
Abstract
Abstract
An important feature of structural data, especially those from structural determination and protein-ligand docking programs, is that their distribution could be mostly uniform. Traditional clustering algorithms developed specifically for nonuniformly distributed data may not be adequate for their classification. Here we present a geometric partitional algorithm that could be applied to both uniformly and nonuniformly distributed data. The algorithm is a top-down approach that recursively selects the outliers as the seeds to form new clusters until all the structures within a cluster satisfy a classification criterion. The algorithm has been evaluated on a diverse set of real structural data and six sets of test data. The results show that it is superior to the previous algorithms for the clustering of structural data and is similar to or better than them for the classification of the test data. The algorithm should be especially useful for the identification of the best but minor clusters and for speeding up an iterative process widely used in NMR structure determination.
1. Introduction
R
An important feature of structural data, especially those from NMR structural determination and protein-ligand docking, is that their distribution could be mostly uniform, and thus may not be properly described by a Gaussian mixture model. Traditional clustering algorithms developed specifically for nonuniformly distributed data may not be adequate for their classification. In this article, we present a novel geometric partitional algorithm that could be applied to both uniformly and nonuniformly distributed data. The algorithm is a top-down approach that recursively partitions all the data points of a previously generated cluster into c new clusters where c is a user-specified number. It stops and then outputs a final set of clusters that satisfy the classification criterion that no metric distances between any pair of data points in any cluster are larger than a certain value. Compared with the previous clustering algorithms, the salient features of our geometric partitional algorithm are (a) it uses the global information in the beginning, (b) it can handle both uniformly and nonuniformly distributed data, and (c) it is deterministic.
We have applied the algorithm to the classification of a diverse set of data: the intermediate structures from an NMR structure determination project, poses from protein-ligand docking, and MD trajectories from an ab-initio protein folding simulation (data not shown), as well as six sets of test data that have been used widely for the evaluation of clustering algorithms. We have also compared the algorithm with the following five different clustering algorithms: common nearest-neighbor, bipartition, complete-link, average-link, and k-medoids, on both real structural data and test data. The results show that our algorithm classifies the structural data with a higher accuracy than a k-medoids does. For the structural data sets, though the final set of clusters from our algorithm may be similar to those from a hierarchical algorithm such as complete-link or average-link, and to those from a nearest-neighbor or bipartition algorithm, the structures assigned to the same cluster by our algorithm are more uniform in terms of their structural and physical properties.
More importantly, our algorithm outperforms the previous ones in singling out the minor clusters with “good” properties (the best or correct clusters) that are often to be overlooked or even discarded by other criteria used for the selection of representative structures. Furthermore, the comparisons of our algorithm with the above five algorithms on the test data sets confirm its generality: the algorithm performs as well as or better than the previous ones in their classification. The rest of the article is organized as follows. In section 2 we first present the algorithm and then describe the real structural data sets. In section 3 we present the results of applying both our algorithm and five previous algorithms to the structural data sets for the identification of the clusters with good scores, and discuss the significance of the geometric algorithm for speeding up the iterative NMR structure determination process and for the selection of accurate docking poses. In section 4 we compare our clustering algorithm with the previous ones from both the theoretical and practical perspectives. Finally, we conclude the article with a section on the challenges of structural data classification.
2. The Algorithm and Data Set
In this section, we first present our novel geometric partitional algorithm for the clustering of structural data. Then we describe the data sets used for assessing the performance of the new and five previous clustering algorithms.
2.1. The geometric partitional algorithm
where
In the following we present the key steps of the algorithm at recursive step s with an input one of clusters
1. S
2. I
S
3. I
4. S
5. I
(a) F
A
(b) F
G
6. S
7. S
8. I
(a) A
(b) F
G
9. E
(a) S
(b) A
(c) F
G
where dmax is a user-defined maximum RMSD such that all the structures in the same cluster must have their pairwise RMSDs less than dmax. This condition will be called the cluster restraint criterion. In step 2 if the largest pairwise RMSD among all the points in a cluster is less than dmax, no more partition is required, thus stops the recursive procedure.
Please see the Supplementary Materials for their proofs.
2.2. Running time
Let the number of structures be n. It takes O(n2) to populate the set
2.3. Structural data set
The structural data to which we have applied our algorithm as well as nearest-neighbor, bipartition, hierarchical (both complete-link and average-link), and k-medoids algorithms include (a) two sets of intermediate structures from an NMR structure determination project, and (b) twenty-two sets of poses from protein-ligand docking. In the following we describe both the data and the computational processes to generate them.
2.3.1. NMR data set
The two sets of intermediate structures chosen for the comparison of clustering algorithms are from the structure determination project of the protein SiR5 with 101 residues. Its NMR structure was determined by one of the authors using an iterative procedure of automated/manual nuclear Overhasuer effect (NOE) restraint assignment followed by structure computation using CYANA/Xplor with conformational sampling achieved by simulated annealing (SA). A large number of intermediates need to be generated during the iterative process in order to properly sample the huge conformational space defined as the set of all the structures that satisfy the experimentally derived restraints to the same extent. In contrast to the final set of 20 structures deposited in the PDB (2OA4), the intermediates especially those from an early stage of the iterative process are less uniform in terms of structural similarity, molecular mechanics energy, and restraint satisfaction. The pairwise RMSDs are computed only for C α atoms of residues 20–70 since almost no long-range NOEs were observed for the rest. The dmax for both geometric and complete-link hierarchical clustering algorithms are either 1.0Å or 1.5Å. Each cluster is assessed by its average van der Waals (VDW) energy, NOE restraint violation (the NOE violation per structure is defined as the number of NOE restraints with violation ≥0.5 Å), and its average da (da is the pairwise RMSD between two structures within a cluster), and average df (df is the RMSD between a structure in the cluster and the centroid of the 20 structures in 2OA4).
2.3.2. The set of poses from protein-ligand docking
Structural clustering plays an increasingly important role in both protein-ligand docking and virtual screening (Downs and Barnard, 2002) since a large amount of poses or library hits are typically generated during either a docking or virtual screening process. To demonstrate the importance of clustering to protein-ligand docking, we have performed rescoring experiments on 22 sets of poses † generated using GOLD software suit (version 1.2.1) (Jones et al., 1995). Several rounds of docking are performed using a binding site specified by a manually picked center with a 20.0Å radius. GOLD requires a user to pick a point that together with a user-specified radius defines a sphere inside, which poses are searched for using a genetic algorithm (GA). We use the default parameters as provided by GOLD except the requirement that any pair of the generated poses must have its pairwise RMSD >1.5Å. Only the ligand-heavy atoms are used in the pairwise RMSD computation. The 3D starting conformation for each ligand was generated by Corina (Sadowski et al., 1994). A set of 500 poses are saved for each protein-ligand complex.
A well-known difficulty with the current scoring functions for protein-ligand docking is that they often fail to rank in the top positions the poses that are most similar to the experimentally determined one. To investigate whether clustering could provide the guarantee that the top-ranked clusters have high probability to be composed of the poses that are most similar to the experimental one, we first perform a series of clustering experiments with decreasing dmax values using our geometric partitional algorithm. We then rank the most populated clusters whose combined number of poses either exceed 90% of the total number of poses for larger dmax or 75–50% for smaller dmax. The ranking is based on their cluster-wide average values of both the GOLD scoring function Sg that consists of three items—ligand internal energy Gi, intermolecular VDW energy Gw, and intermolecular hydrogen bond energy Ghb—and our newly developed scoring function St that also has three items—Gi; Ee, the electrostatic energy computed using the partial charge assigned by Corina and the electrostatic potential from APBS (Baker et al., 2001); and Saa, the change in solvent accessible surface area (SAA) of the ligand before and after its binding.
where ge, gs, ke, and ks are weighting factors. The details of our scoring function, its rationale and practical performance, will be described elsewhere. Here we only briefly state the rationale behind our scoring function and compare it with the GOLD scoring function. The key difference between our scoring function and the GOLD scoring function is that we have replaced the GOLD's Gw and Ghb terms with our Ee and Saa terms. Our analysis of protein-ligand complex (data not shown) as well as the results presented in this article suggest to us that neither Gw nor Ghb term has much discriminatory power for pose selection. In GOLD, they have been used mainly for pose generation. Our new term Ee, computed using the electrostatic potential from APBS, has been found to be the dominating term for a protein-ligand system with a net charge on the ligand. The goal of our second term Saa is to approximate, to some extent, the protein-ligand binding entropy and desolvation effect in particular. For some protein-ligand systems, the entropic change before and after the ligand binding dominates the binding affinity.
3. Results and Discussion
To evaluate the performance of our algorithm, to compare it with the previous algorithms for structural data classification, and to demonstrate the importance of clustering to structural analysis, we have applied them to a diverse set of data including two sets of intermediate structures from an NMR structure determination project and twenty-two sets of poses from protein-ligand docking. In the following, we first present the results and then discuss the significance of clustering to the selection of correct representative structures and the identification of best poses.
3.1. NMR structural ensemble
In theory the computation of structures using sparse and inexact geometric restraints derived from NMR experiments is an
The process stops when the computed structures converge according to certain criteria. During the iterative process, a large number of intermediate structures are generated in order to properly sample the conformational space. However, all but a small subset of intermediates must be discarded in the next cycle due to time and space limitation. There exists no well-established criteria for such a selection though in practice it is typically achieved using a user-specified threshold for a scoring function used in the structure determination. Such a selection assumes that there exists only a single or a few large clusters of structures that satisfy the restraints, a condition that may be difficult to meet especially in the early stages when only a small number of restraints per residue are used. A different selection of representative structures in the iterative process may lead to different ensembles of structures in the PDB as demonstrated by an investigation into two ensembles of NMR-derived structures of the protein Sox-5 HMG-box reported by two different groups (Adzhubei et al., 1995). In this article, we have applied four algorithms to two sets of intermediates to assess how the distribution of intermediates could affect the selection of representative structures and which algorithms are most suitable for such a task. The first set has 301 intermediates from an early stage of the SiR5 project while the second has 159 intermediates from a late stage. The clusters are analyzed in terms of the number of structures Ns per cluster, their average da, df values, VDW energies, and NOE violations. In the following we only present the clusters obtained with dmax=1.5Å. Similar but a larger number of clusters each with smaller number of structures are generated with dmax=1.0Å.
The listed are the most populated clusters with the number of structures NS≥3 from geometric, common nearest-neighbor, bipartition, and complete-link algorithms generated with a dmax=1.5 Å, and all the non-singletons from average-link and k-medoids algorithms. The cluster shown with the boldfaced font has the smallest df among all the clusters. The three numbers are respectively the range and average. For k-medoids the number of initial clusters is 10, with the initial centers to be selected randomly. Please refer to the Supplementary Material for the implementation of the nearest-neighbor, bipartition, complete-link, average-link, and k-medoids algorithms.
3.2. Protein-ligand docking
A well-known difficulty with the current scoring functions for protein-ligand docking is that they often fail to rank the docked poses correctly (Warren et al., 2006) (Figs. 1 and 2). Because both the correct and incorrect poses are similarly ranked, it greatly reduces the value of the computational results to the practitioners such as medicinal chemists for either lead identification or optimization. One reason for improper ranking is that the scoring functions themselves have errors. From an algorithmic viewpoint, the failure also originates from the formulation of the docking problem as a global optimization problem that seeks to find the minimum in a scoring function with many variables. The complexity of the scoring functions forces the current docking programs to rely on heuristics such as GA or MC to search for the minimum. However, such a formulation is not consistent with the statistical mechanics conclusion that an experimentally measured pose corresponds to the ensemble average, not necessarily the global minimum of a scoring function (Landau and Lifshitz, 1980). Assuming that a cluster represents a statistical ensemble, a good scoring function should be able to identify the best (or correct) cluster with high probability, though it may fail to assign the best score to the pose that is the closest to the experimental one. Here a best cluster means the cluster whose average RMSD, df, to the experimental pose is the smallest among all the clusters. Using our geometric partitional algorithm, we have applied both GOLD and a newly developed scoring function (Eqs. 2 and 3) to 22 sets of poses to assess which one is better suited to the identification of the best clusters. In the following we describe in detail the results on two sets of poses that represent the extreme cases among the 22 sets; our scoring function works well for the first but no 100% guarantee is provided for the second. However, even in the latter case, our scoring function still outperforms the GOLD scoring function.
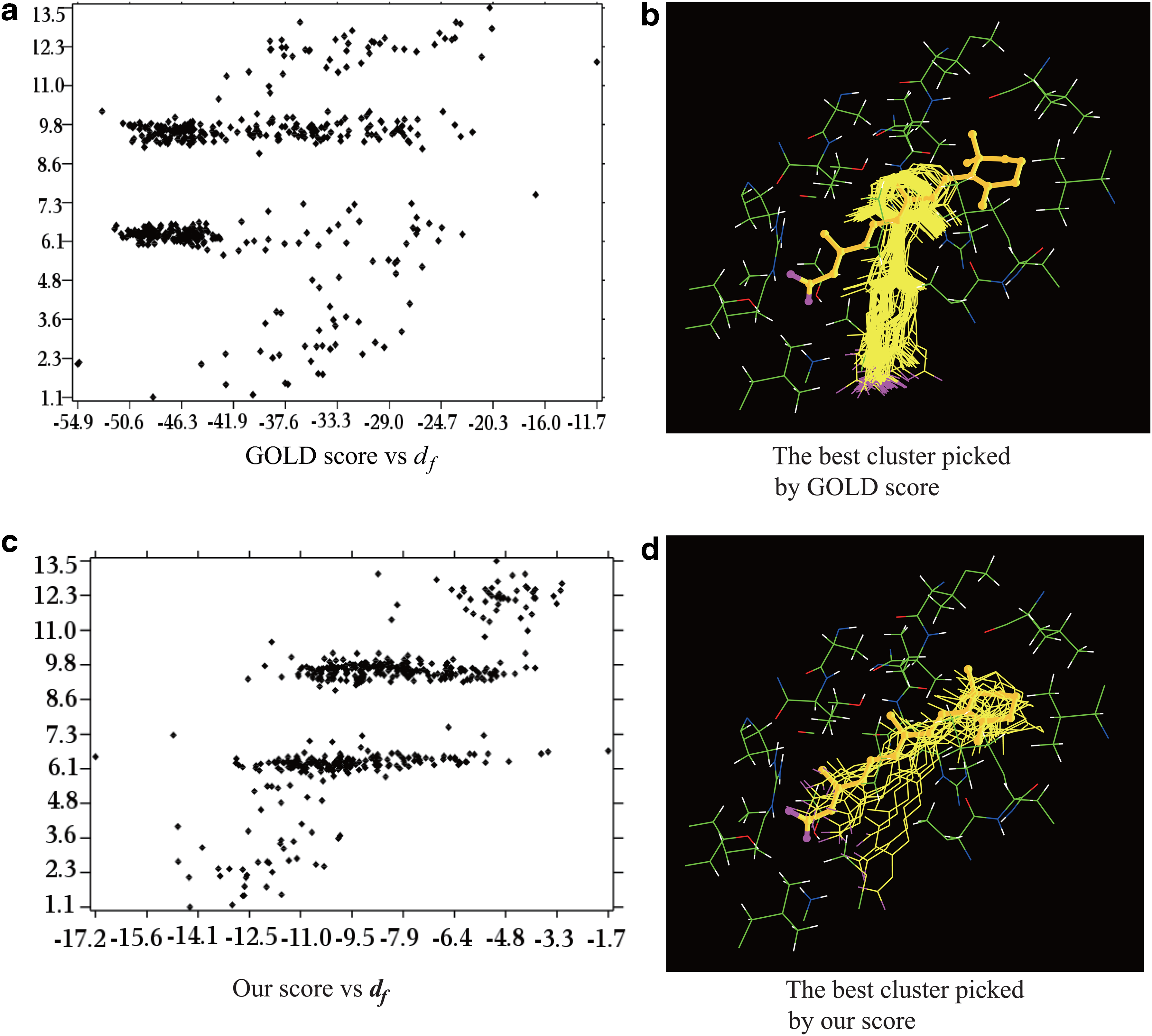
A comparison of GOLD and our scoring functions for best cluster selection for 1CBS. The x-axis and y-axis in
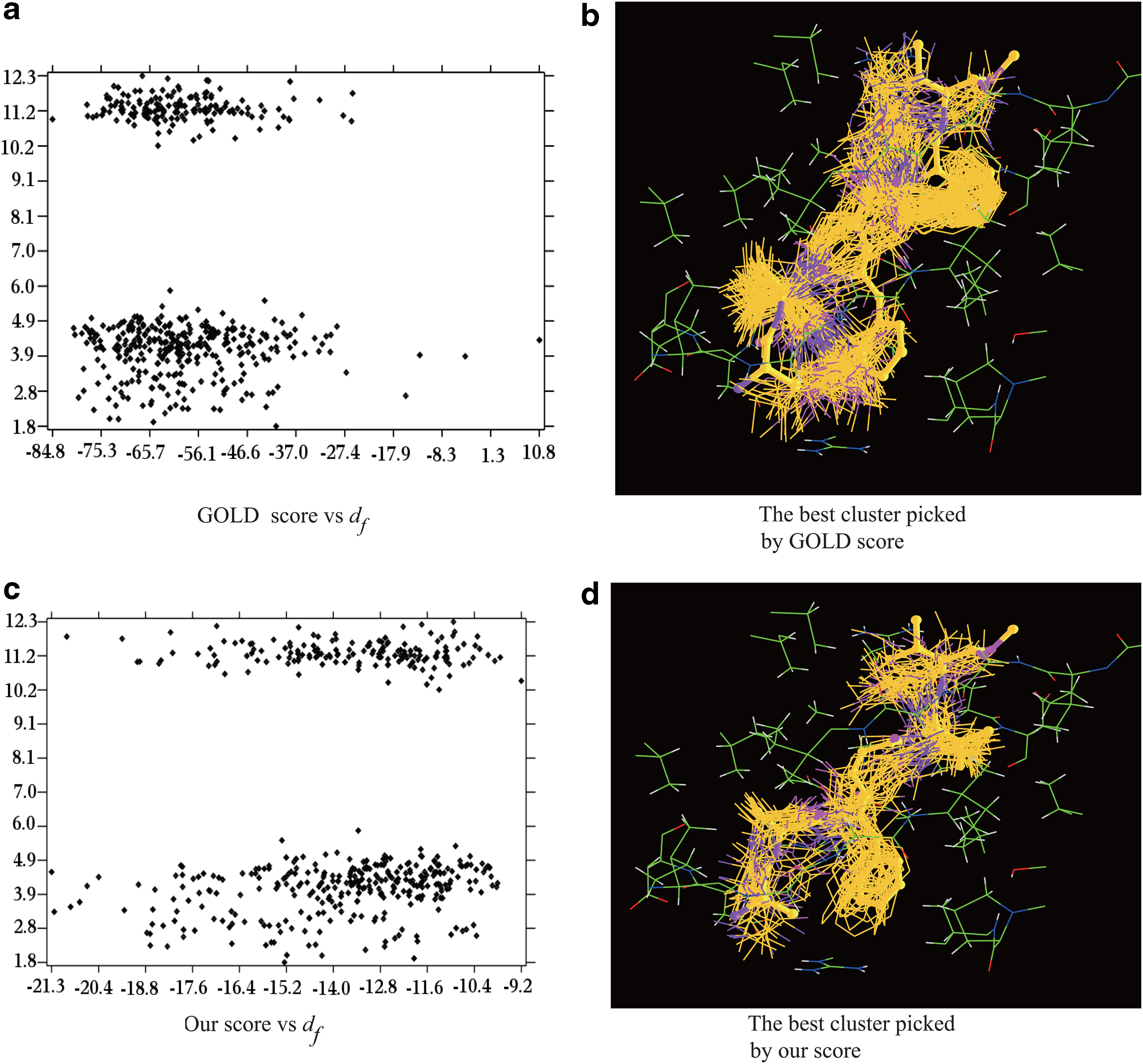
The comparison of GOLD vs our scoring function for best cluster selection for 1AAQ. The x-axis and y-axis in
The first example is human CRABP2 complexed with an RA analog (1CBS). We first generate three sets of clusters with decreasing dmax values (dmax=5.0Å, 4.0Å, 3.0Å), the average scores are then computed for each cluster (Table 2). Smaller dmax generates smaller but more accurate clusters. With dmax=5.0Å, there are four major clusters, while the poses in each of them distribute rather uniformly (Fig. 1a and c). Both GOLD and our scores could correctly select the most populated cluster as the best cluster. However, with dmax=4.0Å, GOLD picks wrongly the third cluster as the best one while our score identifies correctly the second one. With dmax=3.0, GOLD still selects the wrong cluster (the third cluster with 91 poses) (Fig. 1b) while our score identifies correctly the sixth cluster (15 poses) as the best one, with df=2.2Å (Fig. 1d). The main reason for the failure of the GOLD scoring function is that it does not include any term that accounts for the contribution of the intermolecular electrostatic interactions to the binding affinity. For CRABP2, it is well known that the electrostatic interaction between the carboxylic group of the RA analog and the two arginine residues (R111 and R132) contributes greatly to the binding (Wang et al., 1997).
The clusters are generated using three decreasing dmaxs. The listed clusters include more than 90% of the total poses. Ns, ST, SG, and df are respectively the number of structures in a cluster, the average score computed using our and GOLD scoring functions, and the average RMSD between the GOLD generated poses and the experimental pose. The lower a score, the better. The three columns with the boldfaced numbers have the lowest average score as computed by our scoring function. RMSD, root-mean-square deviation.
The second example is an HIV protease complexed with a peptide analog (1AAQ). We first generate three sets of clusters with decreasing dmax values (dmax=4.5Å, 3.5Å, 3.0Å), and the average scores are then computed for each cluster (Table 3). We start with dmax=4.5Å, since only a single large cluster is generated with dmax=5.0Å. With dmax=4.5Å, there are four major clusters while the poses in each of them distribute very uniformly (Fig. 2a and c). GOLD wrongly picks the third cluster as the best one while our score identifies the second as the best, though the most populated one has slightly smaller df. With dmax=3.5Å, GOLD still picks wrongly the third (76 poses) as the best (Fig. 2b) while our score identifies the 4th (47 poses) (Fig. 2d), 5th (33 poses), and 7th (11 poses) clusters as the best ones with respective df of 2.7Å, 3.5Å, and 11.5Å. With dmax=3.0Å, GOLD again selects the wrong cluster (the 15th cluster with 5 poses) as the best, while our score identifies correctly the 14th cluster (6 poses) as the best, with df=2.1Å. For the HIV protease, the exclusion of electrostatic interaction in the GOLD scoring function may still contribute to its failure though the latter likely plays a small role. Though our scoring function outperforms the GOLD function in all the 22 cases tested it remains challenging for our function to select the correct cluster with 100% confidence. In this case, a dozen outliers with very low electrostatic energy or ligand internal energy must be removed, otherwise, with small dmax, both our and GOLD score may mistake the wrong clusters as the best ones. A systematic approach for outlier detection and for minimizing their ill-effects are under development.
The clusters are generated using three decreasing dmaxs. With dmax = 3.0Å the clusters whose number of poses is ≤1.0% of the total number of poses are not shown. The listed clusters include more than 85% of the total. The symbols have the same meanings as those in Table 2.
The complexity of the scoring functions forces almost all of the current docking programs to rely on heuristics for optimization. However, a heuristic search may not cover the pose space adequately, as being demonstrated in the above two examples; the poses with small df to the experimental one are the minority: accounting for less than 5% of the total poses. Another noticeable feature of the set of poses generated by GOLD is that the poses inside the first few largest clusters have similar GOLD scores though their df values differ greatly. Their large variations in df contribute to the improper ranking of the best clusters by the GOLD scoring function. In contrast, the combination of our scoring function with the geometric algorithm that is capable of classifying both uniformly and nonuniformly distributed data is capable of singling out the best clusters. In other words, the geometric clustering algorithm is ideally suitable for the identification of these minor clusters populated with the best poses.
4. Algorithmic Comparison
In this section, we first describe the six data sets used widely for the performance evaluation of clustering algorithms. Then we compare our algorithm with previous five algorithms from both theoretical and practical perspectives.
4.1. The test data sets
The six test data sets (Fig. 3) are generated using the statistical language R and a machine learning benchmark database program
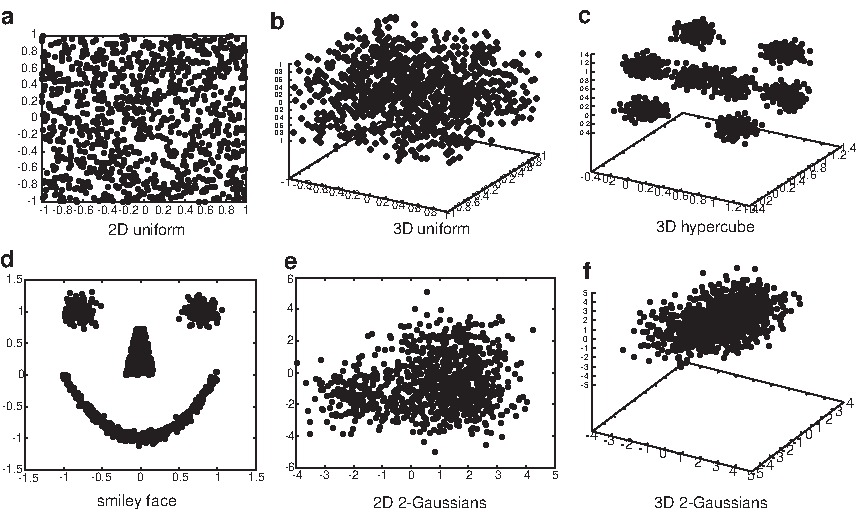
The six test data sets.
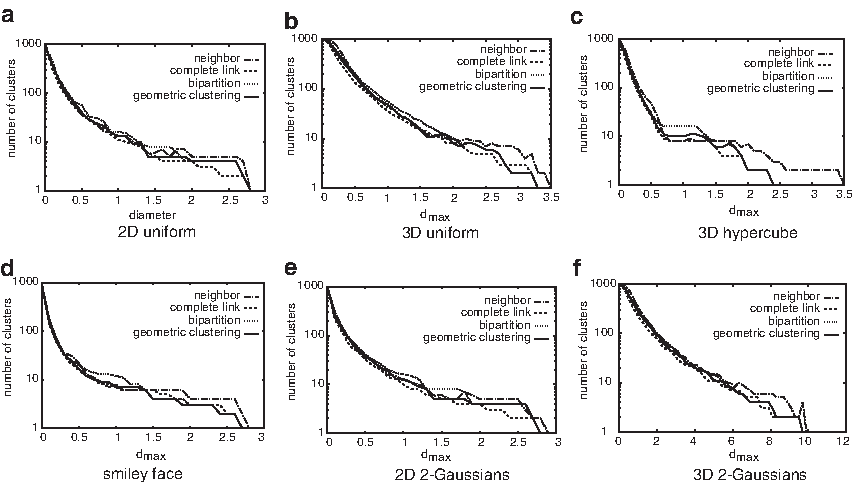
A comparison of our algorithm with the nearest-neighbor, complete-link, and bipartition algorithms on the six test data sets. The x-axis is dmax; the y-axis is the corresponding number of clusters (shown in log scale).
4.2. Algorithmic comparison
Data classification by means of clustering is a natural exploratory process for knowledge discovery, and thus clustering algorithms have found wide applications in many different areas. The clustering algorithms themselves could be classified into two groups based on their goals. Those in the first group (e.g., k-medoids and average-link) share the goal to minimize the metrics (e.g., the metric to the center of the cluster) of all the data points assigned to the same cluster, while those in the second group aim to minimize the number of clusters and simultaneously to satisfy the classification criterion that any pairwise metric in a cluster is less than a certain value. Our algorithm belongs to the second group as are the nearest-neighbor, bipartition, and complete-link algorithms. In our algorithm the seeds for new clusters are those data points that form the largest polyhedron. These points are likely to be labeled as “outliers” by the algorithms of the first group, but our algorithm initializes the clustering process with them and thus ensures them and together with their neighbors to be in different clusters.
Consequently, the representatives of the clusters from our algorithm sample the data space more uniformly than those from an average-link algorithm and much more uniformly than those from a k-medoid do. The geometric algorithm, differs largely from k-mean or k-medoid algorithms, and thus have no problems associated with them such as (a) the tendency to find hyperspherical clusters, (b) the danger of falling into local minimal, and (c) the variability in results that depends on the choice of the initial seeds. Because our algorithm classifies the data by iteratively separating them into smaller clusters according to their distances to the seeds, it will not be affected by an irregular or nonuniform distribution, as it is for a density-based clustering algorithm such as the k-medoids. The results from the applications to the clustering of both the intermediate structures and poses suggest that the k-medoids algorithm are not suitable for structural data classification.
The geometric algorithm shares some critical features with the other algorithms in the second group. However, unlike a hierarchical algorithm such as the complete-link that only optimizes an objective function locally, our algorithm takes into consideration the global information in the very beginning. The average time complexity of our algorithm is O(n2 log n) that is the same as the complexity of the agglomerative hierarchical algorithm implemented with a priority queue Day and Edelsbrunner (1984). Furthermore, the implementation suggests that our algorithm is faster than the hierarchical algorithms, most likely because the base in the logarithmic function is ≥4 rather than 2 as in a typical hierarchical algorithm. In a sense, the geometric algorithm could be looked upon as an extension of a bipartition algorithm except that the geometric algorithm may use up to any number of seed points at each step while a bipartition algorithm could use only two. An algorithm runs faster with more seeds at each step.
The geometric algorithm is somewhat similar to the minimum-diameter divisive hierarchical algorithm by (Guenoche et al., 1991). The key difference lies in how a previous cluster is divided into new clusters: in the minimum-diameter hierarchical algorithm, two new clusters are generated by an expensive search for the two balls with the minimum diameters while in our algorithm up to four new clusters are initialized with the seeds computed in linear time in terms of the number of data points in the previous cluster. In summary, the applications to both real structural data and test data have demonstrated that the performance of our algorithm is either similar to or better than that of a nearest-neighbor, bipartition, or complete-link algorithm. A possible drawback of our algorithm as well as the other algorithms in the second group is that prior knowledge is required to specify a dmax value, and several dmax values may need to be tried in order to find the best classification for a data set. As far as the structural data is concerned, it is not difficult for the practitioners to find a reasonable dmax based on the quality of the data or the required precision in the final clusters.
5. The Challenges Of Structural Data Classification
Though at present many clustering algorithms are available, as shown by Kleinberg (2002), there exists no best or universal clustering algorithm that could be applied to any type of data with equal success. The classification of structural data, especially those computed using restraints, poses particular challenges because one must take into consideration their unique features, such as the distribution of data may be both uniform and nonuniform or both regular and irregular because of the sparseness of the input restraints, the errors in the scoring function, the limited sampling provided by heuristics, and the extreme energy level degeneracy of biomolecules in solution (Landau and Lifshitz, 1980). The lack of a solid theoretical foundation for a general clustering algorithm and the difficulty in structural data clustering have led to quite some confusion about the classification of the protein global folds in the PDB (Orengo et al., 1997; Murzin et al., 1995) and make it rather tricky to compare different protein active sites. An objective classification of global folds and active sites should be based solely on a structural clustering algorithm without any manual intervention. Though, as shown in the article the geometric algorithm is both efficient and more suitable than the previous algorithms for structural data classification, much efforts are required for the design of a robust clustering algorithm with a solid theoretical foundation that could be relied on for the objective classification of global folds, active sites, and ligand poses.
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.