
Abstract
Zona-free somatic cell transfer (SCT) and embryo aggregation increase throughput and efficiency of cloned embryo and offspring production, respectively, but both approaches have not been widely adopted. Cloning efficiency is further improved by cell cycle coordination between the interphase donor cell and metaphase-arrested recipient cytoplast. This commonly involves inclusion of caffeine and omission of calcium to maintain high mitotic cyclin-dependent kinase activity and low calcium levels, respectively, in the nonactivated cytoplast. The aim of our study was to integrate these various methodological improvements into a single work stream that increases sheep cloning success. We show that omitting calcium during zona-free SCT improved blastocyst development from 6% to 13%, while caffeine treatment reduced spontaneous oocyte activation from 17% to 8%. In a retrospective analysis, morula aggregation produced high morphological quality blastocysts with better in vivo survival to term than nonaggregated controls (15% vs. 9%), particularly after vitrification (14% vs. 0%). By combining cytoplast cell cycle control with zona-free embryo reconstruction and aggregation, this novel SCT protocol maximizes the benefits of vitrification by producing more cryoresilient blastocysts. The presented cloning methodology is relatively easy to operate and further increases throughput and efficiency of cloned embryo and offspring production. Integration of additional reprogramming steps or alternate donor cells is straightforward, providing a flexible workflow that can be adapted to changing experimental requirements.
Introduction
To successfully produce animals by cloning, the cell cycles of the donor cell and recipient cytoplast must be coordinated to maintain the correct ploidy in the reconstructed embryo. The standard approach for somatic cell transfer (SCT) is to match a diploid G0/G1 donor with a recipient arrested at metaphase of the second meiotic division (MII), which can result in viable offspring (Wilmut et al., 1997).
After transfer of the interphase nucleus, the MII environment with high levels of mitotic cyclin-dependent kinase (M-Cdk) complexes causes nuclear envelope breakdown (NEBD) and premature chromosome condensation (Lee and Campbell, 2006), while also reprogramming the donor epigenome to a totipotent state. After the diploid G0/G1 donor cell nucleus has condensed from high M-Cdk activity, artificial activation (e.g., with the calcium ionophore ionomycin) decreases M-Cdk activity and initiates formation of a single pseudo-pronucleus (Dinnyes et al., 2016). This is dependent on subsequent treatment with protein kinase inhibitor 6-dimethylaminopurine (6-DMAP), which ensures correct transition into interphase, while also preventing the extrusion of a pseudo-polar body (Szöllösi et al., 1993). Thus, the entire donor cell genetic material is maintained, and cloned offspring can be produced.
This cell cycle coordination has been difficult to achieve in some species due to activation upon donor cell electrofusion or spontaneous oocyte activation. The former was resolved by fusing in the absence of calcium (Boquest et al., 2002), while the latter was prevented by treatment with a proteasome inhibitor to maintain M-Cdk activity (Zhou, 2003). Another approach to increase M-Cdk activity is treatment with caffeine (Smythe and Newport, 1992). Treating sheep oocytes with caffeine increased M-Cdk activity, occurrence of NEBD, and blastocyst cell numbers, and mitigated oocyte age-related changes in cloned embryos (Lee and Campbell, 2008).
In primates, both removing calcium from the fusion buffer and caffeine treatment were important for the production of cloned blastocysts (Mitalipov et al., 2007; Tachibana et al., 2013). Both electrical activation (Peura et al., 2003) and spontaneous oocyte activation (Campbell et al., 1994) have been observed in sheep, but mitigating treatments have not been routinely incorporated into sheep SCT protocols.
Improved ease of operation and increased throughput have been achieved with zona pellucida-free SCT in cattle (Oback et al., 2003), sheep (Ritchie et al., 2005), and horse (Lagutina et al., 2005). Zona removal simplifies and accelerates enucleation and electrofusion, overall doubling the throughput of cloned embryo and offspring production (Oback et al., 2003). Another commonly used assisted reproductive technology is embryo cryopreservation by vitrification (Mandawala et al., 2016). Vitrification eases the logistical constraints for generating offspring since recipient ewes do not need to be concurrently synchronized when producing embryos. Vitrification was successfully used for sheep cloning in one study (Peura et al., 2003) and recently in goats (Hajian et al., 2020), but overall, there are few reports on its effect on embryo survival for cloned sheep embryos.
Even with improvements to cloned embryo reconstruction, somatic cloning efficiency remains low due to poor embryo survival in vivo. This is particularly relevant for sheep, where the survival of transferred blastocysts into healthy, viable offspring is only around 1% in large-scale studies (Deng et al., 2013; Fan et al., 2018; Loi et al., 2006; Peura et al., 2003; Wilmut et al., 1997), compared to ∼30% for in vitro-fertilized blastocysts (Ishwar and Memon, 1996).
A promising approach to increase cloning efficiency is embryo aggregation, which originally increased embryo survival fourfold to eightfold in mice (Boiani et al., 2003). For cattle, aggregating at the one-cell stage showed no improvement for somatic clones, but cloning efficiency more than doubled for embryonic clones (Misica-Turner et al., 2007). The effect was confirmed in pigs with aggregation at the four-cell stage, improving embryo survival from 0% to 3% (Siriboon et al., 2014). Another approach that may increase cloning efficiency is to use high-quality in vivo-matured oocytes. For embryonic clones in sheep, this resulted in threefold to fourfold higher embryo survival (Wells et al., 1997).
The aim of our study was to combine the various methodological improvements into a single work stream to test their added effect on sheep cloning efficiency. We show that preventing premature activation improved development to the blastocyst stage, while morula aggregation improved postvitrification viability and blastocyst survival to term. Overall, our novel SCT protocol integrates several methodological improvements to produce live lambs with increased efficiency.
Materials and Methods
Animal ethics
Investigations complied with the New Zealand Animal Welfare Act 1999 and were approved by the Ruakura Animal Ethics Committee, covered by approvals #14696, #14697, #14693, #14641, #14258, #14395, and #14396.
Experiment 1: oocyte premature activation and embryo development
Effect of calcium and electrical stimulation on parthenogenetic development
Slaughterhouse ovaries were collected from mature ewes (>1 year old) in the New Zealand breeding season from February to July (latitude ∼37°S, Waikato, New Zealand). Follicles between 1 and 5 mm were aspirated and oocytes were in vitro matured as described (Oback and Wells, 2003). Oocytes with a polar body were collected at 19 hours post onset of maturation (hpm) in H199 + 3 mg/mL bovine serum albumin (BSA) (SCT base medium) and kept in SCT base medium until 22 hpm, when they were randomly allocated into four groups.
To quantify oocyte activation by electrical stimulation in the presence of calcium, the first two groups received an electrical stimulus (two direct current 10 μsecond pulses at 2 kV/cm, mimicking the fusion pulse) in either (i) calcium-containing fusion buffer (260 mM
To determine maximal and baseline activation rates, the other two groups were kept in SCT base medium until 26 hpm before either (iii) incubating them for 4.5 minutes in 5 μM ionomycin in HEPES-buffered SOF + 1 mg/mL BSA, followed by 3–4 hours in 2 mM 6-DMAP in ESOF or (iv) keeping them in ESOF for 3–4 hours. Activation was assessed by cleavage in all groups at 51 hpm (n = 3 independent experiments).
Effect of caffeine on parthenogenetic embryo development
Nonactivated oocytes were exposed to different concentrations of caffeine (0 vs. 5 vs. 10 mM) in SCT base medium from 20.5 to 26.5 hpm and transferred into ESOF. Cleavage, as a result of spontaneous activation, was assessed at 51 hpm (n = 9 independent experiments comparing 0 mM caffeine to 5 mM caffeine [n = 6], or 10 mM caffeine [n = 2], and both 5 and 10 mM caffeine [n = 1]).
Effect of calcium on SCT embryo development
A detailed step-by-step procedure and media description, based on a modified protocol for cattle zona-free embryo reconstruction (Oback and Wells, 2003), can be found on protocols.io (Mclean et al., 2020).
Briefly, G0 ovine fetal fibroblasts (“OFF3,” Supplementary Table S1) were obtained by culture in medium containing 0.5% fetal bovine serum (FBS) for 5 days and harvested by trypsinization. Zona-free MII oocytes were enucleated ∼19 hpm. Serum-starved donor cells were attached to a cytoplast in 10 μg/mL phytohemagglutinin. Couplets were automatically aligned using a few seconds of ∼80 V/cm alternating current and fused with two direct current 10 μs pulses of 2 kV/cm in 270 mOsm calcium-containing or calcium-free fusion buffer (runs evaluated for fusion: +Ca [n = 9] and −Ca [n = 30] and total reconstructs: +Ca [N = 457] and −Ca [N = 1873]). For double cytoplast SCT, a second cytoplast was attached to the fused reconstruct in phytohemagglutinin and fused at 1.5 kV/cm (Delaney et al., 2007).
Caffeine (5 mM) was included in the base medium from 20.5 to 26.5 hpm. Activation by ionomycin (5 μM; 4.5 minutes)/6-DMAP (2 mM; 4 hours) was carried out 3–4 hours postfusion (∼27 hpm). Groups of 10 embryos were cultured in 20 μL drops of ESOF at 7% oxygen, each embryo occupying a manually prepared microwell. Half the media was changed on day 2, and after 4 days, the media were replaced with late SOF (LSOF). On day 5, the embryos were transferred into single in vitro culture (IVC) in individual 5 μL LSOF drops. Blastocysts were morphologically graded on day 6 or 7 according to the International Embryo Technology Society guidelines (Robertson and Nelson, 1998) (number of runs evaluated for blastocyst development: +Ca [n = 9] and −Ca [n = 12]; number of embryos placed into in vitro culture [IVC]: +Ca [N = 394] and −Ca [N = 528]).
Experiment 2: SCT embryo development after aggregation
Aggregation
As SCT donors, we used four different parental fibroblast cell lines from fetal and adult sources of different sheep breeds, including both genetically modified cell strains and nonmodified parental cells (Supplementary Table S1).
Aggregation was carried out with either precompacting morulae showing little fragmentation, regular sized blastomeres, and advanced numbers of blastomeres (days 5–6 of IVC) or with compacted morulae. Morulae were incubated in a disaggregation medium (HEPES-buffered SOF [HSOF] without calcium and BSA, containing 0.1 mg/mL PVA, 0.2 g/mL ethylenediaminetetraacetic acid, and 5 μg/mL cytochalasin-B) for 2 minutes, followed by a wash through HSOF, and then transferred to LSOF. The embryos were disaggregated by blowing up and down with a pipette (inner diameter of 50-70 μm) until the embryo was broken into two to three fragments.
Two disaggregated embryos of similar developmental stage were combined by repeated pipetting inside a mouth pipette (approximately the diameter of the reconstituted embryo). Each aggregate was placed in a microwell, allowing the pieces to compact into a single morula. After 5–6 hours, morulae were removed from the microwell and transferred into a fresh culture drop. This was done before the blastulating embryo could adhere to the edges of the microwell, resulting in damage to the trophoblast during transfer. Blastocysts were morphologically graded as above.
In vitro- versus in vivo-matured oocytes
In vitro-matured oocytes were prepared as described above. In vivo oocytes were obtained after superovulation of donor ewes and oviductal flushing. A controlled internal drug release (CIDR) device for sheep (Zoetis, Kalamazoo, MI) was inserted 17 days before cloning and 125 μg estroPLAN® (cloprostenol sodium, prostaglandin F2α analogue; Parnell Technologies Pty Ltd., Australia) was injected 10 days before SCT. Starting 5 days before SCT, injections of 32 mg Folltropin®-V (porcine pituitary follitropin extract containing follicle-stimulating hormone; Bayer, Germany) were given at 8 a.m. and 5 p.m., with the final injection 2 days before SCT (eight dosages in total). The CIDRs were withdrawn at 5 p.m., 3 days before SCT (64 hours before flushing). An injection of 150 μg Ovurelin® (gonadotropin-releasing hormone analogue; Bayer) was given at 5 p.m. the day before SCT (16 hours before flushing) and the oocytes were flushed from the oviducts at 8–10 a.m. on the day of SCT.
The zona pellucida of ovulated oocytes was removed by a combination of pronase treatment and mechanical dissection. The oocytes were first treated with 2% pronase solution (dissolved in H199-FBS, 0.1 mg/mL PVA) for 5 minutes to loosen the zona pellucida and then washed with 0.2 M sucrose in Embryo Hold (modified LSOF with 20 mM 3-morpholinopropane-1-sulfonic acid (MOPS) and 5 mM NaHCO3, and without BSA and 2,4-dinitrophenol), including 5 μg/mL cytochalasin B and 0.1 mg/mL PVA, to osmotically shrink the oocyte and increase the perivitelline space for microsurgery.
Zona-enclosed oocytes were immobilized by adherence to a Petri dish in the same solution without PVA. A zona incision was made using an ultra-sharp splitting blade (AB Technologies, Australia) mounted to a three-axis joystick oil hydraulic micromanipulator (MO-188; Narishige, Japan). FBS was gently pipetted onto the oocytes until they were released from the dish.
Oocytes were then transferred to 0.2 M sucrose in SCT base medium with caffeine and aspirated from the broken zona with a flame-polished glass pipette (inner diameter 120 μm). If oocytes had inadvertently separated into fragments during zona removal, the enucleated parts were fused by first sticking them together in 10 μg/mL phytohemagglutinin (in SCT base medium with caffeine), followed by a direct current electrical stimulus at 1.5 kV/cm (two pulses, 10 μseconds each) to reconstitute the original volume. The cytoplasts were then used for SCT as described in Experiment 1.
Experiment 3: aggregate SCT embryo development after vitrification and thawing
Vitrification and thawing
Aggregate and nonaggregate blastocysts were vitrified and thawed as described previously (Wei et al., 2018). Only blastocysts from Experiments 1 and 2 that were fused in the absence of calcium were included in this study. The blastocoel expansion and morphological grade were reassessed 2 hours post-thawing.
In vivo development
The effect of aggregation and vitrification on embryo survival was assessed in an observational study from transfers over 2 years. This only included blastocysts from Experiments 1 and 2 that were fused in the absence of calcium. Groups of 2–4 blastocysts were transferred into recipient ewes by laparoscopic surgery. Embryo survival on day 30–45 was confirmed by observation of embryonic heartbeat with transrectal ultrasound (L52x, 10-5 MHz transducer; SonoSite). Survival at term was defined as lambs that were delivered or surgically recovered with heartbeats. Five fetuses from vitrified aggregates were recovered for different purposes after the first scan; hence, those fetuses were not included in the analysis of survival to term.
Statistics
All of the statistical analyses were carried out in R (R Core Team, 2018) and the graphs were produced with the “ggplot2” package (Wickham, 2016). Exact p-values are given in the text, except when they were very small (p < 0.0001). Following statistical best practice and accepting uncertainty, we have not indicated arbitrary thresholds for p-values and abandoned declarations of “statistical significance” (Wasserstein et al., 2019). Instead, we provide exact p-values and display 95% confidence intervals where possible (Ganesh and Cave, 2018).
For the oocyte electrical activation experiment (Experiment 1), the categorical data for the different groups were compared with Fisher's exact test and the p-values adjusted for multiple comparisons by the Benjamini–Hochberg method. The 95% confidence intervals for the proportion were calculated by bootstrap resampling using the “infer” package in R (Bray et al., 2019).
For the fusion and blastocyst development in the presence and absence of calcium in Experiment 1, the data were retrospectively evaluated with Wilcoxon rank sum test. The data are reported as the median ± half the bootstrapped 95% confidence interval.
For the aggregation experiments (Experiment 2), the total across experiments were retrospectively evaluated with Chi-squared test. Fisher's exact test was used for the in vivo-derived oocytes due to the smaller sample size.
The vitrification data in Experiment 3 were analyzed with Fisher's exact test and the p-values adjusted for multiple comparisons by the Benjamini–Hochberg method. The 95% confidence intervals for the proportion were calculated by bootstrap resampling.
The in vivo embryo survival in Experiment 3 was retrospectively modeled using a generalized linear (logistic) mixed model for embryo survival. Embryo survival was first modeled with embryo stage, aggregation, vitrification, and the interaction between aggregation and vitrification as fixed effects. Recipient ewes were included as random effects. After fitting this model with the “lme4” package (Bates et al., 2015), the group of vitrified nonaggregates had “complete separation” since none of the transferred embryos survived. To solve this problem, we used a Bayesian approach of applying weak priors to the fixed-effect parameters with the “blme” package (Bolker, 2018; Chung et al., 2013).
The effect of additional fixed terms that may affect embryo survival was added to the model and compared with likelihood ratio tests. None of the additional following variables helped explain embryo survival after controlling for the above fixed effects: (i) donor cell line (p = 0.4), (ii) donor cell sex (p = 0.3), (iii) target gene modification (p = 0.2), and (iv) individual cell strain (p = 0.9). Consequently, only the main effects of embryo survival using the simplest model of embryo stage, aggregation, vitrification, and the interaction between aggregation and vitrification as fixed effects are discussed. The “emmeans” package was used for multiple comparison p-values and odds ratios (ORs), as well as Tukey-adjusted p-values (Lenth, 2020). The 95% confidence intervals for the proportion in the Figure 3 were calculated by bootstrap resampling.
Results
Experiment 1: calcium-free conditions and caffeine reduce premature activation
We sought to minimize spontaneous activation of MII oocytes that may be caused by SCT. First, we assessed the effect of electrical stimulation in the presence or absence of calcium ions on the cleavage of sheep oocytes (Fig. 1a). In the absence of electrical stimulation, all chemically activated oocytes had cleaved within 1 day (77/77 = 100%) and there was a low baseline cleavage rate (9/75 = 12%) for nonactivated oocytes. Applying an electrical stimulus in the presence of calcium ions increased cleavage of nonactivated oocytes (33/53 = 62%, p < 0.0001), indicative of premature activation.
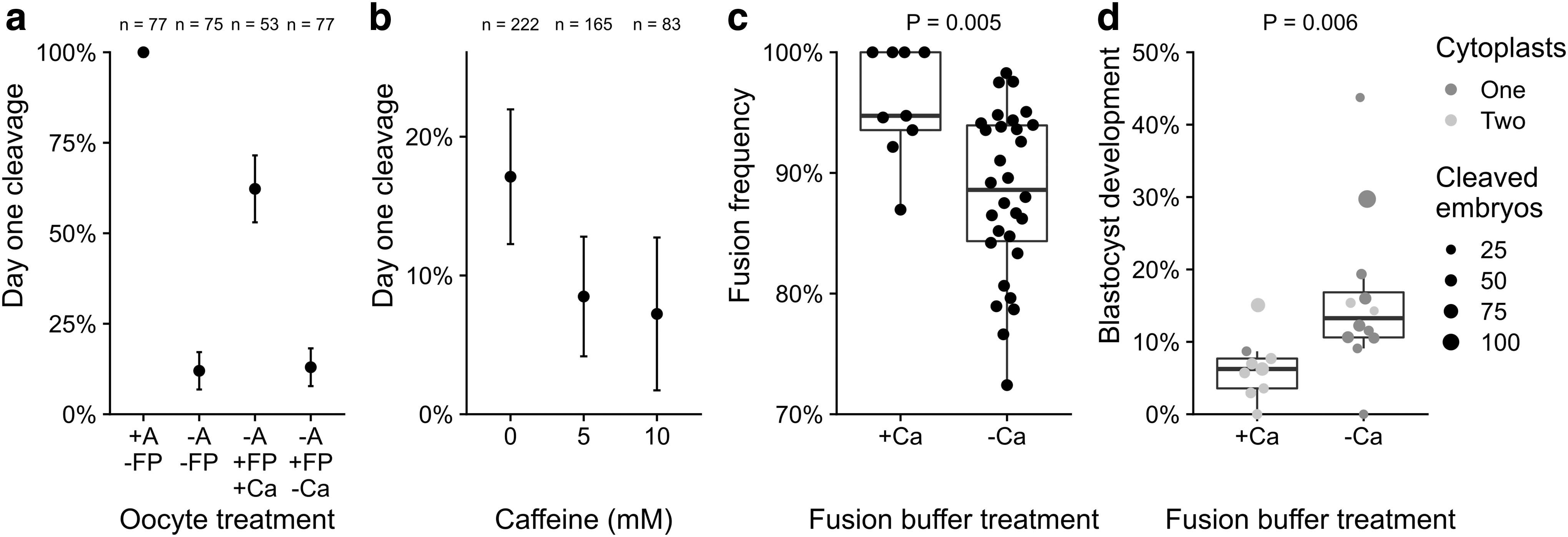
Calcium ions and caffeine affect cleavage, donor cell fusion, and cloned blastocyst development.
By contrast, nonactivated oocytes, electrically stimulated in the absence of calcium, did not cleave above baseline levels (10/77 = 13%, p = 1). Chemically activated, cleaved oocytes had higher blastocyst development than electrically stimulated ones in the presence of calcium (28/77 = 36% vs. 2/33 = 6%, respectively, p = 0.0009). In the absence of calcium and electrical stimulation, cleavage was reduced at caffeine concentrations of both 5 mM (14/165 = 8%, p = 0.02) and 10 mM caffeine (6/83 = 7%, p = 0.03) compared to no caffeine (38/222 = 17%), indicating a reduction in spontaneous activation (Fig. 1b).
We next assessed the effect of performing electrofusion without calcium ions on SCT embryos (Fig. 1c). Across various fetal fibroblast cell lines, fusion frequency was higher in the presence of calcium (94.7 ± 5.2% vs. 88.6 ± 4.3%, p = 0.005). However, development into cloned blastocysts was doubled following fusion in calcium-free versus calcium-containing buffer (13.3 ± 4.4% vs. 6.2 ± 2.7%, respectively, p = 0.006), both for single and double cytoplast transfer (Fig. 1d). Calcium-free conditions and inclusion of 5 mM caffeine were used for all subsequent experiments.
Experiment 2: morula aggregation efficiently produced blastocysts
Using the above optimized embryo reconstruction methodology, we compared development of aggregate versus nonaggregate embryos across numerous cloning experiments over several years (Table 1). From the embryos that were identified as morulae on embryonic day 5 or 6 (1196/5510 = 22%), 599 aggregated embryos were generated. Development into aggregate blastocysts was high (588/599 = 98%), with the majority forming a single blastocyst (481/599 = 80.3%). A small number of aggregation events resulted in two (49 events, 98 blastocysts) or three (3 events, 9 blastocysts) blastocysts forming.
Summary of Zona-Free Cloned Embryo Development Under Optimized Conditions that Prevent Premature Activation and Enable Morula Aggregation
Number (n) of embryos placed into IVC (nIVC).
Cleavage normalized on nIVC.
Blastocyst development normalized on number of aggregates.
Blastocyst development normalized on >1-cell embryos.
Combined number of blastocysts, normalized on >1-cell embryos.
Overall, 49 aggregates resulted in two blastocysts each and three aggregates resulted in three blastocysts each.
IVC, in vitro culture.
In aggregation experiments, some embryos had already blastulated before day 5 and a few viable morulae were missed during our screen and subsequently developed into nonaggregate blastocysts (115/5510 = 2%). In total, a slightly lower proportion of cleaved embryos developed into blastocysts under aggregation versus nonaggregation conditions (703/5510 = 13% vs. 186/1192 = 16%, p = 0.01).
Following SCT, the proportion of morulae identified for aggregation was 1.8-fold higher from in vivo- versus in vitro-derived oocytes (22/101 = 22% vs. 32/274 = 12%, p = 0.02) (Table 2). Aggregation success was similar for both in vivo- versus in vitro-derived groups (11/11 = 100% and 17/16 = 106%), with the majority of aggregates producing a single blastocyst (two in vivo and one in vitro aggregation resulted in two blastocysts). The higher proportion of morulae from in vivo-derived oocytes translated into a 1.4-fold higher total blastocyst output (18/101 = 18% vs. 36/274 = 13%, p = 0.3). In experiments without aggregation, in vivo- versus in vitro-derived blastocyst development was similar (26/138 = 19% vs. 22/139 = 16%, p = 0.5).
Cloned Embryo Development from In Vitro- Versus In Vivo-Derived Oocytes
Number (n) of embryos placed into IVC (nIVC).
Cleavage normalized on nIVC.
Blastocyst development normalized on number of aggregates.
Blastocyst development normalized on >1-cell embryos.
Combined number of blastocysts, normalized on >1-cell embryos.
One aggregate resulted in two blastocysts.
Two aggregates resulted in two blastocysts each.
Experiment 3: aggregate SCT embryos develop better after vitrification and thawing
Vitrification and thawing
Aggregate and nonaggregate blastocysts were vitrified when recipient ewes were not available for embryo transfer. Upon thawing, both groups were functionally and morphologically compared. Blastocoel re-expansion frequency within 2 hours post-thawing was higher for aggregate versus nonaggregate blastocysts (211/240 = 88% vs. 28/44 = 64%, p = 0.0002) (Fig. 2a). Blastocyst quality was morphologically graded before and after vitrification (Fig. 2b–d). The proportion of good-quality blastocysts decreased 1.5-fold for aggregates (146/240 = 61% to 99/240 = 41%, p < 0.0001) and 6.9-fold in nonaggregates (21/44 = 48% to 3/44 = 7%, p < 0.0001). After vitrification, this resulted in a larger proportion of good-quality aggregate blastocysts compared to nonaggregates (p < 0.0001).
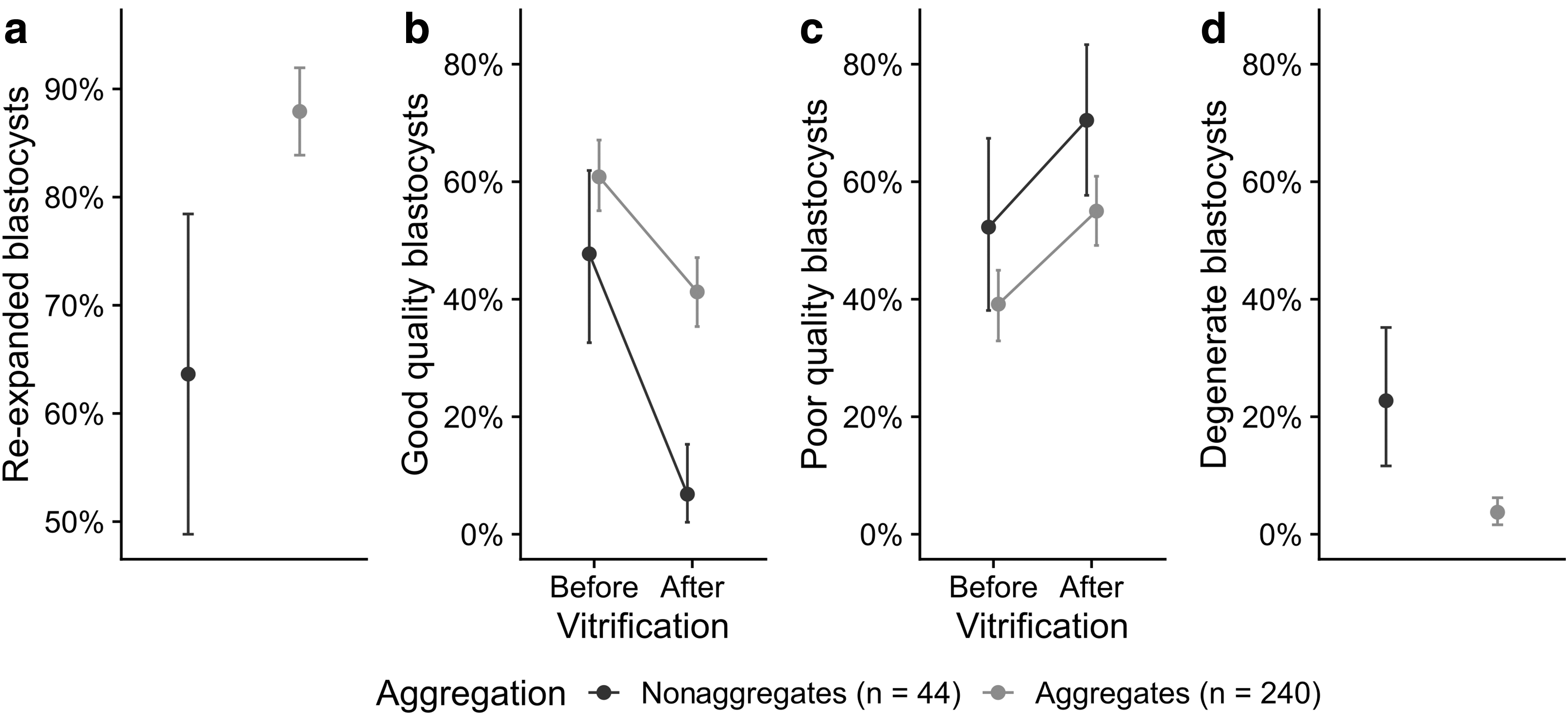
Aggregation improved blastocyst development after vitrification.
With the reduction in good-quality blastocysts, the proportion of poor-quality blastocysts increased 1.4-fold for aggregates (94/240 = 39% to 132/240 = 55%, p = 0.002) and 1.3-fold for nonaggregates (23/44 = 52% to 31/44 = 70%, p = 0.2). Thawed aggregate blastocysts showed less signs of degeneration (collapsed blastocoel and lysed blastomeres) than nonaggregate blastocysts (9/240 = 4% vs. 10/44 = 23%, p < 0.0001). Some collapsed blastocysts that had not re-expanded within 2–3 hours were put back into IVC instead of transferring them into recipients. On these occasions, all aggregates re-expanded within 24 hours compared to only few nonaggregates (5/5 = 100% vs. 2/10 = 20%, p = 0.014). We conclude that aggregation consistently improved cryoresilience, the capacity to recover quickly from the challenges of cryopreservation.
In vivo survival
We investigated the combined effect of aggregation and vitrification on in vivo embryo survival. For non-cryopreserved “fresh” control embryos, the odds of successful embryo development were 2.4-fold higher for aggregates compared to nonaggregates (OR = 2.4, p = 0.045) at both day 30–45 of gestation (43/138 = 31% vs. 20/120 = 17%) and term (21/138 = 15% vs. 11/120 = 9%) (Fig. 3a).
Vitrified aggregate embryos developed to day 30–45 of gestation at a similar rate as their fresh equivalents (53/188 = 28% vs. 43/138 = 31%, p = 0.9) (Fig. 3b). Vitrified nonaggregate development (0/28 = 0%) was poorer than vitrified aggregates (OR = 0.035, p = 0.02) and fresh aggregates (OR = 0.029, p = 0.01), but not necessarily when compared to fresh nonaggregates (OR = 0.068, p = 0.1). At term, the same trend was seen with vitrified aggregates surviving better than vitrified nonaggregates (24/175 = 14% vs. 0/28 = 0%, p = 0.02).
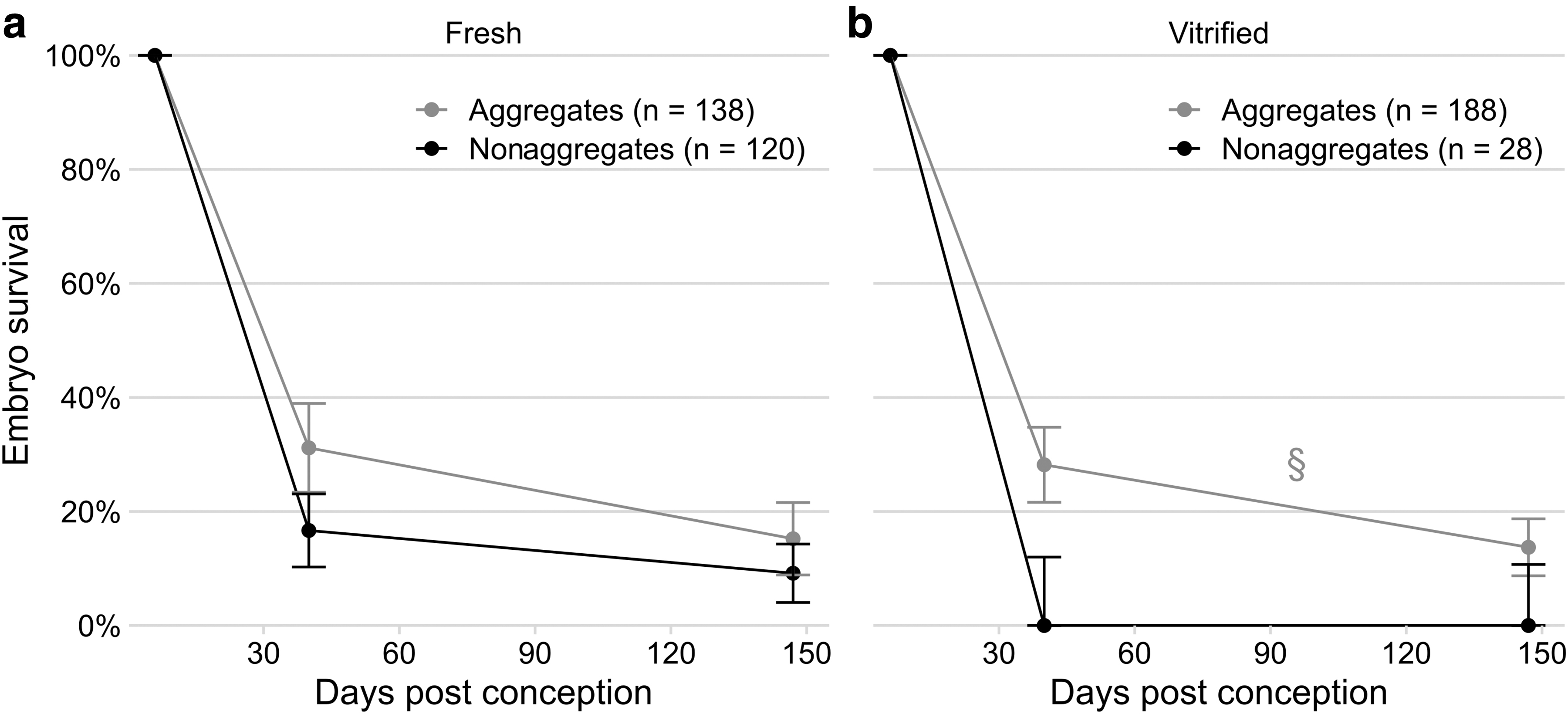
Aggregation improved in vivo embryo survival for
After taking aggregation and cryopreservation status into account, the embryo loss from day 30 until 45 to term had an OR 0.38 (p < 0.0001), indicating a high loss of pregnancies from mid-late gestation. The false-negative rate for ultrasound detection (i.e., ewes that had more lambs at term than fetuses at day 30–45) was 7/158 (4%), five of which were carrying vitrified aggregates, one carrying a fresh aggregate, and one carrying a fresh nonaggregate. The number of true positives (ewes with the same number of lambs at term as fetuses at day 30–45) and true negatives (ewes still non-pregnant at term) was 35/158 (22%) and 68/158 (43%), respectively. The remaining 48/158 (30%) had fewer lambs at term, which could be embryo/fetal death postday 30–45 or false positives.
In summary, our sheep cloning protocol combined reduced cytoplast activation with high-throughput zona-free embryo reconstruction and aggregation to improve blastocyst development and embryo survival, particularly after cryopreservation (Fig. 4).
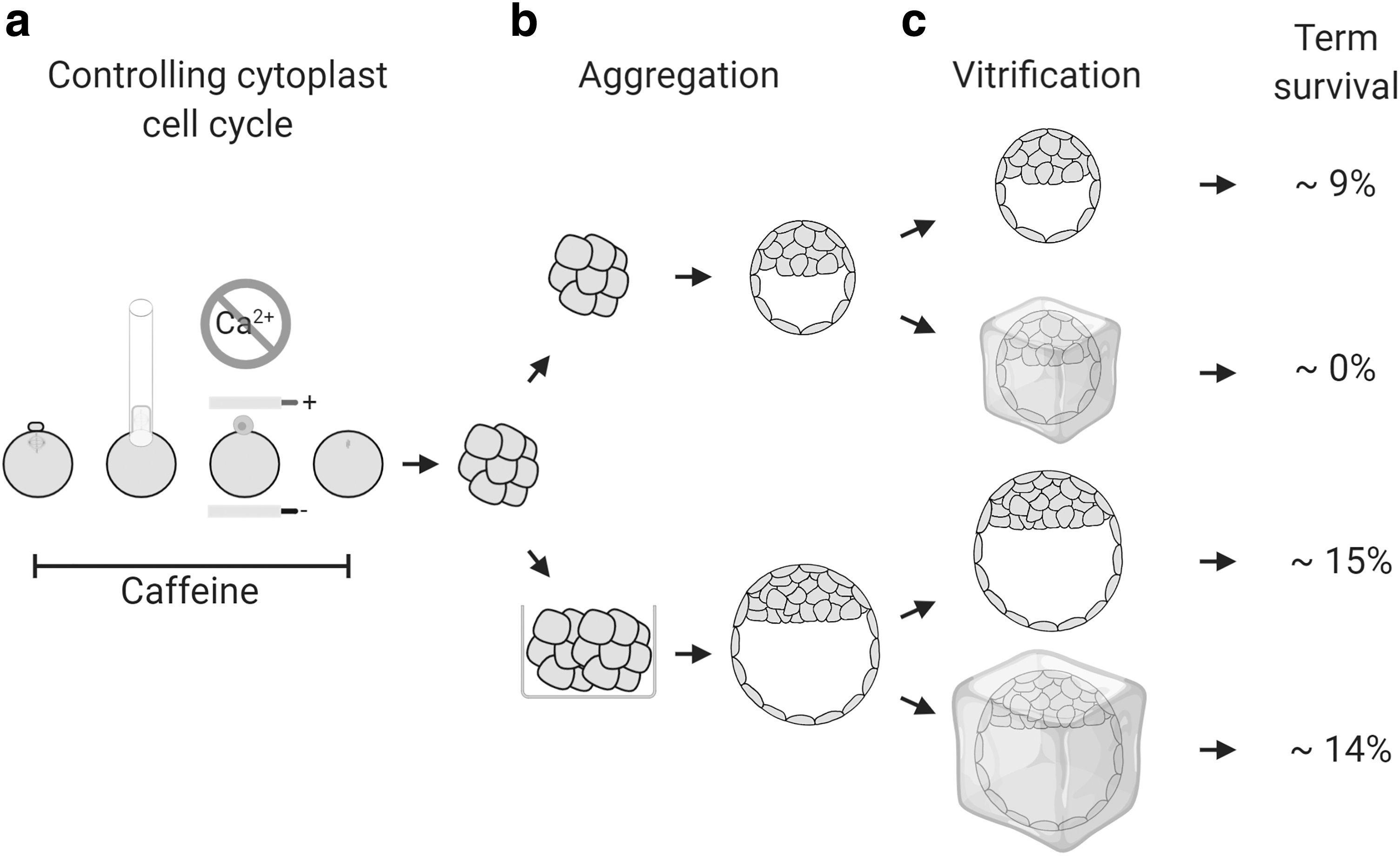
Summary of the optimized sheep cloning methodology. (
Discussion
We show that integrating several methodological advancements improves sheep cloning. Preventing premature activation improved development to the blastocyst stage, while morula aggregation improved postvitrification viability and blastocyst survival to term.
Cell cycle coordination during embryo reconstruction
Despite sheep being the first animals cloned from adult somatic cells, both the in vitro and in vivo efficiencies have remained poor. One problem may have been unintentional premature oocyte activation. During normal fertilization, sperm activates the oocyte through both extracellular calcium influx across the plasma membrane and release from intracellular calcium stores (Miao et al., 2012). Electrofusion can artificially stimulate extracellular calcium influx and cause premature activation, which reduces blastocyst development. Accordingly, we found more than 50% of sheep oocytes activated in the presence of calcium ions when electrically stimulated.
This is supported by a previous report in sheep, which found that across all attempted fusion parameters to trigger cell fusion, the oocyte activation rate was consistently greater than 60% (Peura et al., 2003). This is also true in pig (Boquest et al., 2002), but not cattle where only 1% prematurely activated (Wells et al., 1999). Peura et al. (2003) were unable to identify electrical parameters that triggered cell fusion without prematurely activating sheep oocytes. However, all their fusion experiments were conducted in the presence of calcium ions, and as we show in this study, acceptable fusion rates were possible in sheep with standard electrical parameters that do not cause premature activation. Overall, fusing reconstructs in the absence of calcium improved development to the blastocyst stage in our system.
The vast majority of sheep cloning publications in the past decade have reported fusing donor cells in the presence of calcium ions (Fan et al., 2018; Fu et al., 2012; Hajian et al., 2011; Heidari et al., 2010; Hua et al., 2013; Khan et al., 2018; Ma et al., 2014, 2015; Moulavi et al., 2013; Rutigliano et al., 2017; Tang et al., 2012; Wen et al., 2014; Ye et al., 2010), rather than in the absence (Choi et al., 2010, 2013; Deng et al., 2013; German et al., 2015; Rathbone et al., 2010). Most of the publications that report fusing in the presence of calcium ions treat the reconstructs with ionomycin/6-DMAP within 1 hour, so correct ploidy may have been maintained. However, fusing in the absence of calcium better controls cell cycle coordination and ploidy, while also allowing for delayed chemical activation, which is a standard practice in numerous species.
In mice, when the donor cell nucleus was exposed to the oocyte's MII cytoplasm for 1 and 3 hours, it increased blastocyst development rate and the production of live offspring, suggesting the importance of reprogramming in this period (Wakayama et al., 1998). This effect has also been observed for full-term viability of dogs (Hossein et al., 2009), pigs (Yin et al., 2003), and for improved blastocyst development rate in cattle using somatic (Wells et al., 1998) and embryonic (Stice et al., 1996) donor cells. There are conflicting reports of the effect in sheep with two publications showing no effect on blastocyst development (Campbell et al., 1996; Lee and Campbell, 2008), but another showing an improvement (Deng et al., 2013).
The increased embryo development we observed could have been due to the improved reprogramming from the activation delay; however, we also must consider the effect of the delayed 6-DMAP treatment on prematurely activated reconstructs. In bovine oocytes, activated with ionomycin and then treated with 6-DMAP after a delay of 0–4 hours, the proportion of those that had correctly formed a single pronucleus decreased with time (Susko-Parrish et al., 1994). Delaying 6-DMAP treatment after ionomycin incubation increased the proportion of oocytes that extruded a second polar body, compromising blastocyst development in both parthenogenotes and SCT embryos (Appleby, 2015). Therefore, inadvertently activating SCT reconstructs upon fusion may result in the extrusion of a pseudo-polar body, incorrect ploidy, and reduced blastocyst development.
Furthermore, rapid degradation of M-Cdk activity would prevent NEBD of the somatic donor nucleus, preventing efficient chromatin remodeling and in vitro development (Tani et al., 2001, 2003). In line with these predictions, we observed lower blastocyst development in electrically stimulated oocytes compared to oocytes that were exposed to the standard ionomycin/6-DMAP chemical activation procedure.
The other improvement to cell cycle coordination involved caffeine treatment. Around 15% of nonactivated oocytes spontaneously cleaved, which we speculated was due to suboptimal in vitro maturation and low M-Cdk activity. It has been reported that treatment with caffeine increases M-Cdk activity in sheep oocytes (Lee and Campbell, 2006). When we treated nonactivated MII oocytes with caffeine at 5 or 10 mM, the rate of spontaneous activation approximately halved, suggesting that the spontaneous activation we observed was due to low M-Cdk activity. This is an additional improvement caffeine makes to SCT along with increased occurrence of NEBD and the total number of cells at blastocyst stage (Lee and Campbell, 2008).
When we used in vivo-matured oocytes for cloning, which had optimal maturation conditions, but an undetermined range of M-Cdk activity, the blastocyst development was only marginally improved. Therefore, further improvements to cloned embryo development may come from exogenous epigenetic reprogramming factors like histone lysine demethylases (Antony et al., 2013; Liu et al., 2018; Matoba et al., 2014).
Benefits of aggregation
Embryo aggregation is a straightforward approach to improve embryo viability. We observed a 2.4-fold increase in the odds of embryo survival after morula aggregation with fresh transfers. This supports the original findings in mice (Boiani et al., 2003), although the improvements in embryo survival that we observed were not as large as the murine fourfold to eightfold increase. Since the embryo survival of nonaggregated mouse clones was an order of magnitude lower than in sheep clones, this may indicate that the developmental compensation provided by each aggregation partner may be more pronounced when the overall efficiency is very low.
The timing of embryo aggregation may also be important. Previously, we aggregated cattle somatic clones at the one-cell stage, which did not improve development to term (Misica-Turner et al., 2007). In this study, we refined our approach and aggregated as late as possible by selecting competent morulae, which may have increased the chance of both embryos contributing to embryogenesis.
After selecting a pair of embryos for aggregation, the improvement in survival to term approximately offsets the decrease in the total number of blastocysts. This has the benefit of using fewer recipient ewes, which is a positive outcome for animal welfare and reduces the costs of cloning experiments. Aggregation is also easily adopted within the zona-free cloning system since the embryos are already zona free at the point of aggregation and were cultured in microwells. Similar observations were made in cattle, where aggregating zona-free embryonic and somatic cell cloned embryos at the eight cell to morula stage improved pregnancy rates following transfer of both fresh and vitrified embryos (Akagi et al., 2011; Peura et al., 1998; Vajta et al., 2003). Overall, the improvements in efficiency make aggregation a worthwhile addition to cloning.
Benefits of vitrifying aggregates
Without aggregation, we observed a detrimental impact of vitrification on blastocyst quality after thawing and embryo survival after transfer. By contrast, almost all aggregate blastocysts re-expanded within 2 hours of warming after vitrification, which has been used as an indicator of embryo quality (Kaidi et al., 1998; Rizos et al., 2002). The re-expansion rates of cloned aggregate embryos reported here are similar to rates for in vivo- and in vitro-produced embryos (Dattena et al., 2004).
Vitrification compromises both the inner cell mass (ICM) and trophectoderm (TE) lineages in bovine blastocysts, resulting in ∼20% of cells losing their membrane integrity immediately after thawing, which did not recover after 2–3 days in culture (Kaidi et al., 2001). Bovine ICM cells appear to be more susceptible to vitrification damage than TE cells (Gómez et al., 2009), perhaps due to their higher basal rate of apoptosis (Byrne et al., 1999; Gjørret et al., 2003), which may be further activated in response to cryodamage (Park et al., 2006). Over a third of vitrified and warmed in vitro-fertilized (IVF) embryos had no ICM cells at all, compared to only 4% of fresh embryos (Gómez et al., 2009).
The proliferation rate of ICM cells was reported to be about half that of TE cells (Copp, 1978), which makes it more difficult to recover from cell losses within the ICM lineage. Sheep cloned blastocysts, produced using a similar zona-free protocol as ours, had ∼20% fewer cells than IVF-derived blastocysts and comprised ∼100 cells, including ∼13 ICM cells (Moulavi et al., 2013). In nonaggregates, vitrification may have disproportionally depleted ICM cell numbers below a critical threshold required for engendering embryonic development.
By contrast, morula aggregation has been shown to double blastocyst size and cell numbers, without affecting lineage composition within the embryo or ICM (Saiz et al., 2016). Cell numbers were also almost doubled in bovine blastocysts derived from morula aggregation (Akagi et al., 2011). Since aggregates contain more cells, they are expected to be more resilient to ICM cell depletion, which could explain their better embryonic and fetal development to term.
Nonaggregate sheep cloned blastocysts showed poor survival after freeze-thawing, which presents a major biological barrier for successful vitrification. By improving embryo cryoresilience, aggregation overcomes this obstacle and allows to capitalize on the many benefits of cryopreservation.
First, vitrification uncouples in vitro from in vivo development because cloned embryo development and ovulation of recipient ewes do not need to be synchronized in real time. This simplifies experimental planning and benefits animal welfare on several levels. Any last-minute cancellation on the day of SCT can be accommodated (e.g., if slaughterhouse ovaries cannot be sourced) by filling already synchronized ewes with vitrified embryos, which reduces animal manipulations by not having to resynchronize unused recipients.
Before embryo transfer, the exact number of recipient ewes can be accurately matched with the number of cryopreserved embryos, reducing animal numbers and cost. Since most aggregates survive and re-expand after thawing, they can be transferred almost immediately, further facilitating the logistics of embryo transfer. Timing of planned parturition can also be better coordinated, for example, by lambing larger groups on dates that are convenient for staff. This is another important welfare consideration since cloned neonates often require intensive care for several days postpartum.
Second, successful cryopreservation of aggregates reduces embryo wastage. Morulae of different genotypes can be vitrified, facilitating planning of aggregation experiments and maximizing the number of high-quality embryos with desirable genotype combinations. In cases of planned fresh transfers, surplus embryos, which would otherwise be destroyed when there are not enough recipients available on the day of transfer, can be salvaged. This includes embryos with delayed development that do not blastulate until after the day of embryo transfer. On rare occasions, aggregate embryos can even be revitrified and thawed again to obtain viable pregnancies. Overall, aggregation fully realizes the benefits of cryopreservation by adding logistical flexibility and improving the survival of thawed embryos.
Conclusions
The combination of cytoplast cell cycle control, zona-free embryo reconstruction, and morula aggregation improved cloning efficiency, maximizing the benefits of cryopreservation by improving short- and long-term survival of thawed embryos. This integrated cloning methodology provides a reproducible system that is relatively easy to operate and increases the throughput of both cloned embryo and offspring production. Other experimental advances in cell reprogramming, like adding exogenous epigenetic modulators, can be seamlessly added to this system. The presented methodology can be easily adapted for cloning from embryonic donor cells, which offers distinct advantages in multiplying elite embryos.
Footnotes
Authors' Contributions
Z.L.M.: conceptualization, methodology, formal analysis (Experiment 1, 2, and 3), investigation (Experiment 1. Partial contribution to Experiment 2 and 3), data curation, writing—original draft, visualization, and project administration. S.J.A.: conceptualization, methodology, formal analysis (Experiment 1, 2, and 3), investigation (Experiment 1, 2, and 3), data curation, writing—review and editing, and project administration. L.M.F.: investigation (Experiment 2 and 3), writing—review and editing, and project administration. H.V.H.: formal analysis (Experiment 3). J.W.: investigation (Experiment 2 and 3). D.N.W.: conceptualization, investigation (Experiment 2 and 3), and writing—review and editing. B.O.: conceptualization, methodology, writing—original draft, supervision, project administration, and funding acquisition.
Acknowledgments
We thank Stephanie Delaney for Investigation (Experiment 3: ultrasound scanning and animal husbandry); Elyssa Barnaby for Investigation (Experiment 3: embryo transfer and lambing); Dr. Fanli Meng for Investigation (Experiment 2: enucleation); Jan Oliver for Resources (Embryo culture media); Dr. Jennifer Juengel for Writing—Review and Editing; Angela Brennan, Murray Brown, Tony Kemara, Katherine Moors, and Susan Odom for Resources (Ovary collection). Animal Breeding Services Ltd (Hamilton, New Zealand) were contracted for superovulating donor ewes, oviductal flushing of oocytes, and embryo transfer.
Ethics Approval and Consent to Participate
Investigations complied with the New Zealand Animal Welfare Act 1999 and were approved by the Ruakura Animal Ethics Committee.
Availability of Data and Material
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request
Author Disclosure Statement
Z.L.M. was supported by Livestock Improvement Corporation fellowship grant. L.M.F., H.V.H., J.W., D.N.W., and B.O. are employees of AgResearch Ltd. The authors declare no other competing interests.
Funding Information
Z.L.M. was supported by Callaghan Innovation R&D student fellowship grant, AgResearch Ltd. stipend, Livestock Improvement Corporation fellowship grant, Todd Foundation Award for Excellence, and Heritage Incorporated Magnus Grant. S.J.A. was supported by University of Auckland Doctoral Scholarship, AgResearch Ltd. stipend, and Todd Foundation Award for Excellence. This research was supported by funds from the Ministry of Business, Innovation and Employment (C10X1711), AgResearch Ltd. and the University of Auckland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.