
Abstract
Human induced pluripotent stem cells (iPSCs) can capture the diversity in the general human population as well as provide deeper insight in cellular mechanisms. This makes them suitable to study both fundamental and applied research subjects, such as disease modeling, gene-environment interactions, personalized medicine, and chemical toxicity. In an independent laboratory, we were able to generate iPSCs originating from human peripheral blood mononuclear cells according to a modified version of a temporal episomal vector (EV)-based induction method. The iPSCs could subsequently be differentiated into two different lineages: mesoderm-derived cardiomyocytes and ectoderm-derived neuron-astrocyte co-cultures. It was shown that the neuron-astrocyte culture developed a mature phenotype within the course of five weeks and depending on the medium composition, network formation and neuron-astrocyte cell ratios could be modified. Although previously it has been described that iPSCs generated with this EV-based induction protocol could differentiate to mesenchymal stem cells, hepatocytes, cardiomyocytes, and basic neuronal cultures, we now demonstrate differentiation into a culture containing both neurons and astrocytes.
Introduction
Since the demonstration by yamanaka and colleagues (2006; 2007) that mouse and human fibroblasts could be induced into pluripotent stem cells, the application of this technique in human cells holds great promise for many applications for both clinical and research purposes. Generating induced pluripotent stem cells (iPSCs) from patients opens up possibilities for regenerative and personalized medicine, while making iPSCs from a range of people can provide insights into more fundamental questions around gene-environment interactions in cohorts, disease modeling, and screening tissue responses to chemicals, medicinal products, and implants.
While skin fibroblasts are a much-used source of cells, CD34+ peripheral blood mononuclear cells (PBMCs) as starting material are becoming more popular. Contrary to skin fibroblasts, PBMCs do not suffer from somatic mutations caused by UV (Abyzov et al., 2012), can be obtained in a noninvasive manner, are a suitable resource for large quantities of cells (Loh et al., 2009; Staerk et al., 2010), and are typically more readily available in established longitudinal cohort studies than skin fibroblasts.
There are numerous ways to reprogram PBMCs into iPSCs, of which the oriP/EBNA1-based episomal vectors (EVs) (Yu et al., 2009; Zhang, 2013) to carry the Yamanaka factors appear to be the most suitable one. EVs facilitate an integration-free and transient transfection strategy, thereby avoiding disruptive mutations in the endogenous genome and allowing reversion to the same genetic content of the pretransfected primary cell (Zhang, 2013). In addition, EVs are relatively cheap compared to other integration-free methods such as the Sendai virus (Zhang, 2013). The transfection needs to be performed only once, upon which the EVs are introduced by nucleofection, but will gradually be phased out upon cell division (Nanbo et al., 2007).
Su et al., 2013, 2016) have optimized the protocol to reprogram PBMCs into iPSCs by using four oriP/EBNA1-based EV plasmids (pEVs) with a spleen focus-forming virus (SFFV) promotor carrying OCT4-SOX2, KLF4, MYC, and BCL-XL factors, and have also shown that the iPSCs obtained with this method were capable to differentiate into cardiomyocytes, hepatocytes, and mesenchymal stem cells (Su et al., 2013).
In this study, we succeeded in reprogramming PBMCs into iPSCs by using this EV-based nucleofection method (Su et al., 2016). To demonstrate their pluripotent nature, these iPSCs could subsequently be differentiated into cells of different embryonic germ layers. We generated mesodermal cardiac cells, as has been shown before in literature, and as a novel addition differentiated into ectodermal neuron-astrocyte co-cultures based on our protocol that was developed for the differentiation of human embryonic stem cells (hESC; de Leeuw et al., 2020). To our knowledge, this is the first study to show neuron-astrocyte differentiation using the reprogramming method of Su et al.
Materials and Methods
All ingredients were bought at Gibco (Waltham, MA) unless mentioned otherwise.
Isolation and reprogramming of PBMCs to iPSCs
Buffy coat white blood cells were obtained from anonymous donors supplied by Sanquin blood bank (Amsterdam, The Netherlands) under a not-for-transfusion research agreement (NVT0243.02). Isolation and reprogramming of the PBMCs were performed as described previously by Su et al. (2013, 2016) with some adjustments. Briefly, buffy coat was mixed 1:1 with phosphate-buffered saline (PBS) after which Lymphoprep™ (instead of Ficoll; Axis-shield, Dundee, UK) was pipetted on the bottom of the tube underneath the PB/PBS mix. The liquid layers were centrifuged for 30 minutes at 1000g (instead of 400g) with slow acceleration and no brake. Of the resulting gradient, the PBMC layer was harvested, dissolved in PBS, and centrifuged for 8 minutes at 250g (instead 10 minutes at 400g).
After removal of the supernatant, cells were dissolved in Red Blood Cell lysis buffer (containing 2.5 times less ethylenediaminetetraacetic acid) and centrifuged for 5 minutes at 400g. Cells were washed two times with PBS and resuspended in erythroid culture medium (ECM), counted, and optionally cryopreserved before mononuclear cell (MNC) culturing. PBMCs were cultured for 7 days at a density of 5 × 106 cells/mL, changing the medium every 2–3 days.
Cryopreservation was carried out by taking up MNCs in fetal calf serum at a density of 16–24 × 106 cells/mL, aliquoting the cell suspension per 0.5 mL, and putting them on ice. Approximately 0.5 mL ice-cold freezing medium (80% fetal calf serum/20% dimethyl sulfoxide) was added dropwise to each aliquot and mixture was resuspended well before transferring the cells to freezing tubes for storage at −135°C.
One day before nucleofection, 2.6 × 105 cells/mL mitomycin-inactivated murine embryonic fibroblast feeder cells (MEFs; instead of rat fibroblasts) were seeded on 0.1% gelatin (Sigma-Aldrich, Saint Louis, MO)-coated 6-well plates in Feeder Cell Culture Medium. On the day of nucleofection, medium was replaced with ECM. EVs were prepared in the following concentrations (Wen et al., 2016): 2 μg pEV SFFV-OCT4-E2A-SOX2 (pEV-OS); 1 μg pEV SFFV-MYC (pEV-M); 1 μg pEV SFFV- KLF4 (pEV-K); and 0.5 μg pEV SFFV-BCL-XL (pEV-B) (ABMgood, Richmond, Canada). Around 2 × 106 PBMCs were nucleofected with indicated plasmids and cultured at a density of 1 × 105 to 1 × 106 cells in a humidified chamber (37°C, 5% O2, and 5% CO2; instead of a hypoxia chamber).
After two days, ECM was replaced for iPSC generation medium and from day 6, sodium butyrate was added to the medium. Refreshments took place every other day and new MEFs were added every five days.
Colonies were picked on day 21 and expanded in Essential 8 Flex medium according to Gibco's Essential 8 Flex™ protocol on feeder-free Vitronectin (VTN-N)-coated dishes (instead of in iPSC medium on rat fibroblasts as indicated in the original protocol) in a humidified chamber (passage 1, 37°C, 5% O2, and 5% CO2). Medium changes were done every day and passaging was performed every three to four days. Cryopreservation was performed according to the PSC Cryopreservation kit from Gibco. For differentiation experiments, stem cells with passage 25–34 were used.
Determination of plasmid copy numbers
DNA extracts were made of PBMCs with EVs, without EVs, and with iPSCs from passage 1 and 5 to 13 as described previously (Su et al., 2016). Briefly, for quantification of the EV copy number, a standard curve was made by mixing 1 μg DNA of PBMCs with 1.6 pg pEV-OS, which was equivalent to 1 vector copy number per cell, and diluted four orders of magnitude. WHP Posttranscriptional Response Element (WPRE) and Epstein–Barr nuclear antigen (EBNA) were used to track residual DNA and were normalized against Beta-actin (ACTB; Table 1). Corresponding Ct-values from the samples were matched with the copy numbers of the standard curve for both genes.
Overview Primers Used for Quantitative Polymerase Chain Reaction of Episomal Vector Plasmid
Karyotyping
A Giemsa staining was performed to karyotype the iPSC culture. Briefly, medium of confluent iPSCs was replaced with medium containing 20 μL/mL KaryoMAX Colcemid solution and incubated for 1.5 hours in the incubator. Cells were then dissociated using TripLE Select for 7 minutes and centrifuged for 5 minutes at 1000 rpm. Cells were carefully resuspended in 1 mL 0.075 M prewarmed KCl, 2 mL extra KCl was added, and cells were incubated for 10 minutes in the incubator. Three drops of fixative (1:3 acetic acid: methanol) were added and suspension was centrifuged for 5 minutes at 1000 rpm. Cells were washed twice and resuspended in 20–30 drops fixative.
The suspension was dropped onto cold, moist microscope slides and left to dry completely. Giemsa staining (Sigma-Aldrich) was applied onto the slides for 5 minutes, washed in deionized water and imaged on a Leica DMi8 microscope system (Leica, Wetzlar, Germany) using the appropriate Leica Software (LAS X).
Cardiac differentiation of iPSCs
Differentiation into the cardiac lineage was performed according to the PSC Cardiomyocyte Differentiation Kit protocol from Gibco. Briefly, single-cell iPSCs were seeded on Geltrex™ matrix-coated plates at a density of 20–40 × 104 cells/mL in Essential 8 Flex medium. Medium was refreshed every day for four days, after which the medium was replaced by cardiomyocyte differentiation medium A. On day 6 of differentiation, the medium was replaced by cardiomyocyte differentiation medium B. Two days later, the medium was changed for cardiomyocyte maintenance medium. This medium was refreshed every other day until the end of the test on day 14.
Neuronal differentiation of iPSCs
For neuron-astrocyte differentiation, a protocol previously described in de Leeuw et al. (2020) was used. The procedure consisted of four stages: embryoid body (EB) generation, rosette formation, neural progenitor cell (NPC) expansion, and neuron-astrocyte generation.
The first three steps were performed according to a Stemcell neuronal induction protocol (document no. 28782; Stemcell Technologies, Inc., Vancouver, Canada). Briefly, iPSCs were seeded in an AggreWell™800 24-well plate at a density of 3 × 106 cells/mL in STEMdiff™ Neural Induction Medium+SMADi (Stemcell) supplemented with Y-27632 ROCK inhibitor (Stemcell). Partial replacements of the medium were performed every day until day 5, after which the EBs that had formed were washed off and transferred to Poly-L-Ornithine (PLO, 10 μg/mL; Sigma-Aldrich)-/laminin (10 μg/mL; Sigma-Aldrich)-coated dishes in STEMdiff Neuronal Induction Medium (NI)+SMADi (Stemcell) for rosette formation.
After a week with daily medium refreshments, the rosettes were dissociated with Neural rosette Selection Reagent (Stemcell) and transferred to new PLO/laminin dishes for another week, changing the medium every day. On day 19, rosettes were dissociated again and left to grow on new PLO/laminin dishes until confluent, refreshing the medium every day. After about a week, cells were dissociated with StemPro™ Accutase™ Cell Dissociation Reagent and seeded at a density of 1.2–1.5 × 106 cells/mL in STEMdiff Neural progenitor medium (NP) (Stemcell) on PLO-/laminin-coated dishes. These NPCs (passage 1) were passaged every week and medium was refreshed on a daily basis. Cells were frozen after one to five passages in STEMdiff Neural progenitor Freezing Medium (Stemcell) according to the Stemcell protocol until use for differentiation.
Neuron-astrocyte generation was done using four different protocols to optimize culture conditions: medium described as by Pistollato et al. (2017) without and with the addition of 10 ng/mL ciliary neurotrophic factor (CNTF) (hereafter called P− and P+, respectively), and as by Gunhanlar et al. (2018) without and with CNTF (G− and G+). Medium from Pistollato et al. (2017) was composed of neurobasal medium with 1% N2 supplement, 2% B-27 supplement, 1% 5000 IU/mL Penicillin/5000 μg/mL Streptomycin, 1 ng/mL glial cell line-derived neurotrophic factor (GDNF), and 2.5 ng/mL brain-derived neurotrophic factor (BDNF).
Medium from Gunhanlar et al. (2018) comprised neurobasal medium, 1% N2 supplement, 2% B-27-retinoic acid supplement, 1% 5000 IU/mL Penicillin/5000 μg/mL Streptomycin, 1% nonessential amino acids, 20 ng/mL GDNF, 20 ng/mL BDNF, 1 μM dibutyryl cyclic adenosine monophosphate (Sigma-Aldrich), 200 μM ascorbic acid (Sigma-Aldrich), and 2 μg/mL laminin (Sigma-Aldrich). NPCs were seeded 2.56 × 105 cells/cm2 on PLO/laminin-coated on 8-well plate (Ibidi, Gräfelfing, Germany). Medium replacements took place every two to three days.
Immunocytochemistry
iPSC and neuronal differentiation cultures were stained as previously described by de Leeuw et al., 2019. Cardiac differentiation cultures were stained using the Human Cardiomyocyte Immunocytochemistry Kit from Gibco. Primary and secondary antibodies used are listed in Table 2. Samples were imaged on a Leica DMi8 microscope system (Leica) using the appropriate Leica Software (LAS X). Image processing was performed in ImageJ (version 1.51n; Rasband, 1997–2018).
Primary and Secondary Antibodies
RNA isolation and quantitative polymerase chain reaction
Medium was aspirated from the samples and cells (iPSCs/neurons day 14 n = 4, NPCs n = 3, neurons day 7/21/28 n = 5) were directly fixed in QIAzol (Qiagen, Hilden, Germany). Samples were stored until further processing at −80°C. Whole RNA isolation was performed using the RNeasy® mini kit with a DNase digestion step according to the manufacturer's protocol (Qiagen, version October 2019). RNA concentration and quality were determined with the NanoDrop™ 1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE) and the 2100 Bioanalyzer (Agilent Technologies, Amstelveen, The Netherlands), respectively.
Synthesis of cDNA was performed with the high-capacity cDNA reverse transcription kit containing random hexamer primers (Applied Biosystems, Foster City, CA). After cDNA synthesis, quantitative polymerase chain reaction (qPCR) was performed on a 7500 Fast Real-Time PCR system (Applied Biosystems; thermal cycling conditions: 95°C for 20 seconds, 40 cycles of 95°C for 3 seconds, and 60°C for 30 seconds). Primers are listed in Table 3 (all purchased from Applied Biosystems).
Primers Used for Gene Expression with Corresponding Marker Function and Assay ID
Relative gene expression differences were calculated using the 2−ΔΔCt-method (Applied Biosystems, 2001) and normalized against the housekeeping genes Hypoxanthine phosphoribosyltransferase 1 (HPRT1), Glucuronidase beta (GUSB), and RNA Polymerase II Subunit A (POLR2A). Statistical analysis was performed in GraphPad Prism (version 8.4.1) using a one-way analysis of variance (ANOVA) test and post hoc Tukey's multiple comparisons test.
Results
Reprogramming of PBMCs to iPSCs using EVs
Following the protocol of Su et al. (2013, 2016) we were able to generate iPSCs from adult PBMCs within 4 weeks (Fig. 1A). One major adjustment to the original protocol was the shortened culturing on a feeder layer by switching from MEFs to vitronectin coating for picking and expansion of iPSC colonies, which yielded better results than culturing on fibroblasts.
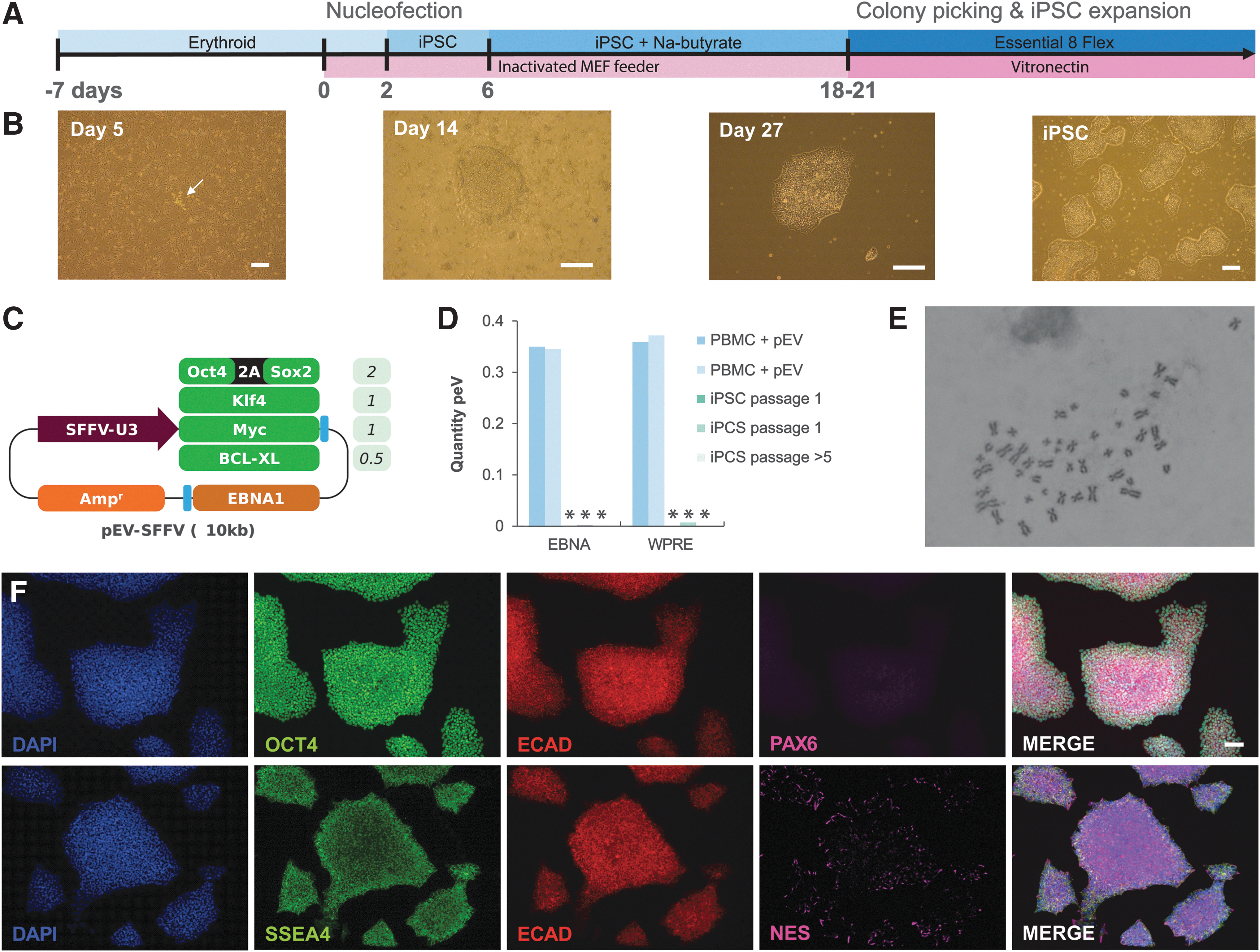
Reprogramming protocol of human PBMCs to iPSCs.
Two weeks after nucleofection, visible iPSC colonies started to form, which were picked and expanded in feeder-free dishes (Fig. 1B). Tight colonies formed and presented the typical morphology of iPSCs (Fig. 1B). Using primer sets for two common loci of the four nucleofected pEVs presented in Figure 1C, there were no detectable levels of residual pEVs present (<0.01 copies per cell after one passage and <0.0001 copies per cell after passaging the iPSCs five times) as determined by qPCR (Fig. 1D).
Karyotyping of the iPSC clone revealed a normal human karyotype of which a representative image is shown in Figure 1E. Immunostaining of iPSCs showed abundant expression of pluripotency markers OCT4 and SSEA4, and tight junction marker ECAD (Fig. 1F). Neural progenitor marker PAX6 was absent and there were only a few NES+ cells on the edges of the iPSC colonies.
Differentiation from iPSCs to beating cardiomyocytes
To explore the potential of the generated iPSCs, cells were differentiated into cardiomyocytes, which has been shown feasible by Su et al. (2013). Starting from single iPSCs, cells were cultured on Geltrex and three optimized media were used to drive differentiation through cardiac progenitors toward beating cardiomyocytes (Fig. 2A, B). After 2 weeks, clusters of cells were positively stained for cardiomyocyte markers TNNT2 and NKX2.5 (Fig. 2C) and aligned beating cardiomyocytes were observed (Supplementary Videos S1, S2, S3).
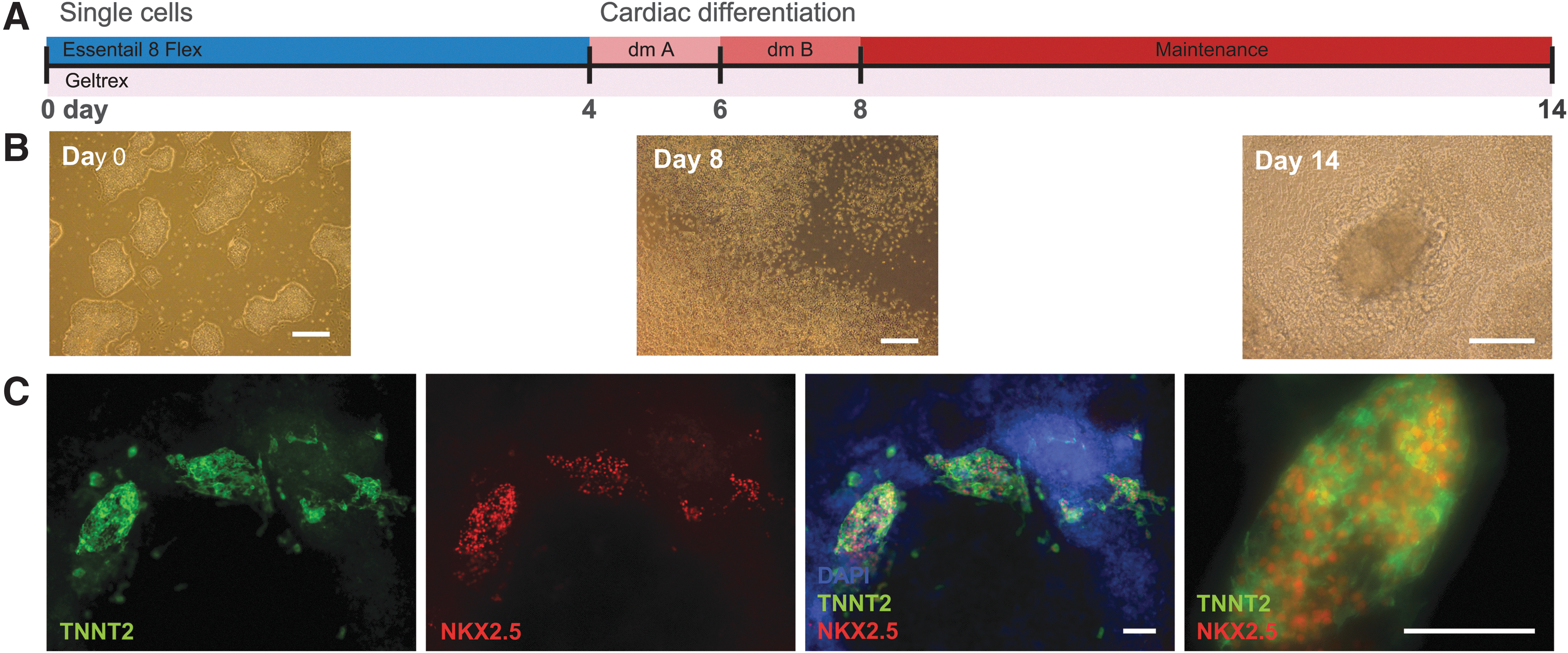
Differentiation of iPSCs to cardiomyocytes.
Differentiation from iPSCs to a neuron-astrocyte co-culture
Subsequently, we differentiated the same iPSC clone to a neuron-astrocyte culture using the protocol we developed for hESCs (de Leeuw et al., 2020). A schematic timeline of the procedure is depicted in Figure 3A. In the initial culture step of ∼26 days, NPCs were generated with clear morphological phases such as EB formation and formation of rosettes and later single-cell NPCs (Fig. 3B), which are suitable for cryopreservation. NPCs were virtually all positively stained for neural progenitor marker NES, but negative for neural progenitor marker PAX6. Some cells already entered neuronal differentiation to some extent (TUBB3 and MAP2) and there were no pluripotency markers present (ECAD and SSEA4). Within the larger cell clusters, there was still a small amount of ZO-1 expression left, which is typical for the previous stage of neural rosette formation (Fig. 3C).
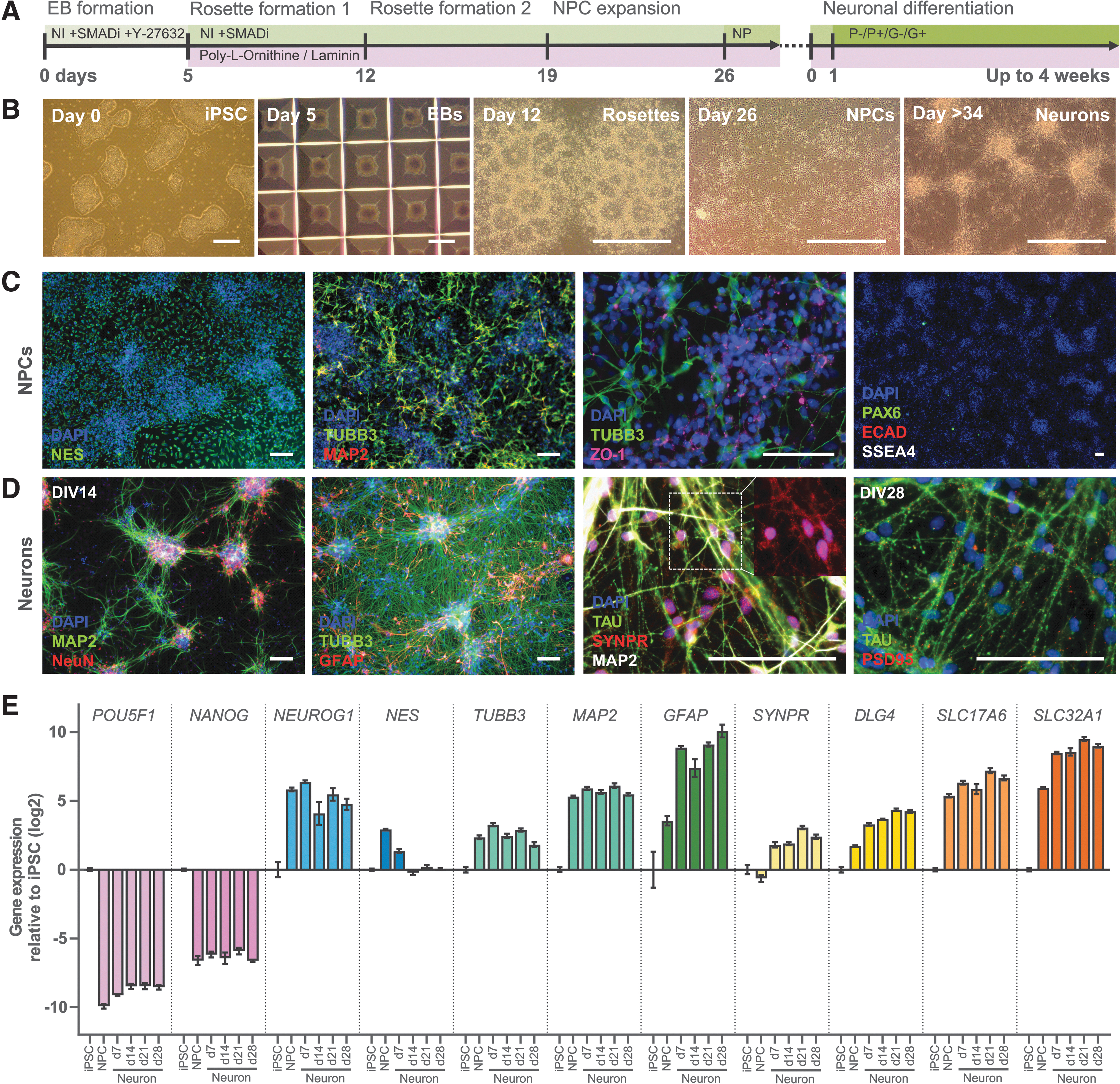
Differentiation protocol of iPSCs into a co-culture of neurons and astrocytes.
Next, in a second culture step of up to four weeks, NPCs were differentiated into a neuron-astrocyte culture (Fig. 3B, D). Regardless of the neuron-astrocyte differentiation protocol used, differentiation of NPCs resulted in a mixed neuron-astrocyte culture, expressing markers for neurons (TUBB3), mature neurons (NeuN), axons (TAU), dendrites (MAP2), synaptic vesicles (SYNPR), and astrocytes (GFAP) on 14 days in vitro (DIV14). On DIV28, post-synaptic marker PSD95 was also abundantly present (Fig. 3D). Quantitative gene expression measurements at different time points along the differentiation path were in agreement with the immunostainings (Fig. 3E; see Supplementary Data S1 for statistical details).
Stem cell marker expression of POU5F1 (which encodes OCT4) and NANOG strongly decreased, while expression of a range of early and late neuronal differentiation markers increased upon neuronal differentiation. Expression of NEUROG1, a marker for early neuronal differentiation, and neuron markers TUBB3 and MAP2 increased and plateaued from the NPC stage on. Neural progenitor marker NES expression peaked only in the NPCs and decreased upon further differentiation. Expression of astroglial marker GFAP, synaptic markers SYNPR and DLG4 (which encodes PSD95), and transporter markers SLC17A6 (which encodes VGLUT2) and SLC32A1 (which encodes VGAT) increased in the maturing neuron-astrocyte culture relative to the NPCs. In summary, both gene and protein expression of neuronal markers increased when neuronal differentiation was induced, which is indicative of a maturing neuronal network.
Neuronal differentiation protocols altered neuron-astrocyte ratio
To find the optimal neuron-astrocyte differentiation conditions, we tested four protocols as originally developed by Pistollato et al. (2017) and Gunhanlar et al. (2018) without (hereafter P− and G−, respectively) and with the addition of Ciliary neurotrophic factor (CNTF; P+, G+). The effect of half medium refreshments and shorter CNTF supplementation was also examined.
The most apparent difference between the four applied protocols was the difference between the protocol according to Pistollato et al. (2017) and the protocol according to Gunhanlar et al. (2018). Regardless of the addition of CNTF, the latter protocol resulted in more cell growth in general and a higher number of astrocytes relative to neurons (Fig. 4A, B, compare top row [P] with bottom row [G]). The addition of CNTF to the medium also enhanced cell growth, but predominantly increased the astrocyte to neuron ratio (Fig. 4A, B, compare left [−CNTF] and right [+CNTF] images within panels). The Gunhanlar-protocol and CNTF supplementation also accelerated the maturity of the neurons, as exemplified by increased MAP2 expression by CNTF (P+/G+) and increased NeuN expression by Gunhanlar (G−; Fig. 5).
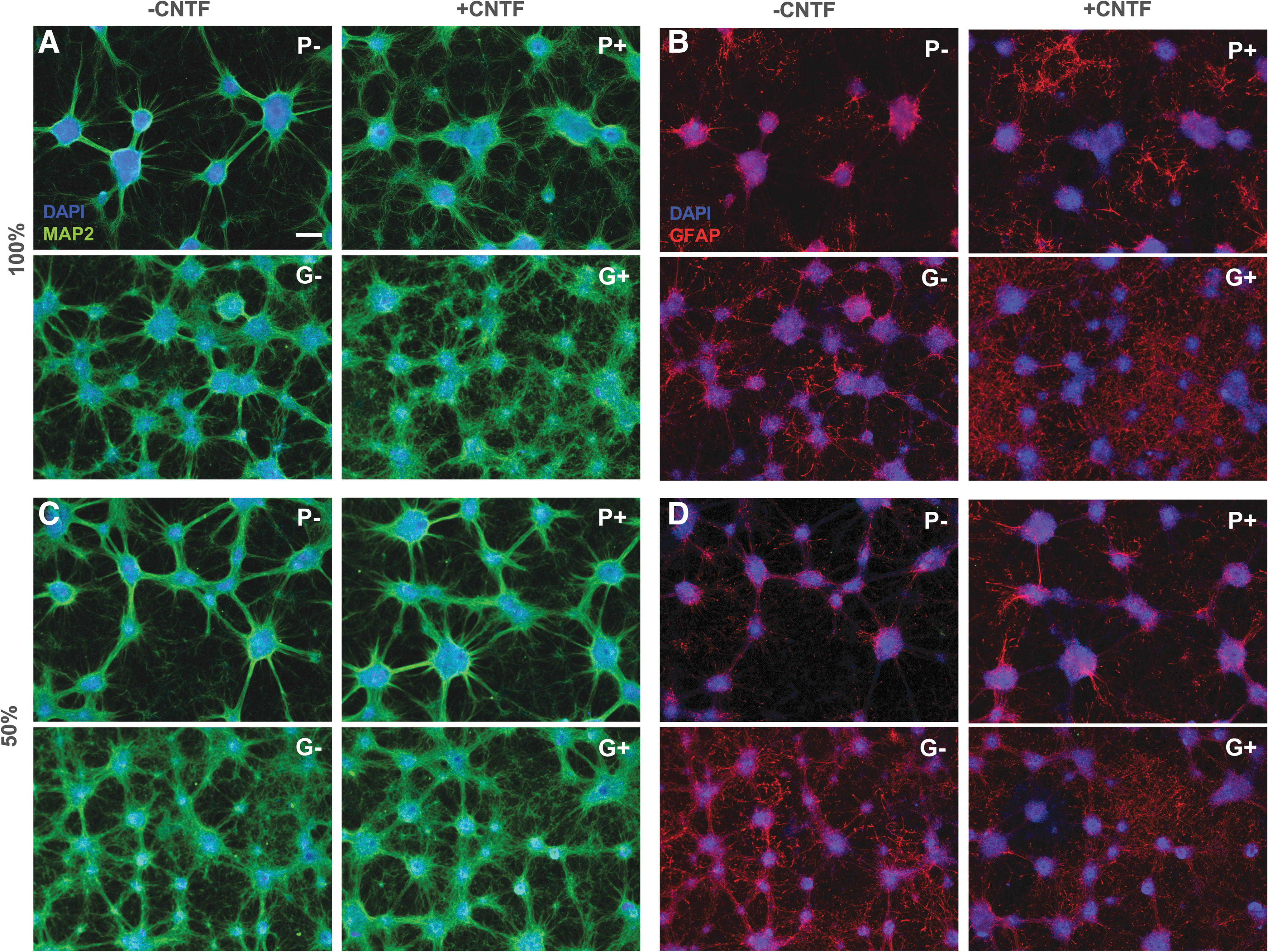
Comparison of four protocols P−, P+, G−, and G+ in terms of neuron-astrocyte ratio.
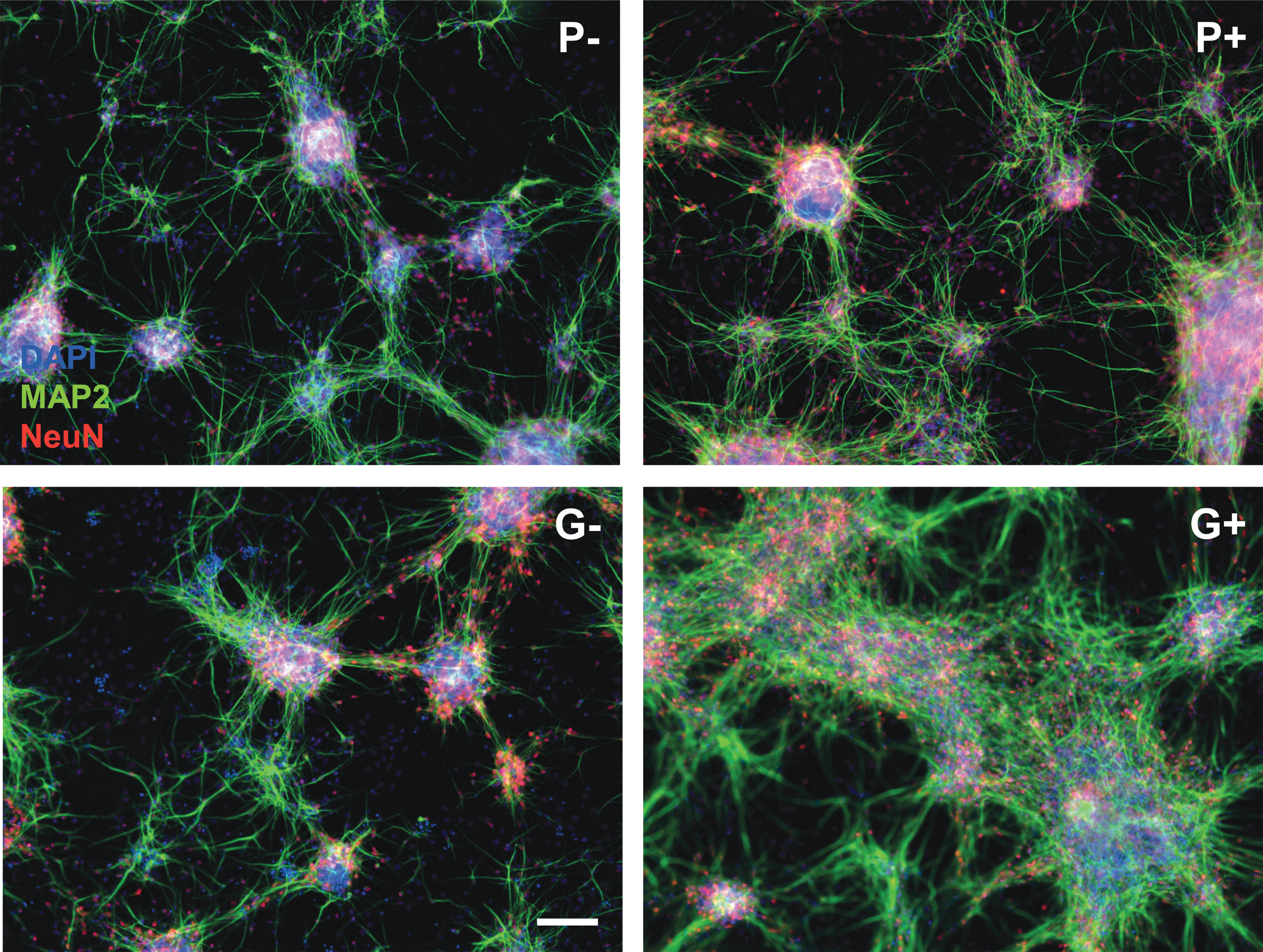
Expression of mature neuron markers MAP2 and NeuN on DIV14 in the four protocols. Scale bar: 100 μm.
Leaving part of the cell culture medium during refreshments may benefit the cell culture as the local environment stays intact, while the nutrients do not get depleted. Also, since in the G+ condition the astrocytes overgrew the neurons, CNTF was added only at the start of the differentiation. The half refreshments changed the shape of the network in both P− and G−, which presented smaller cell colonies (Fig. 4A, C, compare left images). The effect on astrocyte growth was diminished in the cultures with CNTF (P+/G+), while in G−, there were more astrocytes compared to full medium refreshments (Fig. 4B, D, left images).
Shorter supplementation with CNTF did not so much change the network morphology, but did decrease the amount of astrocytes (Fig. 4B, D, right images). Also, the effect of CNTF addition was less extreme than in the full refreshment conditions. In conclusion, it was possible to change the neuron astrocyte ratios by changing medium compositions.
Discussion
In this study, we have shown the ability to reprogram adult human PBMCs into iPSCs using EVs, which could be subsequently differentiated into cardiomyocytes and a neuron-astrocyte co-culture, highlighting the pluripotent nature of the iPSCs and adding ectodermal tissue differentiation to the repertoire of the iPSC method of the Zhang laboratory (Su et al., 2013). We could also change the network formation and the cell-type ratios between neurons and astrocytes in a similar way as shown with hESCs in our laboratory (de Leeuw et al., 2020).
Su et al. (2013, 2016) have established a protocol with which they could obtain iPSC colonies with a relatively high efficiency, and which we have replicated in our laboratory. Improvements of the protocol are still being made in all phases of the reprogramming (Gu et al., 2018), which holds great promise to make this method more efficient and economically feasible. We modified the protocol by replacing fibroblasts by a xeno-free and feeder-free coating for the expansion of the iPSC colonies. In our hands, the prolonged usage of murine feeder layers according to the original protocol did not result in the isolation of viable iPSCs. The modification could be seen as an improvement as it simplifies the culture conditions and eliminates xenogeneic contamination of the iPSCs earlier on.
Recently, we have shown that hESCs could be differentiated to a spontaneously firing neuronal network within the course of five weeks (de Leeuw et al., 2020). Using the same protocol, iPSCs could be differentiated to a similar phenotype containing both neurons and astrocytes, although there were two notable differences. The iPSC-derived NPCs were NES+/PAX6−, while the hESC-derived NPCs were NES+/PAX6+, which suggests that the cells differ in NPC subtype or developmental stage (Chandrasekaran et al., 2017; Molyneaux et al., 2007; Nat et al., 2007; Zhang et al., 2018).
Also, the morphology of the neuron-astrocyte cultures was different: iPSC-derived neurons clustered faster and a larger amount of astrocytes were present in the G− protocol compared to the hESC-derived cultures. Gene expression patterns were highly similar between the two cultures (de Leeuw et al., 2020). These differences are in line with the similar, but not identical gene expression profiles of both cell lines as shown before (Marei et al., 2017). The differentiation potential of both cell types is alike as shown by similar gene and protein expression of neuron, astroglial, and synaptic markers, which implies that iPSC-derived neuron-astrocyte culture matures in a similar manner as hESC-derived culture does. It will be interesting to test whether these iPSC-derived cultures can form functional networks such as we observed in hESC-derived neuronal cultures.
Tang et al. (2016) have shown that it is possible to directly convert PBMCs into NPCs using episomal factors, skipping over the stem cell stage. This has advantages over indirect programming; the culturing time can be greatly reduced and one can obtain a homogeneous cell population (Ladewig et al., 2013). However, iPSCs have higher proliferative potential, yielding higher cell numbers, and true self-replication, although mutations can still occur (Tanabe et al., 2015). The slower differentiation and cell heterogeneity could also be an advantage when mimicking or studying processes in vitro, especially development.
Moreover, to our knowledge, this is the first time it has been shown that iPSCs generated from the Su-protocol could be differentiated into a neuron-astrocyte co-culture instead of only neurons (Liu et al., 2015, 2020; Wang et al., 2018). However, perhaps the most compelling arguments for implementing a protocol based on the generation of stable and expandable iPSC cultures with cryopreservation options are the far more versatile application possibilities, as various cell and tissue types can be generated from such a resource.
Indeed, this method of reprogramming blood cells into stem cells and expanding these cells for differentiation to various organs of interest may provide a useful tool to study patient-specific disease modeling and drug screening. Research groups have already shown examples generating somatic cells from PBMC-derived iPSCs that replicate genetic diseases in all kind of organs, for example, the brain (Frega et al., 2019; Ustyantseva et al., 2020), blood (Lyu et al., 2018), and heart (Perepelina et al., 2020). There are also promising examples of drug and toxicity screening (Chang et al., 2014; Elitt et al., 2018). Another research direction with iPSCs is studying gene-environment interactions in groups of people who are exposed to certain chemicals (Joshi et al., 2019; Prince et al., 2019) or studying cohorts of whom a wealth of information is readily available to study complex processes such as “healthy” aging.
Footnotes
Acknowledgment
We would like to thank Nick Beijer for a critical review of the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This research is funded by the National Institute of Health (NIH)/National Institute of Aging (NIA) (AG017242), the Dutch Ministry of Agriculture, Nature and Food Quality, the Dutch Ministry of Health, Welfare and Sports, and the Dutch NGO Stichting Proefdiervrij.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.