
Abstract
Abstract
Epigenetic landscapes represent how cells regulate gene activity. To understand their effect on gene regulation, it is important to detect their occupancy in the genome. Unlike transcription factors whose binding regions are limited to narrow regions, histone modification marks are enriched over broader areas. The stochastic characteristics unique to each mark make it hard to detect their enrichment. Classically, a predefined window has been used to detect their enrichment. However, these approaches heavily rely on the predetermined parameters. Also, the window-based approaches cannot handle the enrichment of multiple marks. We propose a novel algorithm, called SeqW, to detect enrichment of multiple histone modification marks. SeqW applies a zooming approach to detect a broadly enriched domain. The zooming approach helps domain detection by increasing signal-to-noise ratio. The borders of the domains are detected by studying the characteristics of signals in the wavelet domain. We show that SeqW outperformed previous predictors in detecting broad peaks. Also, we applied SeqW in studying spatial combinations of histone modification patterns.
1. Introduction
T
Identifying enriched regions of histone modification is still challenging because of unique and diverse distribution patterns for each histone modification mark. To deal with it, a predefined window has been used to detect their enrichment (Zang et al., 2009; Heinz et al., 2010; Rashid et al., 2011). Some methods merged the identified narrow peaks within a certain range to obtain the peaks stretched across broad areas in the genome (Micsinai et al., 2012). As previous comprehensive analyses demonstrated, however, their results heavily relied on the predefined parameters (Micsinai et al., 2012).
To identify the regions enriched for histone modification marks without using a predefined window and study their landscapes systematically, we propose a novel wavelet-based method, called SeqW, which employs wavelet transform (WT) and wavelet footprint to implement zooming and border detection. In the wavelet domain (WD), SeqW takes two steps to detect enriched regions. First, it identifies enriched regions roughly by applying downsampling, which implements zooming out the signals in the WD. Second, it determines the edges of the roughly identified enriched regions by applying zero-crossing, which draws the line by connecting characteristic points of the signals over the WD (Nguyen et al., 2010).
Previously, WT has been used to deal with epigenomic data (Day et al., 2007; Zhang et al., 2008). However, their uses of WT were limited to removing noises. Compared with them, SeqW extensively uses the features in the WD to capture the characteristics of the signals. SeqW can deal with the signals of multiple histone modification marks. There have been several integrative analyses that used multiple histone modifications (Ernst et al., 2011; Hoffman et al., 2012; Won et al., 2013; Yu et al., 2013). They were mainly designed to annotate the genome by calling the segments enriched for epigenomic signals. SeqW is different in that it detects the composition and the spatial distribution of histone modification data rather than annotation.
To evaluate the performance of peak detection, we use the simulated data as well as H3K36me3 mark in murine adipocytes (Mikkelsen et al., 2010). We show that the annotation by SeqW well matched with the known gene annotation and that the algorithm SeqW employed is robust to noise. Furthermore, we introduce how SeqW can be used to cluster genes based on their epigenetic landscapes.
2. Methods
SeqW is composed of two steps. SeqW uses Haar Wavelet Moving (HWM) (Nguyen et al., 2014) for rough detection and zero-crossing for fine-tuning of the identified enriched region. HWM is a shift-invariant downsampling wavelet method designed to detect large domains by mimicking a zooming approach. The shift invariance of HWM enables tracing the changes of signals (Mallat, 2009). To predict edges accurately, HWM is combined with the zero-crossing method. Zero-crossing points are where the sign of a function changes, which are equivalent to the characteristic points of a function. To apply the zero-crossing method, we modeled a peak with mixture of Gaussians (MoG). In general, any signal can be represented as a sum of multiple Gaussian signals (Mallat, 2009).
Any signal can be represented as MoG
where μi and σi are the center and the standard deviation of a peak, respectively. To estimate the parameters of MoG, SeqW uses the zero-crossing lines across the multiwavelet scales. A zero-crossing is a point where the sign of a function changes. A zero-crossing line is obtained by connecting the zero-crossing points obtained over the multiwavelet scale s.
Continuous WT converts signals to the WD using a convolution operator.
where s denotes the scale, u is the genome position, and
where
The edges of the MoG can be obtained by taking derivative
where Γ is a gamma function (Mallat, 2009) and n is the order of derivative of the Gaussian wavelet. Replacing
where
Then, the iFFT is
The third derivative of the Gaussians (DOG3) is when n = 3.
The zero-crossing points are the parameters of the Gaussians when
If s is small compared to σi, Equation 17 becomes
In summary, u0(s) in Equation 9 draws a line over the scale s. Equation 10 indicates that the zero-crossing line approximates to
3. Results
3.1. The SeqW procedure
SeqW takes two steps to determine the location and the edges of the enriched regions. To identify the rough location of enriched regions, SeqW uses downsampling in WD. However, downsampling in WD often induces shift variance, which causes large changes in the wavelet coefficients even with small shifts in the time domain signals (Mallat, 2009). Therefore, it is difficult to trace the cause of the changes in the shift-variant WT. To overcome this, we used HWM to downsample data without suffering shift variance (Nguyen et al., 2014). HWM determines the location of enriched regions with significantly reduced computational costs compared with other previous wavelet-based algorithms.
However, the zooming approach that HWM uses makes it difficult to detect the boundary of the enriched regions. To determine the edges accurately, SeqW employed the zero-crossing approach (Nguyen et al., 2010). SeqW modeled an enriched region with MoGs and applied the zero-crossing to obtain the parameters of MoGs. The boundaries of MoGs become the edges of the roughly identified enriched regions.
Figure 1 illustrates how SeqW detects the enriched region of H3K36me3 mark enriched around the NcK2 gene locus. The mapped H3K36me3 data (Fig. 1A) in adipocytes (Mikkelsen et al., 2010) were transformed to WD by HWM (Fig. 1B). Small segments whose intensities are higher than the local background level were identified (Fig. 1C). After downsampling, the neighboring segments were connected to form a broad peak (Fig. 1D). Next, the edges of the enriched regions were obtained using the zero-crossing approach. The middle panel of Figure 1 illustrates the zero-crossing lines for the H3K36me3 signals. Zero-crossing lines over the wavelet scales were obtained by connecting the zero-crossing points. There were hundreds of zero-crossing points in the lower wavelet scales that extended to the zero-crossing points in high-wavelet scales. On the contrary, the signals in high frequencies (usually noise) eventually lose its zero-crossing points in high-wavelet scales. This shows that the border detection using zero-crossing is robust to noise. In this example, the two long zero-crossing lines around the boundary of the large segment obtained by HMW were selected as the edge of the large segment.
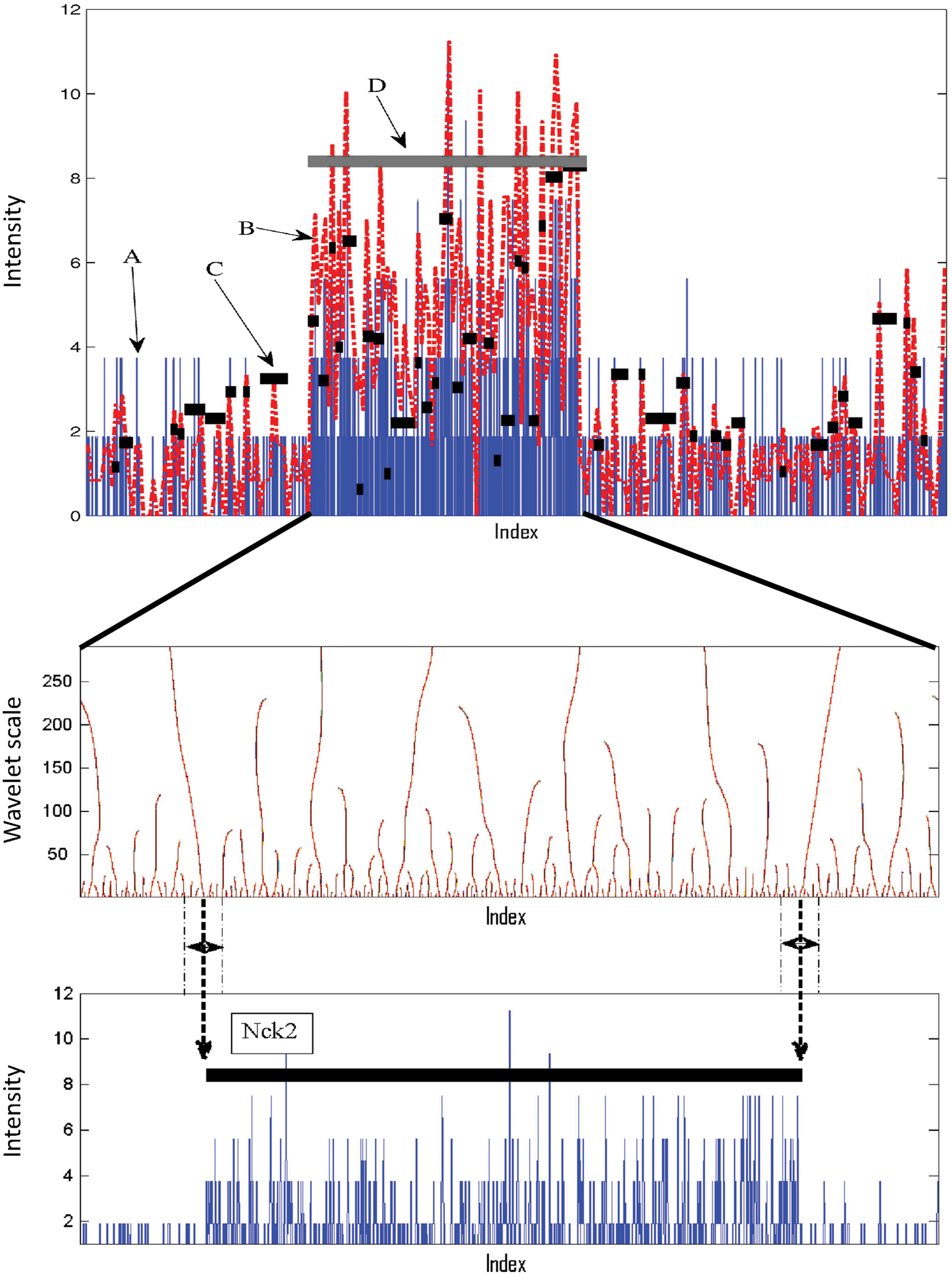
Peak detection procedure using SeqW. Top: (A) the input data (H3K36me3) in blue, (B) the wavelet signals in red, (C) small segments obtained using HMW, and (D) the final prediction after merging neighboring segments using the zooming method. Middle: the zero-crossing lines over wavelet scales. Bottom: the longest zero-crossing lines around the boundary of the large segment become the border of the large segment.
3.2. Performance comparison
We evaluated the performance of SeqW by assessing the gene body detection using epigenomic data. For this evaluation, we included SICER (Zang et al., 2009), RSEG (Song and Smith, 2011), Qeseq (Micsinai et al., 2012), and ChromHMM (Ernst et al., 2010). We chose RSEG (Song and Smith, 2011) and Qeseq (Micsinai et al., 2012) because they showed outstanding performance in the previous comprehensive benchmark (Micsinai et al., 2012). We included ChromHMM because it annotates using multiple epigenetic marks even though it is not designed to detect broad peaks.
We used H3K36me3 in mouse adipocytes (Mikkelsen et al., 2010) for this assessment. As H3K36me3 is relatively depleted at promoters and more enriched toward the 3′ end, we regarded a prediction true if a prediction overlaps with the gene body of the GENCODE (Harrow et al., 2012) genes more than 75% (true positive); otherwise, we regarded it as false negative.
Figure 2 compares the total number of correct predictions against the total number of false predictions. Unexpectedly, the performance of Qeseq was worse than that of ChromHMM in our test even though ChromHMM is designed to annotate enriched regions. RSEG showed better performance than SICER and Qeseq. Our test demonstrates that SeqW performed best in this assessment. We also applied control input by subtracting it from the H3K36me3 signals. Interestingly, SeqW performed better without using control input. This indicates that our scheme obtaining background levels from the given data (H3K36me3) performed effectively.
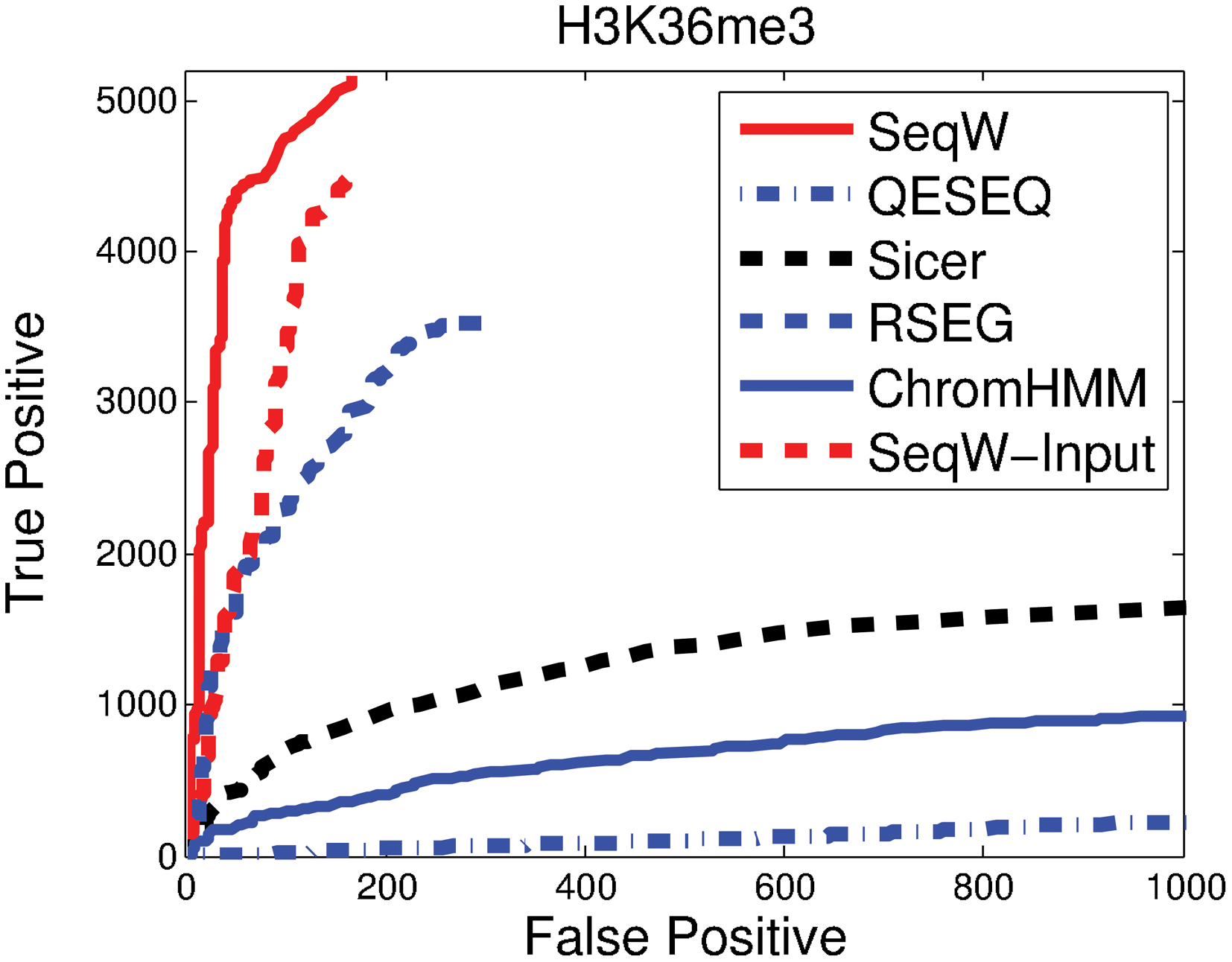
Performance comparison of various methods using H3K36me3. We compared the number of true predictions against the number of false predictions for SeqW, Qeseq, SICER, RSEG, and ChromHMM. Additionally, we also tested SeqW when we applied input control. H3K36me3 of 3T3L1 cells was used.
It is also noteworthy that SICER and Qeseq took more than 30 minutes to obtain the results, while SeqW and RSEG took around 15 minutes for this test (Intel Core CPU 870@2.93 GHz, 16 GB RAM). ZINBA implemented a mixture regression approach using covariate values such as G/C content, mappability score, and local background to detect peaks (Rashid et al., 2011). We could not include the result of ZINBA (Rashid et al., 2011) because of its long running time (ZINBA did not produce any result after 3 days in our system).
Figure 3 compares how each predictor detects the broad peaks of H3K36me3. The enriched H3K36me3 mark is associated with the body of the annotated Refseq genes. The SeqW results correctly matched with the gene annotation when they were associated with enriched H3K36me3 signals stronger than local background. SICER and Qeseq correctly predicted the body of Mrps9 and Uxs1, but their annotations were highly truncated. Qeseq missed Nck2, which has relatively lower H3K36me levels. Compared with SeqW, SICER only predicted a small portion of the gene body. RSEG showed similar results with SeqW except for a possible false prediction upstream of Fhl2. In the genomic regions we tested, Tgfbrap1 and Fhl2 are located close. SeqW and RSEG predicted them as one segment. However, SICER and Qeseq predicted them with more than four segments. Note that a depleted region of H3K36me3 is located inside the body of Tgfbrap1, not in the intergenic region. This may tell that it is not easy to predict the body of Tgfbrap1 and Fhl2 separately using only the H3K36me3 mark. These results as well as the genome-wide comparison (Fig. 2) show that SeqW's broad peak detection is accurate and well matches with the known annotation.
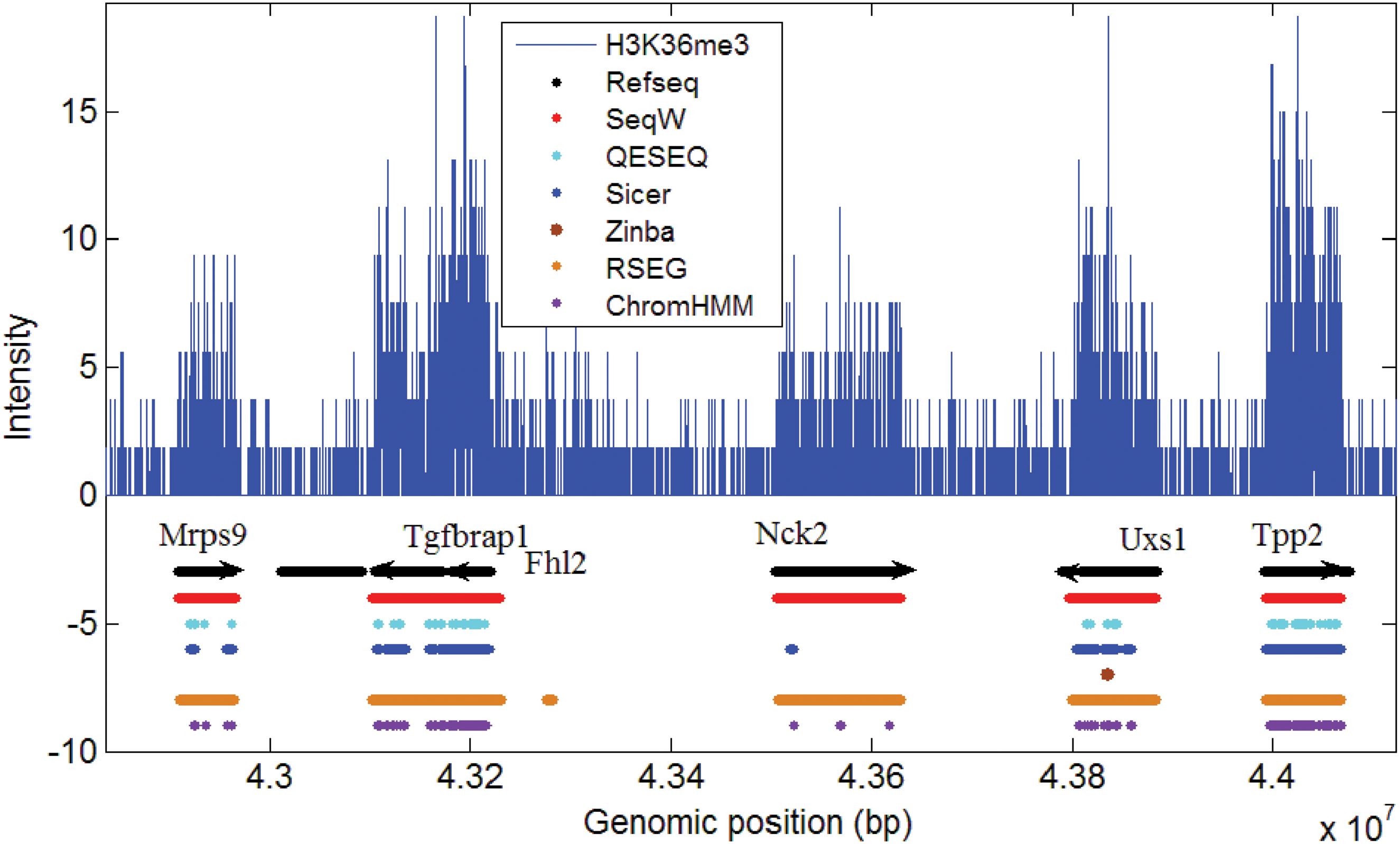
Evaluation of enrichment calling from various methods against the annotated genes. The annotation results of various methods were shown in the tracks. Refseq annotations were shown in black arrowed line. The annotation and the border of SeqW well matched with the known genes.
3.3. SeqW is robust against noise
To understand the algorithmic advantages of SeqW, we performed assessments by comparing the performance of the predictors in various noise conditions. For this, we used the simulated data by collecting H3K36me3 from the top 80% of the highly expressed genes and adding Poisson noises. We included the results of SICER (Zang et al., 2009) and RSEG (Song and Smith, 2011) as they showed good performance in our previous genome-wide test. On the basis of the true and the false-positive rates, we drew receiver operating characteristic curves and obtained the area under curve (AUC).
Figure 4 compares the AUCs of the three methods we tested in various noise conditions. It is expected that performance becomes worse with smaller signal-to-noise ratios (SNRs). When SNR was 6.2, the three methods we tested showed comparable results. Interestingly, SeqW did not lose AUC considerably when noises were added. For example, when SNR was reduced to 1.4 from 6.2, the AUC dropped by 0.44 and 0.99 for SICER and RSEG, respectively. Compared with them, the SeqW has lost AUC by only 0.04. This demonstrates that SeqW is robust against noise.
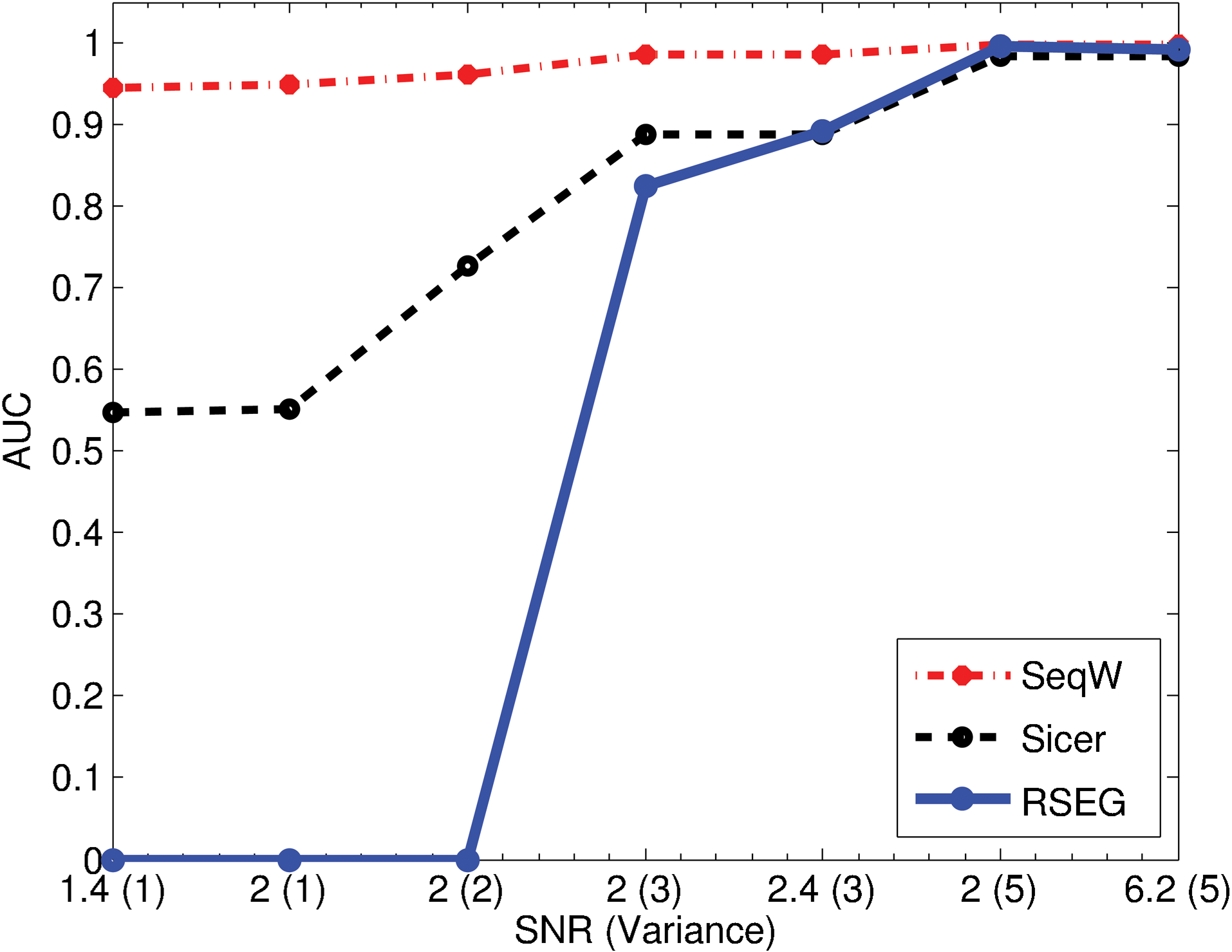
Performance assessment using the simulated data. We compared the performance of SeqW, SICER, and RSEG over various signal-to-noise ratios (SNRs) and the Poisson variances. SeqW was robust to the added noise.
3.4. SeqW clusters genes based on spatial combination of epigenomic pattern
The unique property of SeqW compared with other peak callers is allowing multiple histone marks. This property enabled SeqW to cluster genes based on their epigenomic environments composed of multiple histone marks. To cluster genes based on their epigenomic landscapes, we applied SeqW to four histone modification marks (H3K4me3, H3K27ac, H3K27me3, and H3K36me3) in adipocytes (Mikkelsen et al., 2010).
Using SeqW, we investigated the combination of four histone modification marks around the active genes. SeqW identified 12,338 promoters with strong H3K4me3 levels, among them 8,636 (70.0%) were located at promoters of annotated genes (p<1.0e-130). This number is comparable with previous approaches that predicted promoters using histone modification (Won et al., 2010; Ernst et al., 2011). Among 12,338 predictions, 11,507 (93.3%) promoters were enriched with both H3K4me3 and H3K27ac. Majority of the genes were enriched with H3K36me3 (9,814/12,338 = 59.5%). Around half of them (4,806/9,814) were enriched with H3K27me3 upstream of promoter regions (Table 1).
SeqW classifies genes based on the combination of histone modification. Note that the enriched region of the histone modification marks (H3K4me3, H3K27ac, H3K36me3, and H3K27me3) are different. We studied the composition of histone modification at promoter regions (H3K4me3 and H3K27ac), gene body (H3K36me3), and upstream promoter regions (H3K27me3). Each case is composed of four digits “H1H2H3H4,” which denote H3K4me3, H3K27ac, H3K36me3, and H3K27me3, respectively. Each digit can be 1 (enriched), 0 (depleted), or x (don't care). For example, the case “1110” means that H3K4me3, H3K27ac, and H3K36me3 are enriched, and H3K27me3 is depleted.
We further associated the identified clusters with the gene expressions of four time points during adipogenesis (day -2, 0, 2, and 7) (Mikkelsen et al., 2010). As the identified regions were different depending on the histone marks, we considered three genomic regions separately: promoter region (H3K4me3 and H3K27ac), gene body (H3K36me3), and upstream promoter region (H3K27me3). For simplicity, we denoted the composition with four digits “H1H2H3H4” for H3K4me3, H3K27ac, H3K36me3, and H3K27me3, respectively, even though they do not co-occur at the same location.
The genes with H3K4me3, H3K27ac, and H3K27 (“1110”) have significantly higher expression levels than the genes with all four marks (“1111”) (p < 6.40e-3 at day 7), suggesting that the H3K27me3 upstream promoter has a repressive role (Fig. 5). However, the role of H3K27me3 is not clear when only with H3K4me3 (“1000” versus “1001”; p > 0.6) because of the pausing of RNA polymerase II at the promoter region of these genes (Guenther et al., 2007; Chen et al., 2011; Perez-Lluch et al., 2011). We found that expression of the genes with H3K36me3 and without H3K27ac (case “1010”) is relatively low. This matches with the previous feature selection results showing that H3K27ac has more predictive power than H3K36me3 or H3K4me3 (Karlic et al., 2010; Chen et al., 2011).
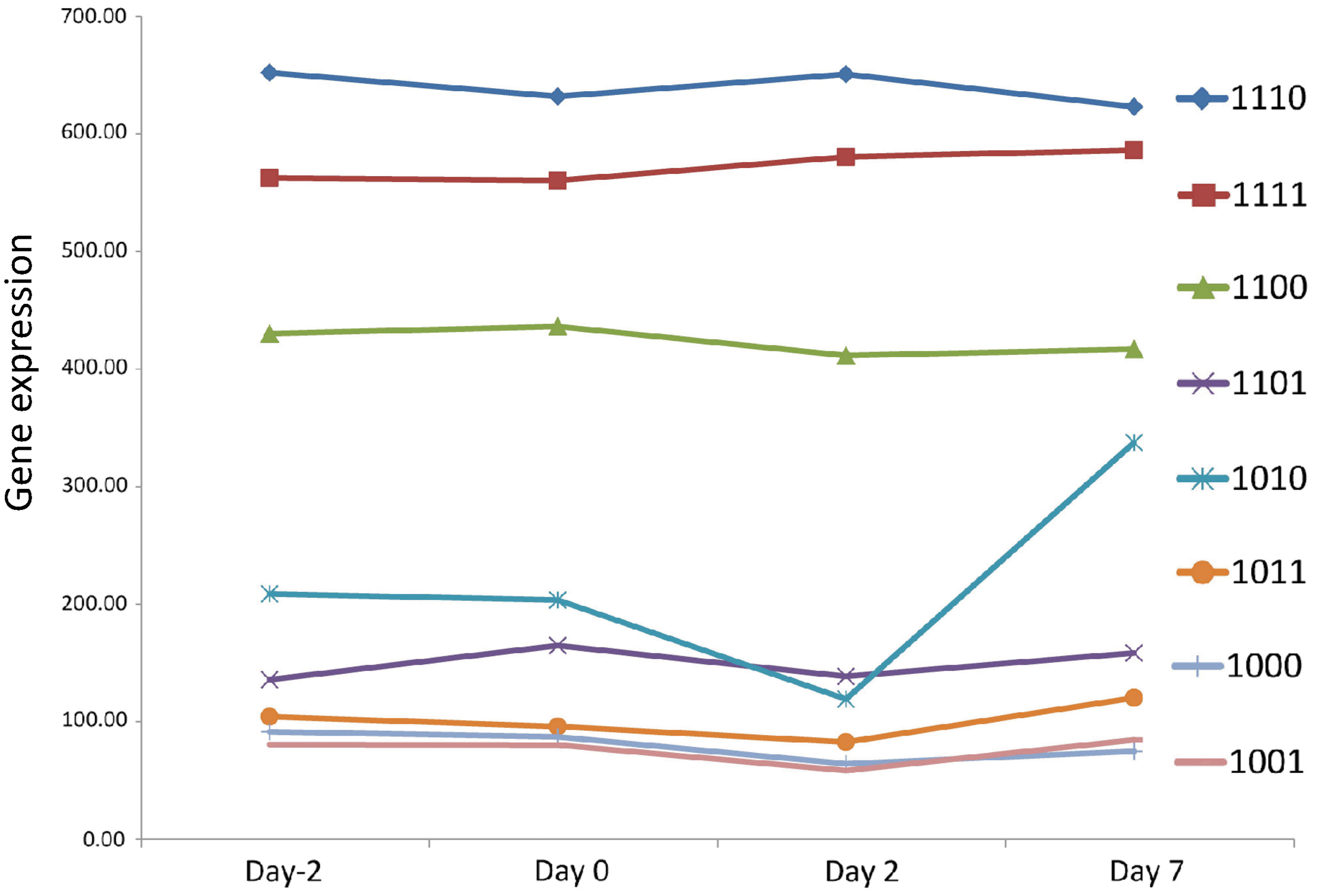
Gene expression associated with the gene clusters that were identified by SeqW using four histone modification marks. Gene expression was compared for each gene cluster based on the combination of histone modification marks. Each case is composed of four digits “H1H2H3H4,” which denote H3K4me3, H3K27ac, H3K36me3, and H3K27me3, respectively. Each digit can be 1 (enriched), 0 (depleted), or x (don't care). For example, the case “1110” means that H3K4me3, H3K27, and H3K36me3 are enriched, and H3K27me3 is depleted.
4. Discussion
Understanding epigenomic regulation is important in studying conditions for gene regulation. We present a systematic way to exploit the epigenomic enrichment of multiple histone modification marks using wavelet. For peak detection, we applied a zooming approach by downsampling the data in WD. Downsampling provided global views of epigenetic landscapes while increasing SNRs. However, downsampling may deteriorate the accuracy of edge detection. To compensate this, we used the zero-crossing approach, which fine-tunes the edge detection while effectively removing the signals in high frequencies.
Our comparisons showed that SeqW correctly annotated the body of the active genes based on the enrichment of H3K36me3 signals, while other predictors identified only part of the enriched regions or produced truncated results. Previous methods, including the methods we tested, used a fixed window and merged neighboring segments (Zang et al., 2009; Qin et al., 2010; Rashid et al., 2011; Micsinai et al., 2012). This shows that the methods using a window can produce truncated results easily in broad peak detection, while our zooming approach detects the peak without suffering too much truncation.
RSEG showed outperforming performance over SICER and Qeseq in our test (Figs. 2 and 3). However, the performance of RSEG was deteriorated in severely noisy conditions (Fig. 4). Interestingly, the performances of SeqW were not affected much by the added noise. This can be because of the advantages of using wavelet, which filters out the unwanted noises with high frequencies.
We also showed a potential use of SeqW in exploiting epigenomic landscapes composed of multiple histone modifications. We applied SeqW in clustering genes based on epigenomic compositions. Instead of using averaged intensities of histone modification marks, we used the identified enriched regions. Because we consider the epigenomic data as multidimensional signals, SeqW obtained the broad peaks of all histone modification marks simultaneously. Using the identified regions, we further studied the composition of histone modification marks in association with gene expression. Our study suggests that signal processing methods can be applied to study the genome. Advanced approaches used in signal processing will help understand the diverse epigenetic languages for gene regulation.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health grant R21DK098769-01 and a pilot award from the DRC at the University of Pennsylvania from a grant sponsored by NIH DK to K.-J.W. We thank the Univeristy of Pennsylvania Diabetes Research Center (DRC) for the use of the Functional Genomics Core Core (P30-DK19525).
Author Disclosure Statement
No competing financial interests exist.