
Abstract
Abstract
The reprogramming of somatic cells into a pluripotent/embryonic-like state holds great potential for regenerative medicine, bypassing ethical issues associated with embryonic stem cells (ESCs). Numerous methods, including somatic cell nuclear transfer (SCNT), fusion to pluripotent cells, the use of cell extracts, and expression of transcription factors, have been used to reprogram cells into ES-like cells [termed induced pluripotent stem cells (iPSCs)]. This study investigated early events in the nuclei of permeabilized murine somatic cells incubated in cytoplasmic extract prepared from Xenopus laevis germinal vesicle–stage oocytes by identifying proteins that showed significant quantitative changes using proteomic techniques. A total of 69 protein spots from two-dimensional electrophoresis were identified as being significantly altered in expression after treatment, and 38 proteins were identified by tandem mass spectrometry. Network analysis was used to highlight pathway connections and interactions between these identified proteins, which were found to be involved in many functions—primarily nuclear structure and dynamics, transcription, and translation. The pluripotency markers Klf4, c-Myc, Nanog, and POU5F1 were highlighted by the interaction network analysis, as well as other compounds/proteins known to be repressed in pluripotent cells [e.g., protein kinase C (PRKC)] or enhanced during differentiation of ESCs (e.g., retinoic acid). The network analysis also indicated additional proteins and pathways potentially involved in early reprogramming events.
Introduction
Several methods other than SCNT have been developed to reprogram cells into ES-like cells [termed induced pluripotent stem cells (iPSCs)], including the fusion of somatic cells to ESCs, embryonic germ cells, or embryonic blastomeres (Bru et al., 2008; Cowan et al., 2005; Do and Scholer, 2004; Egli et al., 2009; Neri et al., 2007; Tada et al., 1997), incubation of somatic cells in oocyte, egg, or ESC extracts (Alberio et al., 2005; Bian et al., 2009; Bui et al., 2008; Miyamoto et al., 2007; Miyamoto et al., 2009; Miyamoto et al., 2008; Novak et al., 2004; Tang et al., 2009; Taranger et al., 2005), and by using transient transduction of the transcription factors, POU5F1 (also called Oct3/4), Sox2, c-Myc, and Klf4 (Meissner et al., 2007; Okita et al., 2007; Park and Daley, 2007; Park et al., 2008; Takahashi and Yamanaka, 2006; Wernig et al., 2007). The reprogramming of cells by these described methods is not always complete or efficient; however, SCNT studies using cells treated with egg extracts have shown an increased rate of blastocyst formation (Ganier et al., 2011; Tang et al., 2009) and studies in mice and sheep using oocyte extracts have shown a significant increase in the production of cloned offspring compared with untreated donor cells (Bui et al., 2008; Rathbone et al., 2010).
Reprogramming somatic cells using extracts from oocytes or eggs has been demonstrated in various mammalian species, including murine, bovine, and porcine (Bui et al., 2008; Miyamoto et al., 2009; Novak et al., 2004; Tang et al., 2009) as well as amphibia (Alberio et al., 2005; Bian et al., 2009; Byrne et al., 2003; Hansis et al., 2004). Many studies using extracts from Xenopus laevis eggs and oocytes have reported various changes in DNA demethylation, histone demethylation, and deacetylation, increases in the gene expression and levels of pluripotency markers, and changes in the nuclear lamina and chromatin (Alberio et al., 2005; Bui et al., 2008; Miyamoto et al., 2007; Miyamoto et al., 2008; Simonsson and Gurdon, 2004; Tang et al., 2009). These studies have also reported the removal of somatic cell proteins such as lamin A/C (LMNA) and chromobox homolog 5 [CBX5, also called heterochromatin protein 1α (HP1α)] (Alberio et al., 2005; Bian et al., 2009), and the incorporation of oocyte/egg components, including Xenopus-specific histone B4 and lamin III (Alberio et al., 2005; Miyamoto et al., 2007; Miyamoto et al., 2008). Subsequent activation of pluripotency genes such as Pou5f1 has also been shown following treatment with amphibian or mammalian oocyte/egg extracts (Bian et al., 2009; Bui et al., 2008; Hansis et al., 2004; Miyamoto et al., 2007; Miyamoto et al., 2008; Tang et al., 2009).
In the current study, murine somatic cells were incubated in X. laevis oocyte extract with the aim of identifying proteins located in the nucleus that were significantly increased or decreased in level to capture a snapshot of early events potentially involved in reprogramming. Subsequent bioinformatic analysis of these proteins highlighted the pluripotency markers Klf4, c-Myc, Nanog, and POU5F1, as well as a range of other protein interactions and pathways potentially involved in reprogramming.
Materials and Methods
Cell culture
Murine STO fibroblasts were cultured in Dulbecco's modified Eagle medium (DMEM), containing 1% minimum essential medium (MEM)–nonessential amino acids, 1 unit/mL penicillin, 0.1 mg/mL streptomycin, and 10% fetal bovine serum (FBS) at 37°C and 5% CO2. For each experiment, cells were cultured until 80–90% confluent; quiescence was then induced by reducing the concentration of FBS to 0.5% for 4 days.
Preparation of Xenopus oocyte extract
Oocytes were isolated from Xenopus laevis ovaries and extract prepared as previously described (Rathbone et al., 2010). In brief, ovaries of euthanized mature female X. laevis were removed and digested using collagenase (8 mg/mL type II collagenase in calcium-free Ringer's solution) for 2–3 h. Following complete digestion, free oocytes were removed and washed in 0.9% saline followed by ice-cold extraction buffer (20 mM HEPES, 100 mM potassium chloride, 5 mM magnesium chloride, 2 mM β-mercaptoethanol, 6.3 μM leupeptin, 0.15 μM aprotinin, and 1.5 μM pepstatin A). The oocytes were centrifuged at 10,000×g for 10 min at 4°C, and the middle ooplasmic layer removed and centrifuged for a further 10 min to remove debris. The supernatant was collected and centrifuged at 100,000×g for 40 min at 4°C. This supernatant was transferred into clean tubes and centrifuged for 30 min. Glycerol was added to the oocyte extract to give a final concentration of 5% (vol/vol), before dividing into aliquots that were snap frozen in liquid nitrogen and stored at −80°C.
Digitonin permeabilization and incubation in Xenopus oocyte extract
Quiescent donor cells harvested by trypsinization were permeabilized for 2 min on ice, at a concentration of 2 million cells per 1 mL of 20 μg/mL digitonin in permeabilization buffer [170 mM potassium gluconate, 5 mM potassium chloride, 2 mM magnesium chloride, 1 mM potassium phosphate, 1 mM EGTA and 20 mM HEPES (pH 7.25) and 2 mM dithiothreitol (DTT), 1% protease inhibitor cocktail (PIC), with an osmolarity of 330–350 mOsm]. Permeabilization was stopped by adding an excess of permeabilization buffer and centrifuging at 700×g for 10 min. Cells were incubated in either supplemented DMEM for controls and permeabilized controls, or Xenopus oocyte extract, at a concentration of 5000 cells/μL for 5 h at 17°C. The cells were washed once in permeabilization buffer and twice in PBS. Cells were either spun onto coverslips for immunocytochemistry or the nuclear proteins extracted for separation by two-dimensional electrophoresis.
Histone and DNA methylation immunocytochemistry
Immunocytochemistry for DNA methylation and H3K9 methylation status was performed using specific antibodies to 5-methylcytosine (5MeC) and trimethylated histone H3K9 (H3K9me3), as previously described (Rathbone et al., 2010).
Labeled cells were examined by epifluorescence at 100× magnification (Leica Microsystems, Germany). Images were captured using a digital camera (Hammamatsu, Japan) and analyzed using SIMPLE PCI software (Compix Inc., USA). Methylation status was calculated as the intensity of fluorescein isothiocyanate (FITC) staining divided by the intensity of nuclear labeling with either 4′,6-diamidino-2-phenylindole (DAPI) or propidium iodide (PI).
Extraction of nuclear proteins
Nuclear proteins were extracted using a NuCLEAR extraction kit (Sigma-Aldrich); the protocol was modified to include a washing step. Cells were incubated in lysis buffer [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT and 1% PIC] on ice for 15 min to allow the cells to swell. A 10% IGEPAL CA-630 solution was added at a final concentration of 1%, and the samples were vortexed vigorously for 2×30 sec to break open the cells. The samples were centrifuged at 10,000×g for 1 min to pellet the nuclei and the supernatant was removed. The nuclei were washed five times in wash buffer [20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 0.2 mM EDTA, 10 mM KCl, 25% glycerol, 0.1% sodium deoxycholate, 1 mM DTT, and 1% PIC], by vortexing and incubating on ice for 5 min before centrifugation. The washed nuclei were resuspended in ice-cold extraction buffer [20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 0.2 mM EDTA, 0.42 M NaCl, 25% glycerol, 1 mM DTT, and 1% PIC] maintained on ice and vortexed every 5 min over a 30-min period. The lysed nuclei were centrifuged at 16,000×g for 5 min at 4°C, and the supernatant containing the soluble nuclei proteins (NP) was transferred to clean chilled tubes. The remaining pellets containing the nuclear envelope, DNA, and DNA-binding proteins (NE) were washed with extraction buffer and these washes added to the NP samples. The pellets were resuspended in extraction buffer, sonicated for 10 sec using a sonic probe (MSE Ltd., UK), and frozen at −80°C.
Two-Dimensional gel electrophoresis
The protein content of permeabilized control and oocyte extract–treated samples was estimated using the Bio-Rad RCDC assay. Samples were prepared using a 2D Clean-Up Kit (GE Healthcare), and the protein was suspended in rehydration buffer [7 M urea, 2 M thiourea, 2% CHAPS, 0.5% ampholytes, 1.2% DeStreak™ (GE Healthcare Ltd.), and 0.01% Bromophenol Blue], at room temperature for 1 h. Samples were sonicated using a sonic probe three times for 10 sec, with incubation on ice for 1 min between each sonication. Then 100 μg of protein was loaded onto pH 3–10 17-cm isoelectric focusing (IEF) strips (Bio-Rad ReadyStrip™), then actively rehydrated (50 V per strip for 12–16 h at 20°C) using the Bio-Rad PROTEAN® IEF system. First-dimension separation was performed using the rapid ramp setting 250 V for 15 min, then a rapid ramp to 10,000 V over 3 h and focusing at 10,000 V for 60,000 V hours. Prior to second-dimension separation, the IEF strips were equilibrated using sodium dodecyl sulfate (SDS) equilibration buffer (50 mM Tris-HCl, 6 M urea, 30% glycerol, 2% SDS) first containing 2% DTT, then with 2.5% iodoacetamide, for 15 min in each buffer. Each equilibrated strip was rinsed briefly in Tris-Glycine-SDS (TGS) buffer and secured on top of a 12% Bis/Tris acrylamide gel with 1% low-melting-point agarose in TGS. Gels were run using the Ettan™ DALTsix electrophoresis tank (GE Healthcare Ltd.) in TGS buffer, then stained with silver using a mass spectrometry (MS)-compatible silver staining method (Yan et al., 2000). pI standards (Bio-Rad) containing proteins with known pI and molecular weight (MW) values were used to estimate pI values of proteins. Gels images were captured using the Molecular imager® GS-800™ densitometer (Bio-Rad).
Three experimental replicates were run for both the NP and NE samples, and each experimental replicate was comprised of triplicate two-dimensional (2D) gels for permeabilized control and extract-treated samples.
2D gel analysis
2D gel analysis was carried out using Delta2D (Decodon, Germany). Expression changes in protein spots were deemed statistically significant over a 95% confidence interval in the Student's t-test. Only proteins that varied in at least two of the three experimental replicates were accepted for further analysis. Spots of interest were excised from the gels and stored at −20°C.
Mass spectrometry
Proteins in excised gel pieces were prepared using the ProteomeWorks MassPREP™ Station (Waters Corp., USA). In brief, the gel spots were destained with 15 mM potassium ferricyanate and 50 mM sodium thiosulfate, then reduced with 10 mM DTT in 100 mM ammonium bicarbonate (AMBIC), alkylated with 55 mM iodoacetamide in 100 mM AMBIC, and dehydrated with acetonitrile (ACN). Proteins were digested into peptides in 30 μL per well of 7.5 ng/μL trypsin (Trypsin Gold, Promega Corp., USA) in 25 mM AMBIC, after chilling for 30 min during absorption of trypsin and incubating for 5 h at 40°C. Samples were made to 0.1% formic acid and stored at −20°C until MS analysis.
Peptides were analyzed on a Micromass Q-ToF2 (Waters Corp.) mass spectrometer after separation on an integrated CapLC system (Waters Corp.). A total of 6.4 μL of the digest was introduced via the autosampler and initially loaded onto a C18 packed precolumn (LC Packings, Dionex Corp., UK) for desalting, and then eluted onto a PepMap C18 reversed-phase, 75 μm inner diameter (i.d.) 15-cm column (LC Packings). Following binding of the sample on the column in 0.1% formic acid, peptides were eluted over 40 min in an increasing gradient of ACN (from 5% to 80% ACN in 0.1% formic acid). Eluted peptides were delivered online at a flow rate of 400 nL/min to the mass spectrometer, which was fitted with a nanolockspray source and operated with MassLynx Version 4.0 acquisition software (Waters Corp.). Data-dependent switching was performed so that whenever a peptide with an associated charge of 2+ or 3+ was detected above a preset threshold signal, the mass spectrometer would automatically switch to MS-MS mode to generate fragmentation data from the precursor peptide.
Raw data files were processed using ProteinLynx Global SERVER Version 2.0.5 (Waters Corp.) to generate a peaklist file of fragment mass data, which was used to search the Swiss-Prot and NCBInr databases using the search engine MASCOT (Matrix Science Ltd., London, UK). Only identifications with probability-based MOWSE scores above a threshold of p<0.05 were accepted. In addition, some fragmentation data was analyzed manually, and de novo sequence elucidation was performed on selected peptides using the PepSeq tool in BioLynx (Waters Corp.).
Analysis of potential protein interactions by network creation
Murine UniProtKB primary accession numbers used for the MS-identified protein data set were analyzed using Ingenuity Pathways Analysis (IPA) (Ingenuity® Systems Inc., USA; www.ingenuity.com) by uploading the proteins and their corresponding expression values into the application. Xenopus UniProtKB numbers were not accepted by the software, so BLAST (Altschul et al., 1990) was used to search Swiss-Prot and TrEMBL for corresponding murine proteins with the highest sequence homology.
The network produced is a graphical representation of the molecular relationships where molecules are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All edges are supported by at least one reference from the literature, from a textbook, or from canonical information stored in the Ingenuity Pathways Knowledge Base. Human, mouse, and rat orthologs of a gene are stored as separate objects in the Ingenuity Pathways Knowledge Base, but are represented as a single node in the network. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation observed from the 2D gel analysis. Nodes are displayed using various shapes that represent the functional class of the gene product.
The UniProtKB number for histone H3 was not accepted by the IPA software and was added manually after creation of the network by using the build tool. Proteins in the network known to be associated or interact with histone H3 were highlighted, and the software was then prompted to grow out from these proteins using direct and indirect interactions. This tool added proteins that only associated/interacted with all of the highlighted proteins. After histone H3 was added, the software was then prompted to reanalyze the network for other known associations.
Results
Effect of permeabilization and incubation at 17°C on donor cells
On average 65% [±5% standard error of the mean (SEM), n=20] of the recovered cells were permeabilized as estimated by fluorescence using FITC-labeled dextran 70000 Mr. The percentage recovery of untreated cells incubated in complete medium for 5 h at 17°C was 91% (±6% SEM, n=3). There was a reduction in the percentage recovery of the number of permeabilized cells after incubation in either complete medium or Xenopus oocyte extract at 17°C, but they were both reduced to a similar extent (86.9% and 83.1%, respectively).
Effect of Xenopus oocyte extract on methylation of DNA and histone H3K9 in donor cells
Analysis of the labeling intensity of FITC-labeled 5MeC in cells, normalized using the intensity of nuclear DNA staining, showed that treatment with Xenopus oocyte extract for 5 h significantly reduced labeling compared to untreated and permeabilized controls [p<0.0001; analysis of variance (ANOVA), n=3]. This was also observed for H3K9me3 (p<0.0001; ANOVA, n=3), as shown in Figure 1. Representative images are presented in Figure S1 (Supplementary Data are available at www.liebertpub.com/cell/). The patterns of labeling observed were similar to those previously reported after exposure of ovine fetal fibroblasts to Xenopus oocyte extract (Rathbone et al., 2010) and mouse embryonic fibroblasts to axolotl oocyte extract (Bian et al., 2009).
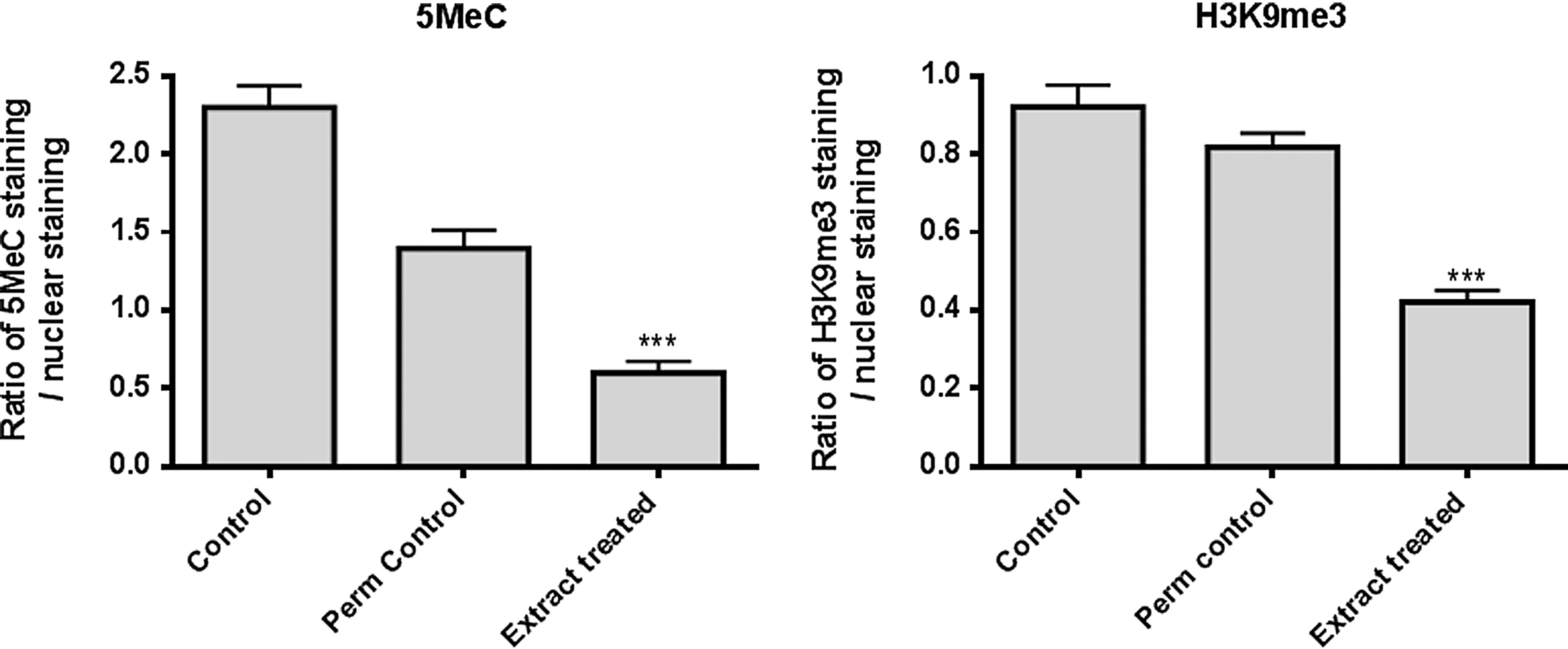
Immunofluorescence labeling analysis of 5-methylcytosine (5MeC) and trimethylated histone H3K9 (H3K9me3) in mouse fibroblasts after no treatment (Control), after permeabilization (Perm control), or after permeabilization and incubation in Xenopus oocyte extract (Extract treated). The intensity of labeling was normalized using the nuclear labeling and the ratios compared for the control, permeabilized control, and Xenopus oocyte extract. Both 5MeC and H3K9me3 labeling was significantly reduced following treatment in Xenopus oocyte extract, when compared to the controls and permeabilized controls. For 5MeC, untreated control 2.30±0.14, permeabilized control 1.40±0.11, treated 0.60±0.07, p<0.0001; analysis of variance (ANOVA) (n=3). For H3K9me3, untreated control 0.92±0.06, permeabilized control 0.82±0.04, treated 0.42±0.03, p<0.0001; ANOVA (n=3).
MS-based identification of proteins showing altered levels after treatment with Xenopus oocyte extract
Delta2D analysis of the 2D gel profiles of nuclei enriched samples revealed that 69 protein spots were significantly altered in levels in the Xenopus extract–treated samples compared to permeabilized controls. In all, 51 of the affected protein spots were located in the soluble nuclei fraction (NP) and 18 were in the nuclear envelope and DNA-binding proteins' fraction (NE). Of these, 28 NP and 16 NE proteins were identified by tandem MS. The functions of the identified proteins, the fraction in which they were located, whether their levels were significantly increased or decreased compared to permeabilized controls, and the number of peptide matches are shown in Table 1. The majority of proteins identified were of mouse origin. Any Xenopus protein matches were confirmed as Xenopus specific using de novo sequence deduction. A total of four proteins were categorized as both Xenopus and/or murine (histone H3, histone H2B, isocitrate dehydrogenase, and vimentin), because the matched peptides had identical sequences in both species and/or both Xenopus- and murine-specific peptides were present.
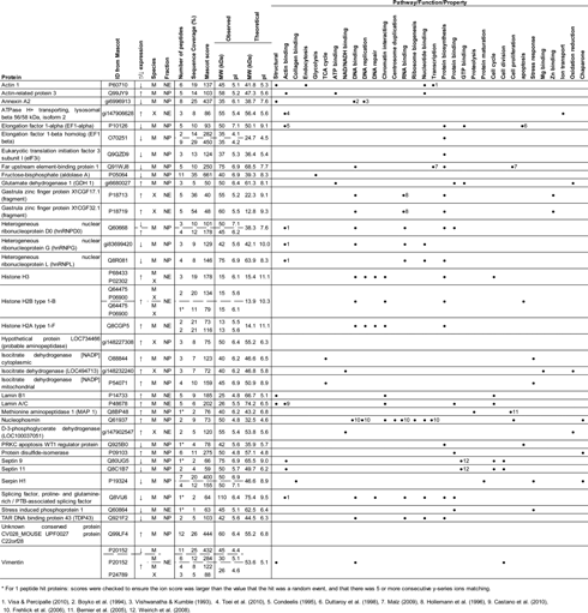
The table shows the direction of protein level change, the species of origin, the fraction it was located in, the observed location on the two-dimensional (2D) gels, the theoretical molecular weight (MW) and pI from UniProtKB entries, the number of identifying peptides from tandem mass spectrometry (MS), the sequence coverage of those peptides on the protein, and the pathway/function the protein is involved in from UniProtKB and the literature.
For 1 peptide hit proteins: scores were checked to ensure the ion score was larger than the value that the hit was a random event and that there were five or more consecutive y-series ions matching.
References: 1. Visa and Percipalle, 2010; 2. Boyko et al. 1994; 3. Vishwanatha and Kumble, 1993; 4. Toei et al., 2010, 5. Condeelis, 1995; 6. Duttaroy et al., 1998; 7. Malz et al., 2009; 8. Hollemann et al., 1996; 9. Castano et al., 2010; 10. Frehlick et al. 2007; 11. Bernier et al., 2005; 12. Weinrich et al., 2008.
Six proteins, heterogeneous ribonucleoprotein D0 (hnRNPD), vimentin (VIM), elongation factor 1-β (EEF1B2), histone H2A, histone H2B, and serpin H1 were found at different locations on the 2D gels. Four of the proteins (EEF1B2, histone H2A, histone H2B, and serpin H1) were each located at the same estimated MW but had different pI values, indicating that these proteins may have been subjected to posttranslational modifications (PTMs). Both hnRNPD and VIM also contain numerous potential PTM sites. Representative 2D gels for each nuclear fraction can be found in Figure S2.
Interaction of identified proteins determined by network analysis
We used the data set of Xenopus oocyte extract–induced altered proteins in a network analysis using bioinformatics software. The Path Explorer tool of IPA was used to explore molecular interactions based on the Ingenuity Knowledge Base, a database comprised of millions of published molecular interactions.
The network produced showed the proteins affected by Xenopus oocyte extracts are involved in the following pathways—gene expression, DNA replication, recombination and repair, and lipid metabolism (see Fig. 2 and Table S1). Pluripotency markers, including Klf4, c-Myc, POU5F1, and Nanog, were also highlighted in these analyses. Proteins with multiple functions and that are involved in multiple pathways were also highlighted; these include follicle-stimulating hormone (FSH), caspase, RNA polymerase II, interleukin-4 (IL-4), and retinoic acid (RA).
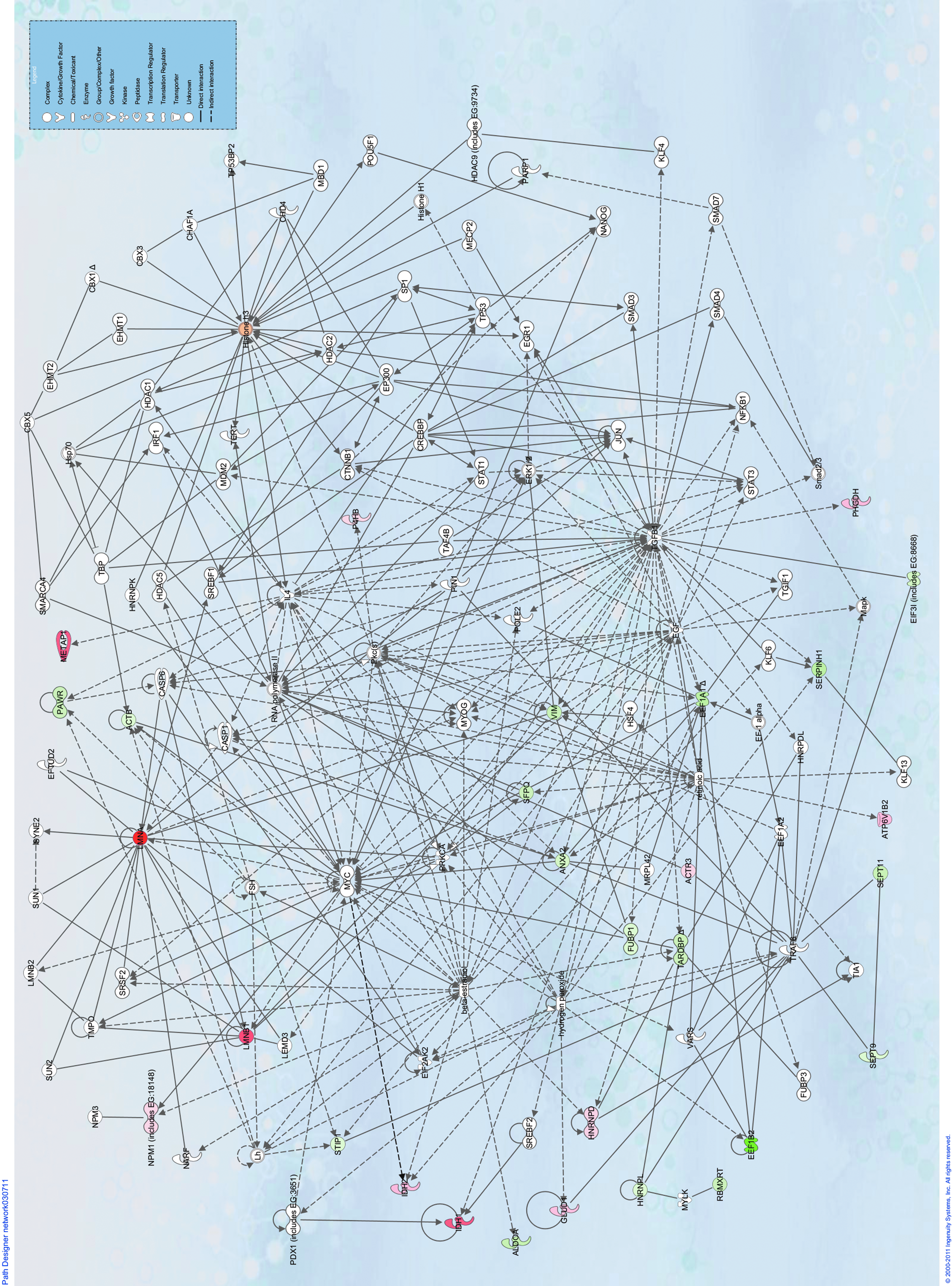
Ingenuity Pathways Analysis (IPA) network analysis of proteins identified as affected by Xenopus oocyte extracts in this study. The network is a graphical representation of the molecular relationships in which molecules are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All edges are supported by at least one reference from the literature, a textbook, or canonical information stored in the Ingenuity Pathways Knowledge Base. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation observed from the two-dimensional gel analysis. Various shapes represent different functional classes of the gene product (see key).
Discussion
This investigation looked at proteins that were increased in concentration in the nuclei of the murine somatic cells due to their migration from Xenopus extract and at mouse nuclear proteins with altered levels caused by exposure to the oocyte extract. Network analysis of the proteins identified indicated pathways involved in early reprogramming events.
Cytoplasm from both mammalian and amphibian GV- and MII-stage oocytes have shown differences in their reprogramming capabilities: Mammalian somatic cells do not undergo DNA replication during incubation in GV-stage oocyte extract, and 99% of these cells show removal of LMNA from the nuclear lamina (Alberio et al., 2005), suggesting considerable nuclear remodeling. When GV-stage oocyte extract–treated cells are subsequently cultured, they form ESC-like colonies and show activation of pluripotent marker genes (including Nanog and Sox2) (Miyamoto et al., 2009). On the other hand, MII oocyte extracts support DNA replication and remove the TATA binding protein (TBP) and LMNA [this is lower (74%) than observed with GV extract] (Alberio et al., 2005; Miyamoto et al., 2009; Miyamoto et al., 2008). In addition, studies using MII extracts have shown conflicting results regarding changes in pluripotency marker genes (Ganier et al., 2011; Miyamoto et al., 2007; Miyamoto et al., 2008; Miyamoto et al., 2009), and in the formation of colonies after cell culture (Ganier et al., 2011; Miyamoto et al., 2009). Taken together, there are indications that GV-stage oocyte cytoplasm is superior to MII-stage oocyte cytoplasm in its reprogramming capability, which was why it was used in this study.
We assessed if the 5-h incubation in oocyte extract was sufficient to initiate reprogramming of murine somatic cells by examining the global levels of demethylation of DNA and of H3K9me3. DNA methylation, a covalent modification that in mammals occurs most commonly at CpG dinucleotides to form 5MeC (Jabbari and Bernardi, 2004; Spivakov and Fisher, 2007), is essential in normal development, and is generally linked to gene silencing in somatic cells (Fulka et al., 2008; Wolffe et al., 1999). Previous reprogramming studies have shown reductions in both global levels of DNA methylation and DNA methylation at promoter regions of key pluripotency genes (Alberio et al., 2005; Bian et al., 2009; Freberg et al., 2007; Simonsson and Gurdon, 2004; Taranger et al., 2005). H3K9me3 is a marker of heterochromatin that is epigenetically stable in somatic cells (Bannister and Kouzarides, 2011; Peters et al., 2003). The loss of methyl groups at this lysine base to form H3K9me2 or H3K9me has been implicated as a marker of ESCs (Atkinson and Armstrong, 2008). Other reprogramming studies have shown global reductions in the level of H3K9me3 (Bian et al., 2009; Ganier et al., 2011), and the complete demethylation of H3K9 has been observed in somatic cells after injection into GV-stage oocytes (Bui et al., 2008). In agreement with the aforementioned studies, the significant reduction in global DNA methylation and H3K9me3 observed in the murine somatic cells in this study indicates that reprogramming has been initiated.
Other proteomic studies have compared proteomic profiles of iPSCs with ESCs (Jin et al., 2011; Kim et al., 2012; Munoz et al., 2011; Wang et al., 2012), or protein expression changes of iPSCs that have been cultured for days or weeks post-reprogramming compared to the somatic cells from which they were derived (Huang et al., 2012; Kim et al., 2012; Munoz et al., 2011; Pewsey et al., 2009). In the current study, we chose to concentrate on global nuclear protein expression changes during the early stages of reprogramming, before the cells express pluripotency markers. We aimed to increase the fundamental understanding of molecular events that underlie the very early events of reprogramming.
In this study, the majority of proteins that were altered in somatic cell nuclei after treatment with oocyte extracts and the proteins from subsequent network analysis fall into three distinct categories—those that interact with pluripotency markers/genes, those that are structural proteins in the nucleus, and those that act as transcription or translation factors/regulators. Other studies have shown changes in the levels of numerous proteins following incubation in oocyte, egg, or ESC extracts, which are summarized in Table 2.
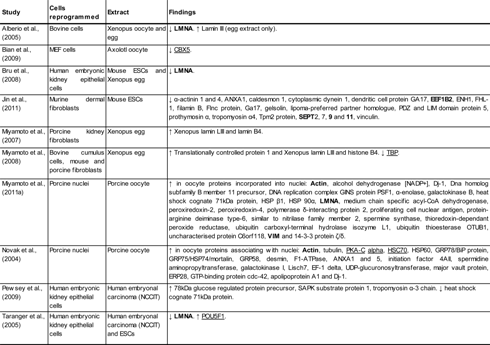
Proteins highlighted in bold were identified by tandem mass spectrometry (MS), and proteins underlined are those highlighted by Ingenuity Pathways Analysis (IPA) network analysis, in this study.
Pluripotency markers and associated factors
Network analysis identified a number of the pluripotency markers/transcription factors known to be important for iPSC formation—Klf4, c-Myc, Nanog, and POU5F1 (Okita et al., 2007; Park et al., 2008; Takahashi and Yamanaka, 2006). POU5F1 and Nanog have been deemed the master stem cell transcription factors for maintenance of the undifferentiated state and self-renewal of ESCs (Li, 2010), and while c-Myc has been shown to be dispensable for inducing pluripotency, significantly more iPSCs are produced with c-Myc expression and the cells generated are consistently of a high quality (Nakagawa et al., 2008). Although not essential for iPSC creation, cells expressing Nanog are more likely to self-renew and are less susceptible to differentiation (Ralston and Rossant, 2010). Klf4 is involved in the regulation of proliferation and differentiation in numerous tissues (McConnell and Yang, 2010).
Numerous proteins highlighted by this study are known to bind to and/or affect the expression of genes related to pluripotency, which include, signal transducer and activator of transcription 3 (STAT3), β-catenin (CTNNB1), chromodomain helicase DNA-binding protein 4 (CHD4), histone deacetylase 1 (Hdac1), heterogeneous nuclear ribonucleoprotein L (hnRNPL), Klf4, poly(ADP-ribose) polymerase 1 (PARP1), transcription activator Brg1 (also called SMARCA4), and SP1 (Li, 2010; Ng and Surani, 2011; Pardo et al., 2010).
Structural proteins
Numerous structural proteins of the nucleus were identified by MS in this study and include β-actin (ACTB), LMNA, lamin B1 (LMNB1), annexin A2 (ANXA2), VIM, septin 9 (SEPT9), and septin 11 (SEPT11). In the nucleus, actin has been shown to be part of chromatin remodeling complexes (Bettinger et al., 2004; Hargreaves and Crabtree, 2011; Olave et al., 2002); it is associated with transcription machineries, becomes incorporated into newly synthesized ribonucleoproteins, influences long-range chromatin organization, and works in conjunction with different types of actin-binding proteins (ABP) to regulate actin function (Rando et al., 2000; Visa and Percipalle, 2010). The polymerization of nuclear actin has been shown to be necessary for transcriptional reprogramming of Pou5f1 by Xenopus oocytes (Miyamoto et al., 2011b), and specifically nuclear ACTB is known to participate in transcription by association with all three RNA polymerases in vitro and in vivo (Hu et al., 2004; Kukalev et al., 2005; Percipalle and Visa, 2006; Philimonenko et al., 2004; Visa, 2005; Wu et al., 2006). The reduction of the ACTB level observed in this study could affect many pathways, including, embryonic development, apoptosis, and cell cycle control (Hargreaves and Crabtree, 2011), and indicates that actin may play a role in the large scale organization of the genome during reprogramming.
The lamins are intermediate filament proteins important for nuclear architecture and are known to be affected during reprogramming. LMNA is also a nonclassical ABP essential for nuclear structural integrity, DNA replication, transcriptional splicing, cell signaling, DNA repair, and cellular proliferation (Castano et al., 2010). Zastrow et al. (2004) have reviewed proteins that bind A-type lamins, which include proteins identified by this study—actin, SREBF1, and protein kinase Cα (PRKCA). PRKCA is a signaling molecule in many pathways that has been shown to interact with the A-type lamins when it translocates to the nucleus. The inhibition of PRKC isoforms have also been shown to maintain mouse ESC pluripotency (Dutta et al., 2011).
The annexins are a family of proteins that bind to phospholipids in a calcium-dependent manner. ANXA2 is required for DNA replication (Vishwanatha and Kumble, 1993); however, during incubation in Xenopus oocyte extract, mammalian somatic cells do not undergo DNA replication (Alberio et al., 2005). The observed decrease in ANXA2 in this study may be an indicator of the loss of DNA replication during reprogramming.
The intermediate filament VIM has many nonstructural roles in the cell, reviewed by Ivaska et al. (2007). Of interest to this study, VIM interacts with and is carried into the nucleus by various DNA structures (Hartig et al., 1998; Tolstonog et al., 2000; Tolstonog et al., 2001). Whether VIM is affected by oocyte extract treatment in relation to any of these specialized functions remains to be elucidated.
Septins are guanosine triphosphatases (GTPases) that form filaments in the cytoplasm, and, in addition to their role in cell division, they have been shown to have roles in membrane trafficking and organizing the cytoskeleton (Lindsey and Momany, 2006; Weirich et al., 2008). In the cytoplasm, SEPT9 and SEPT11 are found in association with actin filaments upon which they are dependent upon for localization (Field and Kellogg, 1999). This may also be the case in the nucleus because ACTB, SEPT9, and SEPT11 levels are all reduced following incubation in Xenopus oocyte extract.
Transcription factors/regulators
Numerous transcription factors/regulators were identified as changing in their protein levels in this study and include ACTB (previously discussed), far upstream element-binding protein 1 (FUBP1), hnRNPD, hnRNPL, nucleophosmin 1 (NPM1), PRKC apoptosis WT1 regulator protein (PAWR), and TAR DNA-binding protein 43 (TARDBP). In the network analysis, 41 proteins were also noted as being involved in transcription regulation (see Table S1).
The heterogeneous nuclear ribonucleoprotein (hnRNP) proteins undertake essential functions in the biogenesis of mRNA and RNA metabolism, and hnRNPD, hnRNPK, and hnRNPL continually shuttle between the cytoplasm and nucleus transporting mRNAs (Krecic and Swanson, 1999). The observed changes in levels of hnRNPD and hnRNPL in this study may be indicators of modifications in mRNA trafficking. TAR DNA-binding protein (TARDBP), a transcriptional repressor classed as an hnRNP (Buratti and Baralle, 2010), has been reported to be crucial for embryonic development, with increasing evidence indirectly implicating its involvement in other cellular processes, including microRNA biogenesis, apoptosis, and cell division (Warraich et al., 2010).
Nucleophosmins are nuclear chaperones required for the proper assembly of nucleosomes and the attainment of proper higher-order chromatin structures. They have been shown to remove linker histones from somatic chromatin leading to a more open and extended chromatin structure (Dimitrov and Wolffe, 1996). NPM1 was observed to be upregulated in extract-treated cells in this study, and both NPM1 and NPM3 (identified in the network analysis) have been shown to be highly expressed in iPSCs compared to mouse embryonic fibroblasts (Huang et al., 2012).
Translation regulators
Elongation factors 1α and 1β (EEF1A1 and EEF1B2) and eukaryotic translation initiation factor 3 (EIF3I, also called TRIP1) levels were reduced in somatic nuclei after oocyte extract incubation. A very abundant protein in eukaryotes (∼1–2% of total protein) and rapidly proliferating cells, EEF1A regulates the rate of polypeptide elongation during translation. In embryos, a large increase in EEF1A mRNA has been observed that relates to the rate of cell growth (Condeelis, 1995). The reduction of EEF1A1 level observed in this study reflects the reduction of proliferation observed in iPSCs.
EIF3I binds to the 40S ribosome during translation initiation to promote binding to mRNA (Asano et al., 1997). EIF3I represses the ability of transforming growth factor-β (TGF-β1) to induce transcription (Choy and Derynck, 1998), and inhibition of TGF-β signaling has been shown to co-operate in the reprogramming of somatic cells (Maherali and Hochedlinger, 2009). The reduction of EIF3I in this study may contribute to TGF-β–mediated changes.
Conclusions
In this study, we have shown proteins whose expression profiles change during the early stages of reprogramming by Xenopus oocyte extracts and the possible pathways involved in the reversal of the differentiated state mediated by oocyte molecules. The proteins identified in this study are involved in pathways that relate to the expression of pluripotency markers and other factors known to repress pluripotency (e.g., PRKC) or enhance differentiation (e.g., RA). Changes seen in the levels of nuclear proteins, such as lamins and histones, confirm the occurrence of extensive nuclear and chromatin remodeling known to take place during reprogramming. Alterations in major cellular structural proteins, such as ACTB, ANXA2, and VIM, were less expected, but these proteins actually have diverse roles in the nucleus related to reprogramming events. Network analysis also revealed many other proteins that are potentially involved in reprogramming events and indicated how these particular pathways interconnect. A number of these proteins warrant further investigation to determine whether they and the pathways in which they are involved are important in the formation or maintenance of pluripotency.
Footnotes
Acknowledgments
We thank Patricia Fisher, Ramiro Alberio, and Paddy Tighe at the School of Molecular Medical Sciences for access to Delta2D software, and Charlie Hodgman at the School of Biosciences for access to Ingenuity Pathway Analysis software. This work was supported by European Science Foundation (EuroSTELLS).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.