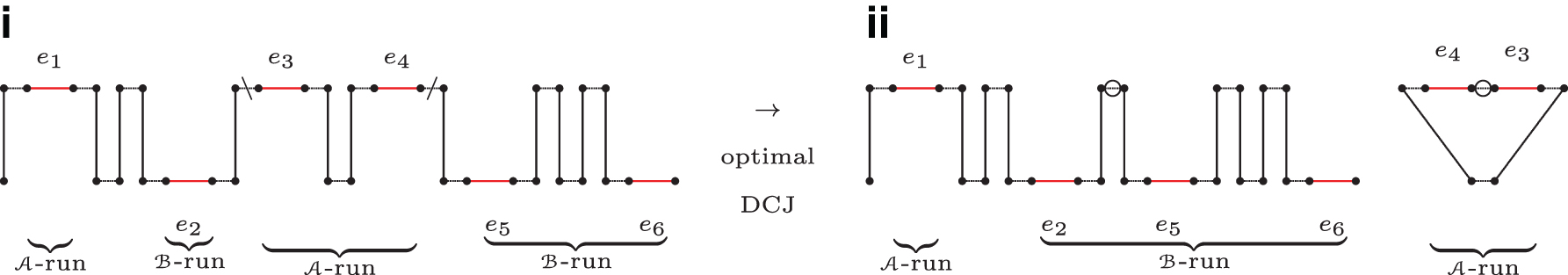
The indel-potential allows to state an upper bound for the DCJ-indel distance.
Let
3.3. Distance of circular genomes
For singular circular genomes, the diagram
3.4. Recombinations and linear genomes
For singular linear genomes, the upper bound given by Lemma 1 is achieved when the components of
3.4.1. Deducting recombinations
Any recombination whose sources are an AA-path and a BB-path is optimal. A recombination whose sources are two different AB-paths can be either neutral, when the resultants are also AB-paths, or counter-optimal, when the resultants are an AA-path and a BB-path. Any recombination whose sources are an AB-path and an AA- or a BB-path is neutral (Braga and Stoye, 2010; Braga et al., 2011).
Let
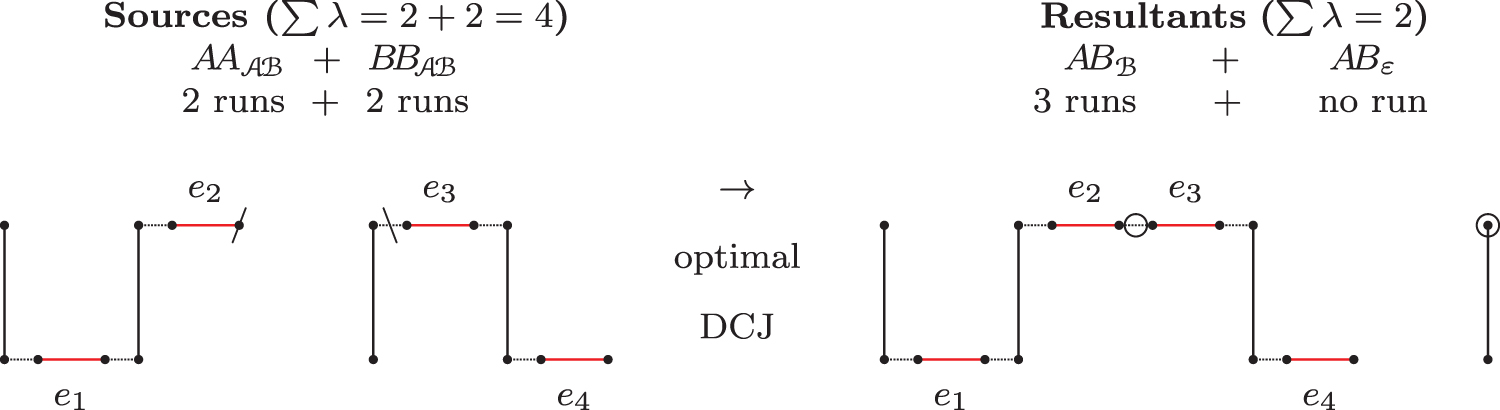
An optimal recombination with
The complete set of path recombinations with
Path Recombinations That Have
Path Recombinations with
The two sources of a recombination can also be called partners. Looking at Table 1, we observe that all partners of
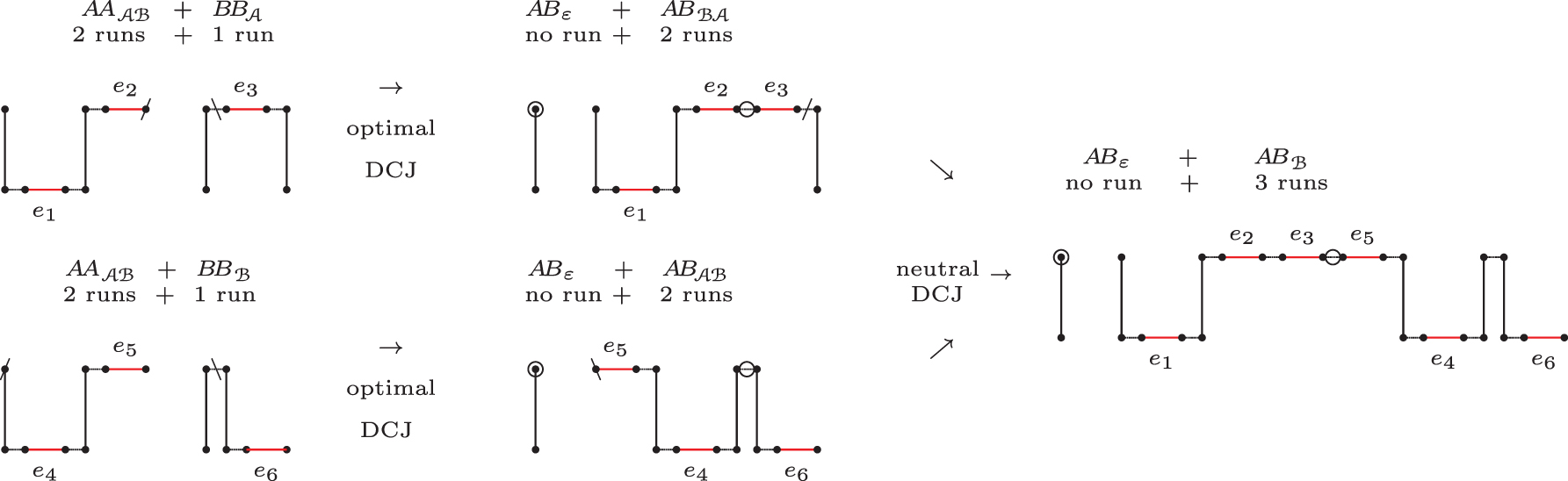
Chained recombinations transforming four paths (
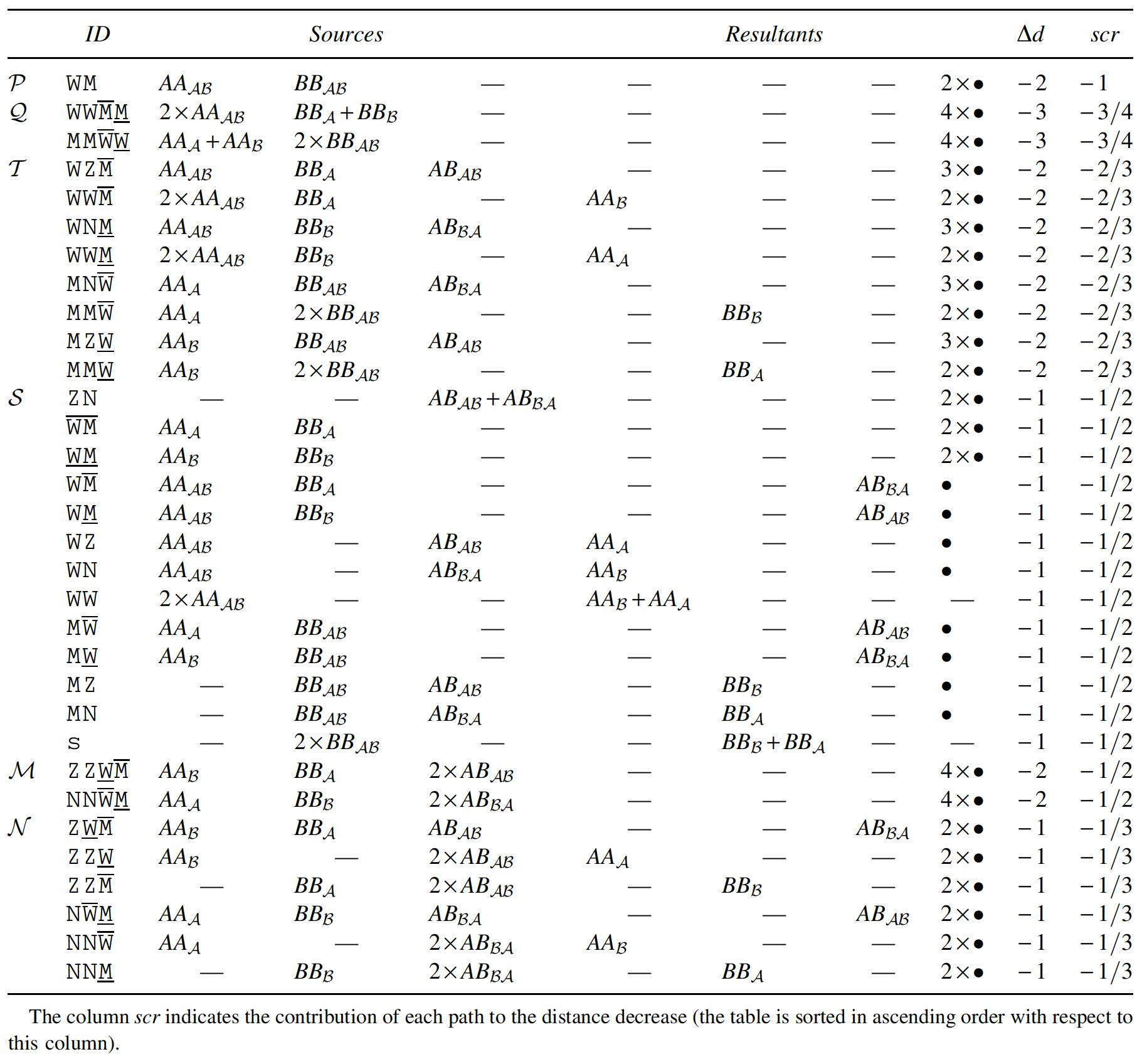
Each group is represented by a combination of letters, where:
Although some groups have reusable resultants, those are actually never reused. (If groups that are lower in the table use as sources resultants from higher groups, the sources of all referred groups would be previously consumed in groups that occupy even higher positions in the table.) Due to this fact, the number of occurrences in each group depends only on the initial number of each type of component.
The deductions shown in Table 3 can be computed with an approach that greedily maximizes the groups in P, Q, T, S, M, and M in this order. The P part contains only one operation and is, thus, very simple. The same happens with Q, since the two groups in this part are exclusive after applying P. The four subparts of T are also exclusive after applying Q. (Note that groups WW¯M, WW_M, MM¯W, and MM_W of T are simply subgroups of Q.) The groups in S correspond to the simple application of all possible remaining operations with
We can now write the theorem that gives the exact formula for the DCJ-indel distance of linear singular genomes.
Given two singular linear genomes A and B, whose relational diagram
where
4. DCJ-indel Distance of Natural Genomes
Based on the results presented so far, we develop an approach for computing the DCJ-indel distance of natural genomes A and B. First, we note that it is possible to transform A and B into matched singular genomes
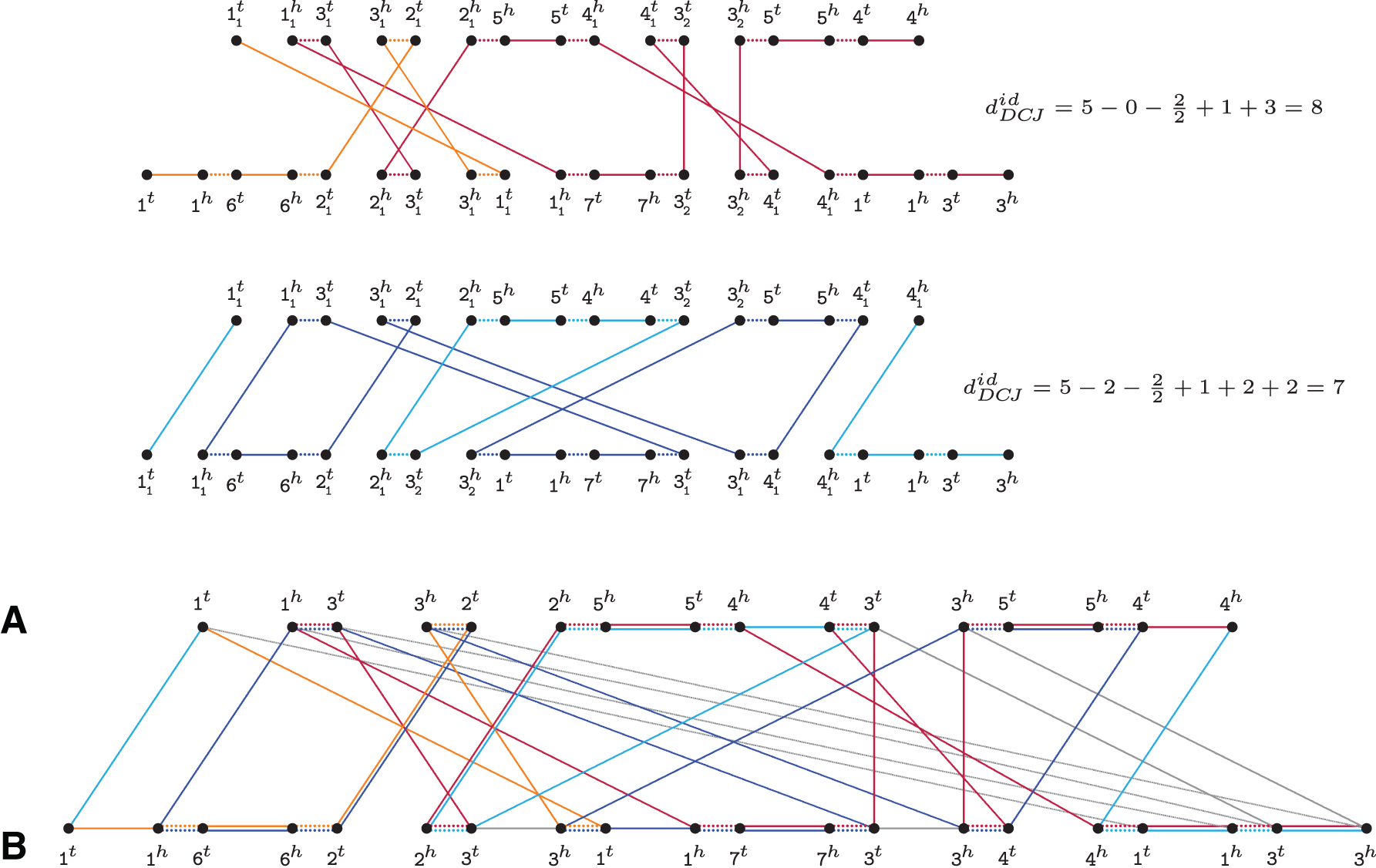
Natural genomes A=1 3 2 ¯5 ¯4 3 5 4 and
Let
4.1. Multi-relational diagram
Although the original relational diagram clearly depends on the singularity of common markers, when they appear in multiple copies we can obtain a data structure that integrates the properties of all possible relational diagrams of matched genomes. The multi-relational diagram
Again, sets
4.1.1. Consistent decompositions
Note that if A and B are singular genomes,
Let
The set of edges
where cD and iD are the numbers of AB-cycles and AB-paths in
Given two natural genomes A and B, the DCJ-indel distance of A and B can then be computed by the following equation:
where
A consistent decomposition
5. Capping
The ends of linear chromosomes produce some difficulties for the decomposition. Fortunately there is an elegant technique to overcome this problem, called capping (Hannenhalli and Pevzner, 1995). It consists of modifying the genomes by adding artificial singular common markers, called caps, that circularize all linear chromosomes, so that their relational diagram is composed of cycles only, but, if the capping is optimal, the genomic distance is preserved.
5.1. Capping of canonical genomes
When two singular genomes A and B have no exclusive markers, they are called canonical genomes.
The diagram
Also, obtaining an optimal capping of canonical genomes is quite straightforward (Hannenhalli and Pevzner, 1995; Yancopoulos et al., 2005; Braga and Stoye, 2010), as shown in Table 4: The caps should guarantee that each AB-path is closed into a separate AB-cycle, and each pair composed of an AA- and a BB-path is closed into an AB-cycle by linking each extremity of the AA-path to one of the two extremities of the BB-path (there are two possibilities of linking, and any of the two is optimal). If the numbers of linear chromosomes in A and in B are different, there will be some AA- or BB-paths remaining. For each of these, an artificial adjacency between caps is created in the genome with less linear chromosomes, and each artificial adjacency closes each remaining AA- or BB-path into a separate AB-cycle.
Linking Paths from
The symbol
Let
We can show that the capping described earlier is optimal by verifying the corresponding DCJ-indel distances. Let the original genomes A and B have n markers and
An example of an optimal capping of two canonical linear genomes is given in Figure 6.
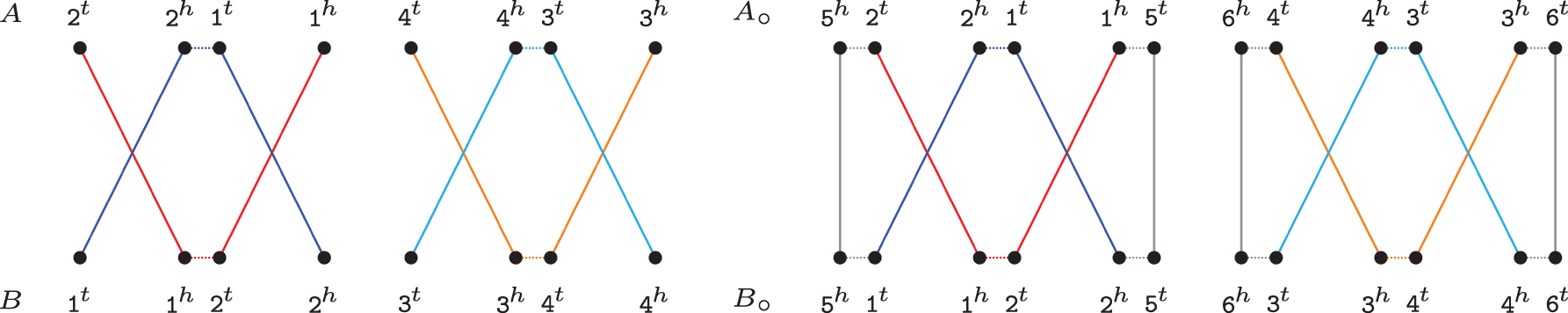
Optimal capping of canonical genomes
5.2. Singular genomes: correspondence between recombinations and capping
When exclusive markers occur, we can obtain an optimal capping by simply finding caps that properly link the sources of each recombination group (listed in Table 3) into a single AB-cycle. Indeed, in Table 5 we give a linking that achieves the optimal
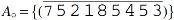
Optimal capping of singular genomes and
Linking Sources of Chained Recombination Groups from Table 3
Further, similar to the case of canonical genomes, the numbers of artificial adjacencies and caps in such a capping are, respectively,
In Table 5, we can observe that there are two types of groups: (1) balanced, which contain the same number of AA- and BB-paths, and (2) unbalanced, in which the numbers of AA- and BB-paths are distinct. Unbalanced groups require some extra elements to link the cycle. These elements can be indel-free AA- or BB-paths (of the type, i.e., under-represented in the group) or, if these paths do not exist, artificial adjacencies either in genome A or in genome B (again, of the genome, i.e., under-represented in the group). We then need to examine these unbalanced groups to determine the number of caps and of artificial adjacencies that are required for an optimal capping.
Proof: It is clear that, after P and until N, we have either only groups
Let us examine the case of group ZZ_W. (1) At a first glance, one could think that this group is compatible with MM_W. However, if all components of these two unbalanced groups would be in the diagram, we would instead have two times the group MZ_W, which is balanced and located before the two other groups in the table (observe that MZ_W has a smaller
With a similar analysis we can show that for all cases either we have only unbalanced groups that are over-represented in genome A or we have only unbalanced groups that are over-represented in genome B.
Proof: First we observe that, after distributing all paths of the relational diagram among the recombination groups, following the top–down greedy approach, there could be AA- and/or BB-paths remaining, which were not assigned to any group, and they might be useful to link unbalanced groups. We will now examine the procedure of linking the unbalanced groups either with those remaining paths or with artificial adjacencies.
A particular case are the unbalanced groups from T. Since all unbalanced groups in T have analogous compositions, without loss of generality, suppose a group over-represented in genome A of type WW¯M is being linked. If, at this point, there is a remaining indel-enclosing BB-path, it cannot be
The unbalanced groups from S or N are easier to analyze: If one of these groups, over-represented in genome A (respectively in genome B), is being linked, there cannot be any remaining indel-enclosing BB-path (respectively AA-path). We can verify this by supposing, without loss of generality, that an unbalanced group over-represented in genome A is being linked. If, at this point, there is a remaining indel-enclosing BB-path, then with the components of the group being linked and the existing remaining path we could form a balanced group that appears in a higher position of the table, with at least the same
Propositions 1 and 2 prove the following result.
caps and
5.3. Capped multi-relational diagram
We can transform
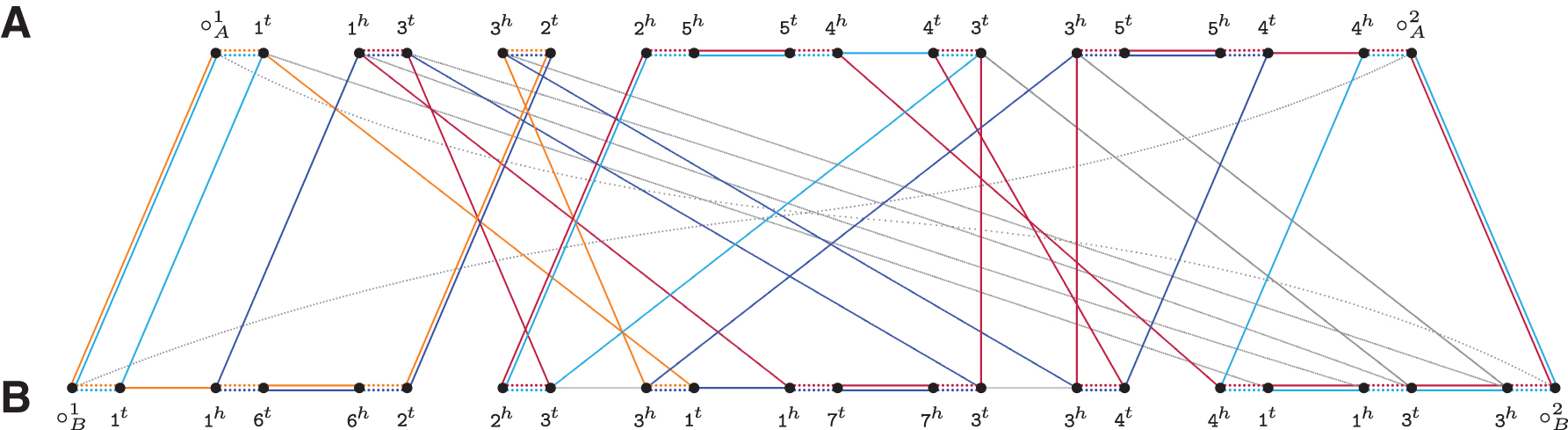
Natural genomes A=1 3 2 ¯5 ¯4 3 5 4 and
A set
Proof: Recall that each maximal sibling set S of
As a consequence of Theorem 3, if
Each decomposition
6. An Algorithm to Compute the DCJ-Indel Distance of Natural Genomes
An ILP formulation for computing the distance of two balanced genomes A and B was given by Shao et al. (2015). In this section, we describe an extension of that formulation for computing the DCJ-indel distance of natural genomes A and B, based on consistent cycle decompositions of
The formula cited earlier can be redesigned to a simpler one, which is easier to implement in the ILP. First, let a transition in a decomposition
Note that
Now, we need to find a consistent decomposition
where
6.1. ILP formulation
Our formulation (shown in Algorithm 1) searches for an optimal consistent cycle decomposition of
In the first part, we use the same strategy as Shao et al. (2015). A binary variable
The second part is our extension for counting transitions. We introduce binary variables
In the third part we add a new constraint and a new domain to our ILP, so that we can count the number of circular singletons. Let K be the circular chromosomes in both genomes and
The objective of our ILP is to maximize the weight of a consistent decomposition, which is equivalent to maximizing the number of indel-free cycles, counted by the sum over variables zi, while simultaneously minimizing the number of transitions in indel-enclosing AB-cycles, calculated by half the sum over variables te, and the number of circular singletons, calculated by the sum over variables sk.

6.1.1. Implementation
We implemented the construction of the ILP as a python application, available at https://gitlab.ub.uni-bielefeld.de/gi/ding.
6.1.2. Comparison to the approach by Lyubetsky et al
As mentioned in Section 1, another ILP for the comparison of genomes with unequal content and paralogs was presented by Lyubetsky et al. (2017). To compare our method with theirs, we ran our ILP by using CPLEX on a single thread with the two small artificial examples given in that article on page 8. The results in terms of DCJ distance were the same. A comparison of running times is presented in Table 6.
Comparison of Running Times and Memory Usage to the Integer Linear Programming in Lyubetsky et al.
6.2. Performance benchmark
For benchmarking purposes, we used Gurobi 9.0 as a solver. In all our experiments, we ran Gurobi on a single thread.
6.2.1. Generation of simulated data
Here, we describe our simulation tool that is included in our software repository (https://gitlab.ub.uni-bielefeld.de/gi/ding) and used for evaluating the performance of our ILP implementation.
Our method samples marker order sequences over a user-defined phylogeny. However, here we restrict our simulations to pairwise comparisons generated over rooted, weighted trees of two leaves. Starting from an initial marker order sequence of user-defined length (i.e., number of markers), the simulator samples Poisson-distributed DCJ events with expectation equal to the corresponding edge weights. Likewise, insertion, deletion, and duplication events of one or more consecutive markers are sampled; however, their frequency is additionally dependent on a rate factor that can be adjusted by the user. The length of each segmental insertion, deletion, and duplication is drawn from a Zipfian distribution, whose parameters can also be adjusted by the user. At each internal node of the phylogeny, the succession of mutational operations is performed in the following order: DCJ operations, duplications, deletions, insertions. To this end, cut points, as well as locations for insertions, deletions, and duplications are uniformly drawn over the entire genome.
In our simulations, we used
6.2.2. Evaluating the impact of the number of duplicate occurrences
To evaluate the impact of the number of duplicate occurrences on the running time, we first keep the number of simulated DCJ events fixed to
Our ILP solves the decomposition problem efficiently for real-sized genomes under small to moderate numbers of duplicate occurrences: Solving times for genome pairs with <10,000 duplicate occurrences (50% of the genome size) shown in Figure 9(i) are with a few exceptions below 5 minutes and exhibit a linear increase, but solving time is expected to increase dramatically with higher numbers of duplicate occurrences. To further exploit the conditions under which the ILP is no longer solvable with reasonable compute resources, we continued the experiment with even higher amounts of duplicate occurrences and instructed Gurobi to terminate within 1 hour of computation. We then partitioned the simulated data set into 8 intervals of length 500 according to the observed number of duplicate occurrences. For each interval, we determined the average as well as the maximal multiplicity of any duplicate marker and examined the average optimality gap, that is, the difference in percentage between the best primal and the best dual solution computed within the time limit. The results are shown in Table 7 and emphasize the impact of duplicate occurrences in solving time: Below 14,000 duplicate occurrences, the optimality gap remains small and sometimes even the exact solution is computed, whereas above that threshold the gap widens very quickly.
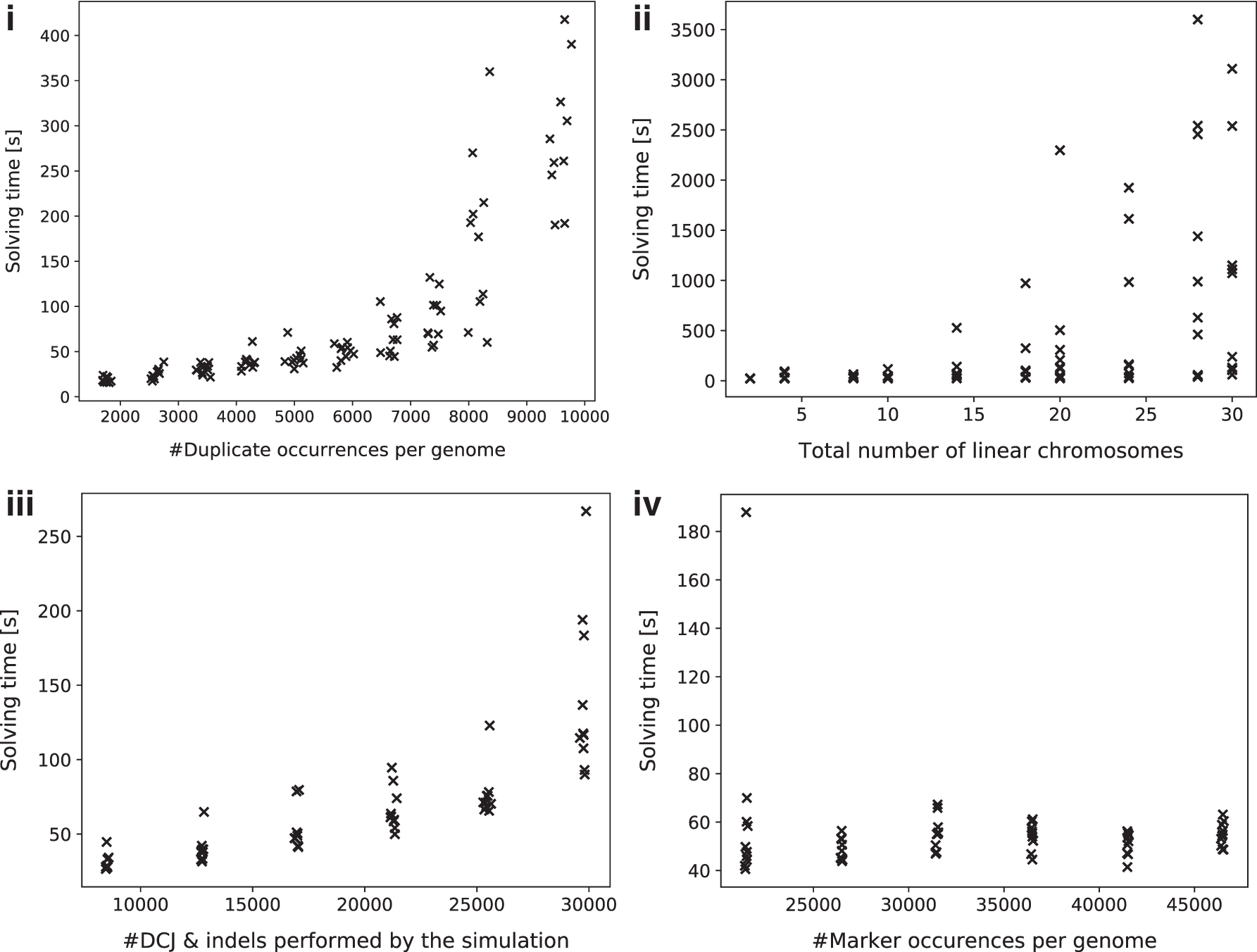
Solving times for:
Average Optimality Gap for Simulated Genome Pairs Grouped by Number of Duplicate Occurrences After 1 Hour of Running Time
6.2.3. Evaluating additional parameters
So far, we have examined only the impact of duplicates on solving times of our program. However, other parameters of our experiment are expected to have an effect on the solving times, too. We ran three experiments, in each varying one of the following parameters while keeping the others fixed: (1) genome size, (2) number of simulated DCJs and indels, and (3) number of chromosomes. The duplication rate was fixed at
The results, shown in Figure 9(ii)–(iv), indicate that the number of linear chromosomes plays a major factor in the solving time. At the same time, solving times vary more widely with increasing chromosome number. The latter has a simple explanation: Telomeres, represented as caps in the multi-relational diagram, behave in the same way as duplicate occurrences of the same marker do. Increasing their number (by increasing the number of linear chromosomes) increases exponentially the search space of matching possibilities.
Conversely, the number of simulated DCJs and indels has a minor impact on the solving times of our simulation runs. However, although initially exhibiting collinearity, the solving times for higher numbers of DCJs and indels divert super-linearly. Lastly, the genome size has a negligible effect on solving time within the tested range of
6.3. Real data analysis
To demonstrate the applicability to real datasets, we compared the genomes of six Drosophila species and reconstructed their phylogeny from pairwise DCJ-indel distances. The species names and the NCBI accession numbers of the assemblies are listed in Table 8.
List of Genome Assemblies Used in Our Experiments
We used two types of markers. In our first experiment, markers correspond to the longest annotated coding sequences (CDSs) per locus obtained from the respective NCBI annotations. Their numbers are listed in Table 8 as well. Subsequently, we inferred hierarchical orthologous groups of these markers, with Drosophila busckii being the outgroup species running OMA standalone version 2.4.1 (Altenhoff et al., 2019) with default settings. As described earlier, we used Gurobi in computing pairwise DCJ-indel distances. As can be seen in Table 9, Gurobi was able to solve most instances within seconds with the exception of one pair, which took about 9 hours to compute, emphasizing again the sensitivity of the ILP's solving time to the number of duplicate occurrences.
Pairwise Comparisons of the Six Drosophila species (busckii [dbus], melanogaster [dmel], pseudoobscura [dpse], sechellia [dsec], simulans [dsim], and yakuba [dyak])
Genomes were constructed by using genes as markers. All instances were solved by Gurobi on a single thread.
Using Neighbor Joining in MEGA X (Kumar et al., 2018), we constructed a phylogeny of the considered species. The tree rooted by D. busckii is shown in Figure 10(i). It is consistent with the knowledge on the Drosophila phylogeny so far, except for the resolution of the subtree containing the taxa melanogaster, sechellia, and simulans. Considering the corresponding Splits diagram constructed by NeighborNet in SplitsTree (Huson and Bryant, 2005) [Fig. 10(ii)], we observe that the distances in this subtree do not behave very tree-like. This suggests that, rather than an erroneous tree being computed, the resolution of the gene-based inference of markers simply does not provide distances for meaningfully clustering any two of the three taxa together.
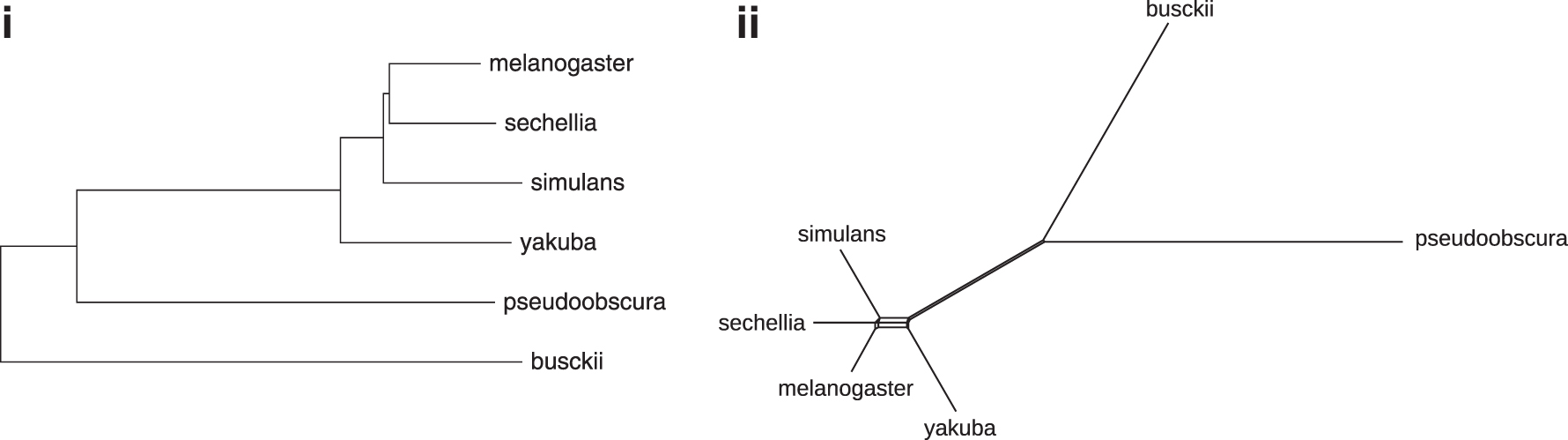
The gene-based distances in Table 9 are used as input to reconstruct Drosophila-Phylogeny:
To increase coverage and resolution, we also generated a second set of markers, directly from the genomic sequences and not restricted to genes or CDSs. We used GEESE (Rubert et al., 2020b) to construct genomic markers of length at least 500 bp. GEESE implements a heuristic for the genome segmentation problem (Visnovská et al., 2013) and takes as input local pairwise sequence alignments that we computed with LASTZ. The parameter settings used for both GEESE and LASTZ are detailed in our software repository. The number of markers in each genome is shown in Table 8. Again using Gurobi on a single thread, we were able to solve all corresponding instances of the ILP within a few minutes. The distances as well as data regarding duplicates and solving times can be found in Table 10.
Pairwise Comparisons of the Six Drosophila Species (busckii [dbus], melanogaster [dmel], pseudoobscura [dpse], sechellia [dsec], simulans [dsim], and yakuba [dyak])
Genomes were constructed by using segmentation. All instances were solved by Gurobi on a single thread.
Using the same procedure as described earlier to construct the Neighbor Joining tree and the Splits diagram (Fig. 11(i) and (ii), respectively), we find that the segmentation-based approach not only produces the correct topology of the tree, but also improves the strength of all correct splits in the previously problematic subtree, including those involving Drosophila yakuba. We notice, however, that the branch length of D. busckii is comparatively short. This is most likely due to the lack of markers, which could be inferred on the D. busckii genome (Table 8), thus leading to some rearrangements being missed. One might attribute the fact that the segmentation did not infer many homologies in this case to more rapid sequence evolution in non-coding regions.
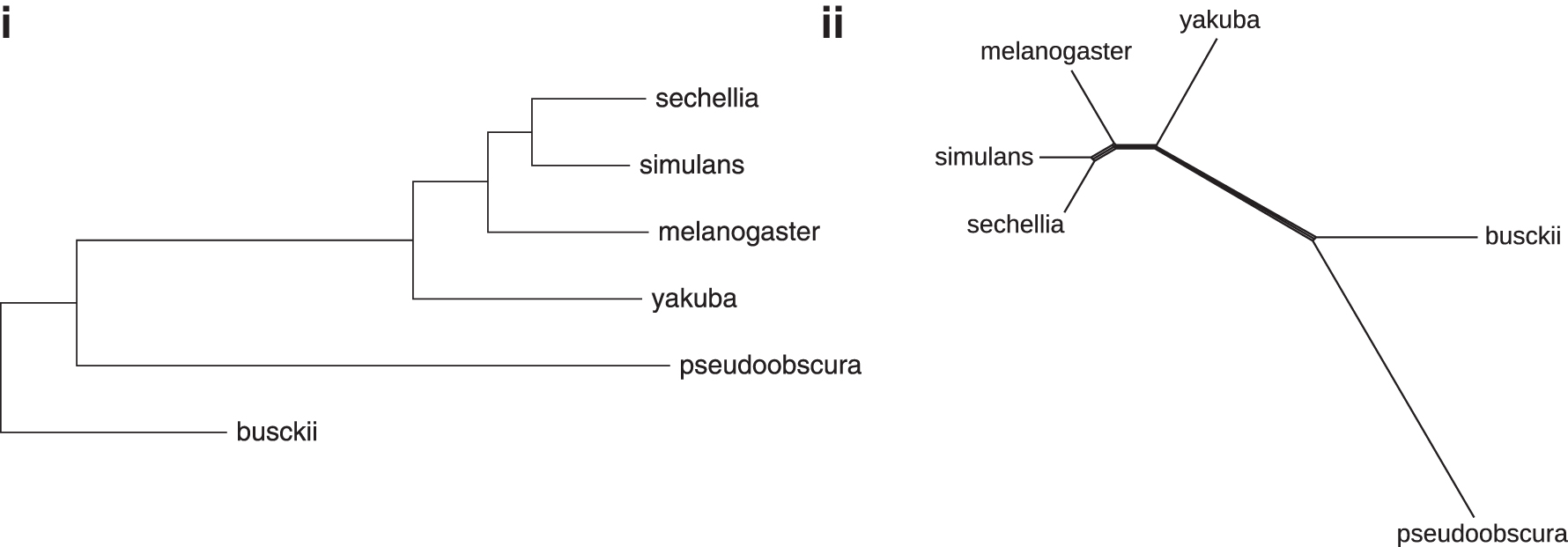
The segmentation-based distances in Table 10 are used as input to reconstruct Drosophila-Phylogeny:
7. Conclusion
By extending the DCJ-indel model to allow for duplicate markers, we introduced a rearrangement model that is capable of handling natural genomes, that is, genomes that contain shared, individual, and duplicated markers. In other words, under this model, genomes require no further processing nor manipulation once genomic markers and their homologies are inferred. The DCJ-indel distance of natural genomes being NP-hard, we presented a fast method for its calculation in the form of an integer linear program. Our program is capable of handling real-sized genomes, as evidenced in simulation and real data experiments. It can be applied universally in comparative genomics and enables uncompromising analyses of genome rearrangements.
Our experiments on real data show that our approach is easily applicable to real-world genomes, with markers generated by different methods. The power of the method, however, depends on the quality of the markers. Genes as markers proved reliable in resolving distances and relations between further related taxa while not being expressive enough to resolve some closer relations. In contrast, segmentation-based markers are better suited to resolve close distances, but they might underestimate larger distances due to lack of markers.
We hope that similar analyses will provide further insights into the underlying mutational mechanisms of other, less well-studied species. Conversely, we expect the model presented here to be extended and specialized in future to reflect the insights gained by these analyses. Follow-up work with a family-free version of our model has just appeared in the Proceedings of the Workshop on Algorithms in Bioinformatics (WABI 2020) (Rubert et al., 2020a).
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
All funding for this work was provided by the authors' home institution, Bielefeld University.