
Abstract
Abstract
Methodologies to reprogram somatic cells into patient-specific pluripotent cells, which could potentially be used in personalized drug discovery and cell replacement therapies, are currently under development. Oct4 activation is essential for successful reprogramming and pluripotency of embryonic stem (ES) cells, albeit molecular details of Oct4 activation are not completely understood. Here we report that endogenous SAF-A is involved in regulation of Oct4 expression, binds the Oct4 proximal promoter in ES cells, and dissociates from the promoter upon early differentiation induced by LIF withdrawal. Depletion of SAF-A decreases Oct4 expression even in the presence of LIF, and results in an increase of the mesodermal marker Brachyury. The overexpression of wild-type human SAF-A rescues the mouse knock-down phenotype and results in increased Oct4 level. We also demonstrate that endogenous SAF-A interacts with the C-terminal domain (CTD) of endogenous RNA polymerase II and that the interaction is independent of CTD phosphorylation and mRNA. Moreover, we show that SAF-A exist in complexes with transcription factors Sox2 and Oct4 as well as STAT3 in ES cells. The number of endogenous SAF-A:Oct4 and SAF-A:Sox2 complexes decreases upon LIF depletion. These discoveries allow us to propose a model for activation of Oct4 transcription.
Introduction
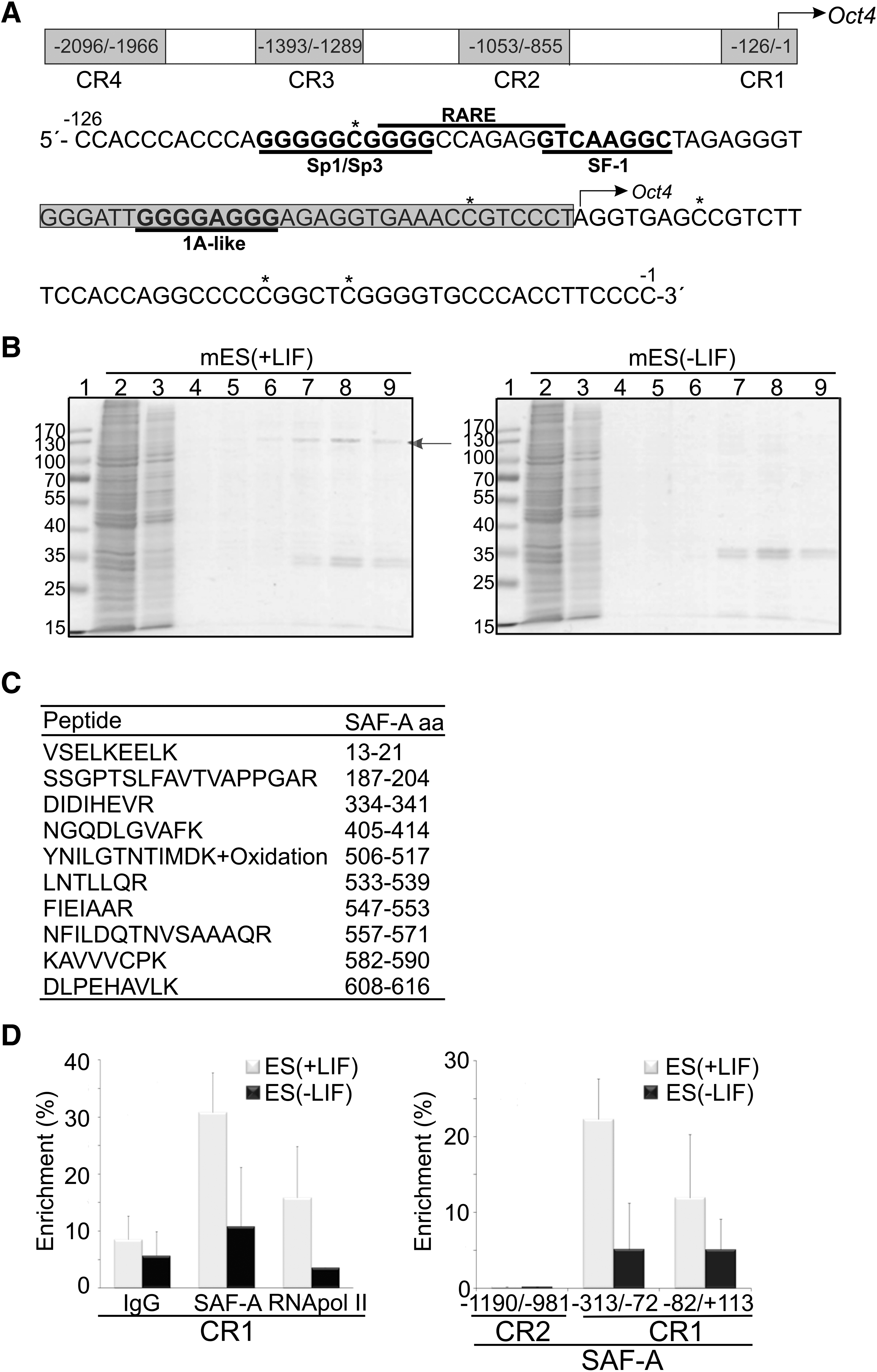
Endogenous SAF-A specifically binds the Oct4 proximal promoter in mES cells. (
Epigenetic modifications control gene expression and cellular differentiation during normal mammalian development. Until recently, the only known epigenetic modification of mammalian DNA itself is methylation of cytosine at position C5 in CpG dinucleotides; however, methylation of non-CG sites has now been reported (Bird, 2002; Lister et al., 2009). Methylation of CpG dinucleotides within the Oct4 promoter has been found to correlate with Oct4 downregulation during early embryonic development (Hattori et al., 2004), whereas Oct4 reactivation during SCNT has been shown to require demethylation of the Oct4 promoter (Simonsson and Gurdon, 2004). Albeit molecular details of Oct4 activation in ES cells are not completely understood, and a factor that specifically recognizes the Oct4 promoter dependent on LIF signaling in ES cells has not been reported. Here we assume that a protein that has the ability to activate Oct4 also physically interacts with the Oct4 promoter. Given that an intact 1A-like (−75 to −70) site is required for Oct4 transcription as well as demethylation using SCNT procedure by DNA injection of mouse sequences into Xenopus oocytes (Simonsson and Gurdon, 2004), we here used the 1A-like sequence in a screen for Oct4 promoter binding proteins dependent on LIF signaling. We identified SAF-A as such and verified its involvement in Oct4 expression.
A better understanding of Oct4 regulation at the molecular level may allow establishment of pluripotent stem cell lines in a more controlled and efficient manner, as well as cell therapies that are independent of human eggs or viruses.
Materials and Methods
Cell cultures
Cell lines were grown at +37°C in humidified atmosphere containing 5% CO2. Murine ES cell lines R1 and RW4 were maintained on mitotically inactivated mouse embryonic fibroblast (MEF) feeder layers in Dulbecco's modified Eagle medium (DMEM) supplemented with 1.0 mM sodium pyruvate, 0.1 mM nonessential amino acids, 2.0 mM L-glutamine, 0.1 mM β-mercaptoethanol, 15% fetab bovind serum (FBS), 20 mM HEPES (pH 7.3), 100 U/100 μg penicillin/streptomycin, and 1000 U/mL LIF (ESGRO, Chemicon, Tumecula, CA). Early differentiation was established by LIF withdrawal from the culture medium for 24–96 h.
Cell extracts
ES cells maintained with LIF [mES(+LIF)] and cells maintained without LIF [mES(−LIF)] for 48 h were washed with ice-cold PBS and harvested by centrifugation (3,500 × g, 10 min). Cell pellets were resuspended in ice-cold extraction buffer (10 mM Tris-HCl, 10 mM MgCl2, 10 mM KCl, and 1.0 mM DTT) containing protease inhibitors (Complete; Roche, Indianapolis, IN) and incubated on ice for 10 min. Following addition of NP-40 to 1% (v/v) the cells were incubated on ice for 10 min and homogenized. NaCl was added to a final concentration of 420 mM (used in DNAase footprint experiments) or 1 M (used in DNA affinity chromatography) followed by incubation on ice for 1 h. The extracts were cleared by centrifugation (19,000 × g, 1 h, +4°C). Clarified lysates were supplemented with 10% (v/v) glycerol, frozen in liquid nitrogen, and stored at −70°C. Protein concentrations were estimated by Bio-Rad Protein Asssay (Hercules, CA) using a bovine serum albumin (BSA) standard (0.25–2 μg/μL).
DNase footprint analysis
DNA probe was prepared by end-labeling 7.0 pmol of EcoRI/BspI 201 (Fermentas, Hanover, MD) cleaved pTZ57R/T vector (Fermentas) containing a double-stranded 300 base-pair (bp) fragment comprising the mouse Oct4 promoter with [α-32P]ATP by 15 units Klenow fragment, exo- (Fermentas). Labeled fragments were methylated in vitro using 4 units SssCpG-methylase (New England Biolabs, Beverly, MA) in 1 × NEBuffer 2 supplemented with 160 μM S-adenosylmethionine at +37°C for 1 h. To test the methylation reaction aliquots of methylated and unmethylated fragments were digested with HpaII (Fermentas). The extent of methylation was >90% effective. Samples for DNase-I footprint analysis were prepared by incubating 100,000 counts per min (cpm) of the probe in a reaction containing 0.1 × MBS, 0.1 μg poly dI/dC as nonspecific competitor and 1.0 μg of protein extract [mES(LIF−) or [mES(LIF+)] for 10 min on ice. Control reactions without protein or with in vitro methylated DNA were prepared simultaneously. A total of 0.001 units DNase I (Invitrogen, Carlsbad, CA) were added and reactions were allowed to proceed for 3 min at room temperature before they were stopped by incubation at +55°C overnight in a solution containing 0.5% SDS, 5.0 mM Tris-HCl pH 8.0, 10 mM EDTA and 0.5 μg/μL Proteinase K (Invitrogen). DNA was phenol-chloroform-isoamyl alcohol extracted twice, ethanol precipitated, resuspended in 1.0 μL TE-buffer and 1.0 μL formamide loading buffer, incubated for 3 min at +95°C and put on ice.
A 20 × 40 cm 6% polyacrylamide sequencing gel was cast. Samples and DNA cleaved at G+A-residues as previously described (Maxam and Gilbert, 1980) were loaded and separated by electrophoresis on Sequi-Gen GT Nucleic Acid Electrophoresis Cell (BioRad) at 40 W and a maximum temperature of +45°C for 2 h. After fixation, gels were vacuum dried at +65°C for 1 h and exposed to X-ray film and/or phosphor storage screens overnight.
DNA affinity chromatography
A total of 220 μg of synthetic 5′-phosphorylated oligonucleotide (MWG-Biotech AG) (Fig. 1A) was annealed and multimerized by ligation. Ligated DNA was covalently attached to cyanogen bromide (CNBr)-activated-Sepharose®4B (Sigma-Aldrich, St. Louis, MO) for DNA affinity chromatography (Kadonaga and Tjian, 1986). Fractions were collected and analyzed by SDS-PAGE. Protein bands were visualized by Bio Safe Coomassie Stain (BioRad) or Sypro Ruby (BioRad), excised, and subjected to nano-LC-FT-ICR mass spectrometry analysis.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using ChIP assay kit (Upstate, Lake Placid, NY) according to the manufacturer's protocol and dimethyl-3,3-dithiobispropionimidate-2HCl (DTBP) (5.0 mM) was added to stabilize protein–protein interactions. Undifferentiated and differentiated R1 cells (5 × 106 cells) were crosslinked with 1.0% formaldehyde for 15 min on ice. Crosslinked DNA/protein was extracted and sonicated [(12AM;5 sec) × 5] to yield DNA fragments with an average size of 500 bp. Chromatin extracts were immunoprecipitated using anti-SAF-A (Abcam, Cambridge, MA) or anti-RNA pol II CTD repeat YSPTSPS (Abcam). ChIP experiments were analyzed by real-time PCR (RT-PCR) using SYBR Green Master Mix with primers mOct4 (−82/+113) Fwd (GGATTGGGGAGGGAGAGGTGAAACCGT) and mOct4 (−82/+113) Rev (TGGAAGCTTAGCCAGGTT CGAGGATCCAC); mOct4 (−313/−72) Fwd (AGCAACTG GTTTGTGAGGTGTCCGGTGAC) and mOct4 (−313/−72) Rev (CTCCCCAATCCCACCCTCTAGCCTTGAC); mOct4 (−1190/−981) Fwd (ACAGGAATGGGGGAGGGGTG) and mOct4 (−1190/−981) Rev (AGTACTCAACCCTTGAAT GGGCCAGGA). The primer pairs yielded a single product as confirmed by dissociation curve analysis, and gave no product in the no-template control. The primers mNanog (−199/−1) Fwd (GGGTCACCTTACAGCTTCTTTTGCA TTA) and mNanog (−199/−1) Rev (GGCTCAAGGCGATAG ATTTAAAGGGTAG) did not render a correct dissociation curve in real-time-PCR; therefore, we present PCR results using Hot Star Taq®Plus DNA polymerase loaded on 2%-EtBr agarose gel.
Sequential chromatin immunoprecipitation (Re-ChIP) assay
Re-ChIP chromatin extracts were prepared using the same protocol as for ChIP assay. Chromatin extracts (15 × 106 cells) were immunoprecipitated using either anti-SAF-A (Abcam) antibody or rabbit normal IgG (Santa Cruz, Santa Cruz, CA). Addition of DTT (10 mM) disrupted the first antibody–antigen interaction and thereafter diluted 20-fold prior to the addition of the second antibody (anti-RNA-polymerase II; Abcam). Re-ChIP experiments were analyzed by PCR. The primers mOct4 (−313/−72) and Oct4 intron [Fwd (GGAG TCCCCTAGGAAGGCATTAATAGTTT) and Rev (GGAT TCTCTCGGCAACAGACAGACTT)] were used. These yielded a single product and gave no product in the no-template control.
Plasmid construction and cell transfection
SAF-A cDNA was amplified by PCR and cloned into a pEPI-eGFP vector (Manzini et al., 2006). Inserts were verified by sequencing.
Approximately 5 × 104 ES cells, cultured in medium supplemented with LIF, were transfected with Lipofectamine™ LTX Reagent (Invitrogen, Carlsbad, CA) 4 h postseeding. The medium was changed 4 h posttransfection and Oct4 and e-GFP-SAF-A proteins were immunostained and analyzed by confocal microscopy 48 h posttransfection.
RNA interference
siRNAs were obtained from Qiagen (Chatsworth, CA). Preparation of siRNA was done according to the manufacturer's protocol. For each siRNA concentration an equivalent amount of negative siRNA control AllStars Neg (Qiagen) was used. 5 × 104 ES cells, cultured in medium supplemented with LIF, were transfected 4 h postseeding with 5 nM, 10 nM, 50 nM, and 100 nM siRNA duplex complexed with different concentrations HiPerfect transfection reagent (Qiagen) according to the manufacturer's protocol. Oct4 and SAF-A proteins were immunostained and analyzed by confocal microscopy 48 h posttransfection. In each experiment the same laser and zoom settings were used.
Four SureSilencing™ shRNA plasmids with SAF-A specific insert sequences 1: ACAGTGTCTTGGCAAGTTTAT; 2: ACAGTGGTTTGTCTTGATACT; 3: AGGCCGTGGAGGAT TCAATAT; 4: GAACTCTCTTATGCGAAGAAT were obtained from SABiosciences. Preparation of shRNA was done as recommended by the manufacturer. ES cells (5 × 104), cultured in medium supplemented with LIF, were transfected 4 h postseeding with 0.4, 0.8, and 1.0 μg shRNA complexed with Lipofectamine™ LTX Reagent (Invitrogen) according to the manufacturer's instructions. For each concentration shRNA an equivalent concentration of negative shRNA control was used. Transfected cells were selected by addition of puromycin to a final concentration of 1 μg/mL (Calbiochem, LaJolla, CA). Forty-eight and 96 h posttransfection cDNA levels of Nestin, Gata4, Brachyury, SAF-A, Oct4, and GAPDH were detected by RT-PCR. Forty-eight and 72 h post transfection global transcription status of the cells was analyzed using immunofluorescence confocal microscopy.
RNAinterference rescue experiments
To perform the rescue experiments, approximately 4 × 104 ES cells were seeded per well in 24-well plates and transfected with 0.8 μg shRNA in combination with pEPI–eGFP–hSAF-A. Puromycin selection was introduced 24 h posttransfection. Oct4 and SAF-A or eGFP, proteins were immunostained and analyzed by confocal microscopy 48 h posttransfection. In each experiment the same laser and zoom settings were used.
Global transcription assay
Global transcription was examined using Click-iT® RNA Imaging Kit (Invitrogen). 5-Ethynyl uridine (EU) at final concentration of 1 mM was added and incubated under normal cell culture conditions for 1 h followed by fixation in 4% paraformaldehyde/PBS for 20 min and permeabilized with 0.25% Triton X-100/PBS for 10 min. EU incorporation was detected according to the manufacturer's protocol with the optional antibody (anti-SAF-A) detection step included. Nuclei were counterstained with 4,6-diamidino-2-phenylidole (DAPI). The fixated cells were analyzed on an inverted Zeiss LSM 510 META confocal microscope equipped with a Zeiss image processing system, using an 63 × /1.4 oil objective and sequential scanning with narrow band-pass filters (420–480 nm for DAPI, 505–530 nm for Alexa 488, and 560–615 nm for Alexa 555). To quantify the proportion of transcribing cells, EU-positive cells were counted manually in SAF-A-depleted cells and compared to control cells that were transfected with SureSilencing™ negative shRNA plasmid.
Immunostaining
For imunofluorescence, cells were trypsinized, counted and 4.0–8.0 × 104 cells were replated onto glass cover slips in 24-well dishes and cultured overnight. Cells were fixed with 4.0% paraformaldehyde/PBS for 20 min, washed, permeabilized with 0.25% Triton X-100/PBS for 5 min, blocked in 0.1% Triton X-100/10% FCS/PBS for 20 min. Primary anti-Oct4 (1:250, BD Biosciences, San Jose, CA), anti-SAF-A (1:500, Abcam), anti-GFP (1:500, Invitrogen), anti-RNA pol II CTD repeat YSPTSPS [4H8] (1:500, Abcam), anti-RNA pol II CTD repeat YSPTSPS [8WG16] (1:500, Abcam), antihistone H3 (tri methyl K4) (1:250, Abcam), anti-STAT3 (1:500, R&D Systems, Minneapolis, MN), anti-CBP (1:200, Santa Cruz), anti-PCAF (1:200, Santa Cruz), anti-Sox2 (1:100, R&D Systems), anti-GCN5 (1:200, Santa Cruz), and secondary antibodies [Alexa Fluor 488 conjugated goat antirabbit IgG and Alexa Fluor 555 conjugated goat antimouse IgG (1:500; Invitrogen)] were diluted in 0.1% Triton X-100/1% FCS/PBS and added for 2 and 1 h, respectively, each followed by washes in 0.1% Triton X-100/PBS. Nuclei were counterstained with DAPI. Coverslips were air dried, mounted, and analyzed on an inverted Zeiss LSM 510 META confocal microscope equipped with a Zeiss image processing system, using an 63 × /1.4 oil objective and sequential scanning with narrow band-pass filters (420–480 nm for DAPI, 505-530 nm for Alexa 488 and 560-615 nm for Alexa 555).
In situ proximity ligation assay (PLA)
Cells (3–4 × 104) were grown on chamber slides overnight. Duolink (Olink Bioscience, Uppsala, Sweden) in situ PLA was performed according to the manufacturer's protocol. Fixation, permeabilization, blockage, and primary antibody incubation were performed as described for immunofluorescence analyzes. PLA probes were diluted in 0.1% Triton X-100/1% FCS/PBS and incubated in a preheated humidity chamber for 1 h at +37°C, followed by hybridization, ligation, amplification, and detection according to manufacturer's protocol. Slides were analyzed by confocal microscopy. In each experiment the same laser settings were used.
Coimmunoprecipitation (Co-IP)
Lysates were prepared from ES cells grown to 80–90% confluence. Cells were washed three times with PBS and 5 mM DTBP was added to stabilize protein–protein interactions before lysis in 50 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM phenylmethyl sulphonyl fluoride, 10 μg/μL aprotinin, 1 μg/mL leupeptin, and pepstatin A. Lysed cells were centrifuged for 10 min at 13,000 × g. In indicated experiments RNase A (Roche) was added to a final concentration of 0.1 μg/μL and incubated for 30 min. The supernatant was precleared and incubated with antibodies and Protein A/G PLUS-Agarose beads (Santa Cruz) overnight at +4°C. Anti-SAF-A and anti-RNA pol II conjugated beads were centrifuged at 1000 × g and the supernatant was discarded. The beads were washed three times with 1 mL of cold lysis buffer, resuspended in 20 μL 2 × Laemli buffer, heated to +95°C for 3 min, and centrifuged for 1 min at 1000 × g. Supernatants were collected and used for Western blot analysis.
Western blotting
Proteins were separated using SDS-PAGE, electrotransferred onto polyvinylidene difluoride (PVDF) membranes for 1 h, 110 mA/gel in transfer buffer (48 mM Tris, 39 mM Glycin, 1.3 mM SDS, 10% MeOH), and immunologically detected. PVDF membranes were blocked with 5% nonfat dry milk/0.1% Tween 20/PBS (PBST) and incubated overnight with primary antibodies (anti-SAF-A (1:1000; Abcam), anti-RNA pol II CTD repeat YSPTSPS [4H8] (1:1000; Abcam), anti-RNA pol II CTD repeat YSPTSPS [8WG16] (1:500; Abcam) diluted in blocking solution. After washing with PBST, blots were incubated for 1 h with secondary antibodies (AP conjugated goat antimouse IgM + IgG + IgA (H + L) and AP conjugated goat antirabbit IgM + IgG (H + L chain specific) (1:1000; SouthernBiotech, Birmkingham, AL). Visualization of proteins was done with NBT/BCIP (Promega, Madison, WI).
RNA preparation/reverse-transcription/real-time PCR analysis
Total RNA was extracted using RNeasy minikit (Qiagen). Contaminating genomic DNA was eliminated with RNase-Free DNase (Qiagen). cDNA synthesis was performed with SuperScript III kit (Invitrogen). Endogenous mRNA levels were measured by RT-Q-PCR analysis based on SYBR Green detection. Briefly, the RT-Q-PCR mixture contained 1 μL of the reverse-transcription reaction product in a total volume of 20 μL, 1 × SYBR Green mix reagent (Applied Biosystems, Bedford, MA), 50 nM forward primer and 50 nM reverse primer. Each sample was analyzed in duplicate using the following oligonucleotide pairs: SAF-A Fwd (GCCGAGGGT ATTTTGAGTACAT) and SAF-A Rev (TGTGTCATCGAAGT GTTCGTCTT); Oct4 Fwd (CACGAGTGGAAAGCAACTCA) and Oct4 Rev (AGATGGTGGTCTGGCTGAAC); Nestin Fwd (AAAGGAAAGGCAGGAGTCCCTGAA) and Nestin Rev (TGG TCCTCTGCGTCTTCAAACCTT); Brachyury Fwd (AGCTC TCCAACCTATGCGGACAAT) and Brachyury Rev (TGGTA CCATTGCTCACAGACCAGA); Gata4 Fwd (ACTCCAAAG TGCTGGGTTCAATGC) and Gata4 Rev (TTGCAGAGGGT AGATGTTCAGGCT); GAPDH Fwd (AATGTGTCCGTCG TGGATCTGA) and GAPDH Rev (GATGCCTGCTTCACC ACCTTCT). All primer pairs yielded a single product, as confirmed by dissociation curve analysis and gave no product in the no-template control.
Results
Endogenous SAF-A specifically binds the Oct4 proximal promoter in ES cells
In vitro DNase I footprinting was employed to screen for sequence specific binding differences at the evolutionary conserved Oct4 proximal promoter region (CR1 in the schematic illustration and sequence in Fig. 1A). Comparisons were made between murine ES (mES) and differentiation induced cells. LIF was used to sustain self-renewal of mES cells (Smith et al., 1988) and differentiation was induced by LIF withdrawal for 2 days. A difference in protection due to differentiation status of the cells was identified in the 1A-like/CTCF region bordering the initiation site of Oct4 transcription [TG nucleotides, red arrows in Supplementary Fig. 1A; see online Supplementary Data at www.liebertonline/cell.com; compare mES(+LIF) and mES(−LIF)]. The 1A-like site has previously been shown to be important for demethylation of the Oct4 promoter and reactivation of the gene during SCNT in Xenopus oocytes (Simonsson and Gurdon, 2004).
In order to mimic binding conditions of differentiated cells, the Oct4 proximal promoter region was in vitro CpG methylated. Comparisons of in vitro CpG methylated (Supplementary Fig. 1A; lane m) and nonmethylated DNA (Supplementary Fig. 1A; lane nm) reveal that the observed protection is independent of the methylation status of Oct4 proximal promoter.
These discoveries encouraged us to enrich and identify factor/factors that bind the 1A-like/CTCF region including downstream sequences until the transcription initiation start site, by DNA affinity chromatography (see box in Fig. 1A for details). A factor with an apparent molecular mass of 120 kDa was enriched merely from pluripotent mES cell extracts (Fig. 1B, arrow); mES [+LIF)], and nano-LC-FT-ICR mass spectrometry analysis uniquely identified this factor as Scaffold Attachment Factor A (SAF-A also known as hnRNP U or HNRPU) (Fig. 1C). Whereas, when using extract prepared from differentiation induced mES cells maintained without LIF (for as short time as 2 days) in the chromatography analysis, no 120 kDa band could be detected on the SDS gel (Fig. 1B, mES(−LIF), right panel) and also no protein was identified with mass spectrometry when the 120-kDa area was excised from the SDS gel.
Given that SAF-A binds to the Oct4 promoter in vitro, we sought to determine whether it is present at the Oct4 promoter in ES cells. To this end we performed chromatin immunoprecipitation (ChIP) experiments. Sonicated chromatin lysates were immunoprecipitated with anti-SAF-A to pull down SAF-A containing nucleosomes. Anti-RNA polymerase II (RNA pol II) was used as positive control and normal IgG as negative control. The Oct4 control region was precipitated from undifferentiated mES cells (Fig. 1D, left graph, gray bars) whereas induction of differentiation by LIF withdrawal for 48 h resulted in substantially lower precipitation yields of both SAF-A and RNA pol II (Fig. 1D, left graph, black bars). To further investigate the binding of SAF-A to different parts of the Oct4 promoter we performed real-time PCR using primers comprising two additional regions: one located in conserved region (CR) 1 (−82/+113) and the other in CR2 (−1190/−981). SAF-A was found more abundantly within CR1 that contains the sequence used in DNA affinity chromatography (Fig. 1D, right graph). In contrast, SAF-A was not bound to the enhancer region of Oct4 even though this region contains 1A-like sequence (Fig. 1D, right graph, CR2). SAF-A thus interacts with the Oct4 proximal promoter when Oct4 is actively transcribed in undifferentiated mES cells.
Because SAF-A has been also found on Klf2 promoter (Ahmad and Lingrel, 2005), we next asked if SAF-A could be detected binding to the promoter region of another pluripotency gene. We found SAF-A bound to the proximal promoter of Nanog by ChIP experiments, and interestingly, when LIF was withdrawn, SAF-A was detached from the Nanog promoter in the same manner as it was dissociated from the Oct4 proximal promoter (Supplementary Fig. 1B).
SAF-A is involved in positive regulation of Oct4 transcription
To explore functional effects of SAF-A on Oct4 expression, mES cells maintained in the medium supplemented with LIF were transfected with SAF-A-specific small interfering RNA (siRNA). Immunofluorescence analysis using confocal microscopy reveals that an siRNA-mediated decrease in SAF-A levels results in decreased Oct4 protein levels (Supplementary Fig. 2A) compared to control mES cells. To explore long-term SAF-A suppression effects we used short hairpin (sh) RNA constructs containing antibiotic selection. In preliminary experiments four SAF-A-shRNA targeting constructs were tested for their ability to knock down SAF-A mRNA. Similar effects were observed for all four independent sequences (Supplementary Fig. 2B) and no off-target effects were observed 48 h posttransfection. At 96 h posttransfection a decrease of housekeeping genes was observed. To address whether SAF-A is involved in general transcription, we measured global transcription in live control and SAF-A knock-down ES cells by incorporation of EU (Fig. 2A, green) which was monitored by immunofluorescence and confocal microscopy. At 72 h posttransfection, RNA transcription could be detected even in cells that lack SAF-A (Fig. 2A, red) and no visible effect could be seen. However, the quantification of the SAF-A knock-down effect on global transcription, accessed by manually counting EU-positive cells, revealed that at 72 h posttransfection the number of transcriptionally active cells (EU positive) when SAF-A is depleted decreases by 54%, which was statistically significant, in comparison to the negative shRNA control. SAF-A depletion for 48 h resulted in the unaffected number (1% decrease) of transcriptionally active cells in comparison to the control cells. These results imply that SAF-A downregulation affects global transcription after 72 h. Therefore, an earlier time point was chosen to study the effect of SAF-A knock down on transcription levels of Oct4. At 48 h posttransfection, SAF-A downregulation decreases Oct4 transcript levels (Fig. 2B and Supplementary Fig. 2B). Although we observed the aforementioned decrease in the transcript levels of housekeeping genes, SAF-A knock down for 96 h correlated with upregulation specifically of the mesoderm differentiation marker, Brachyury, suggesting a loss of pluripotency and directed induced differentiation along the mesodermal pathway. The normal colony morphology of ES cells is also lost in SAF-A–shRNA cells. These results strengthen the concept that SAF-A is involved in regulation of Oct4.
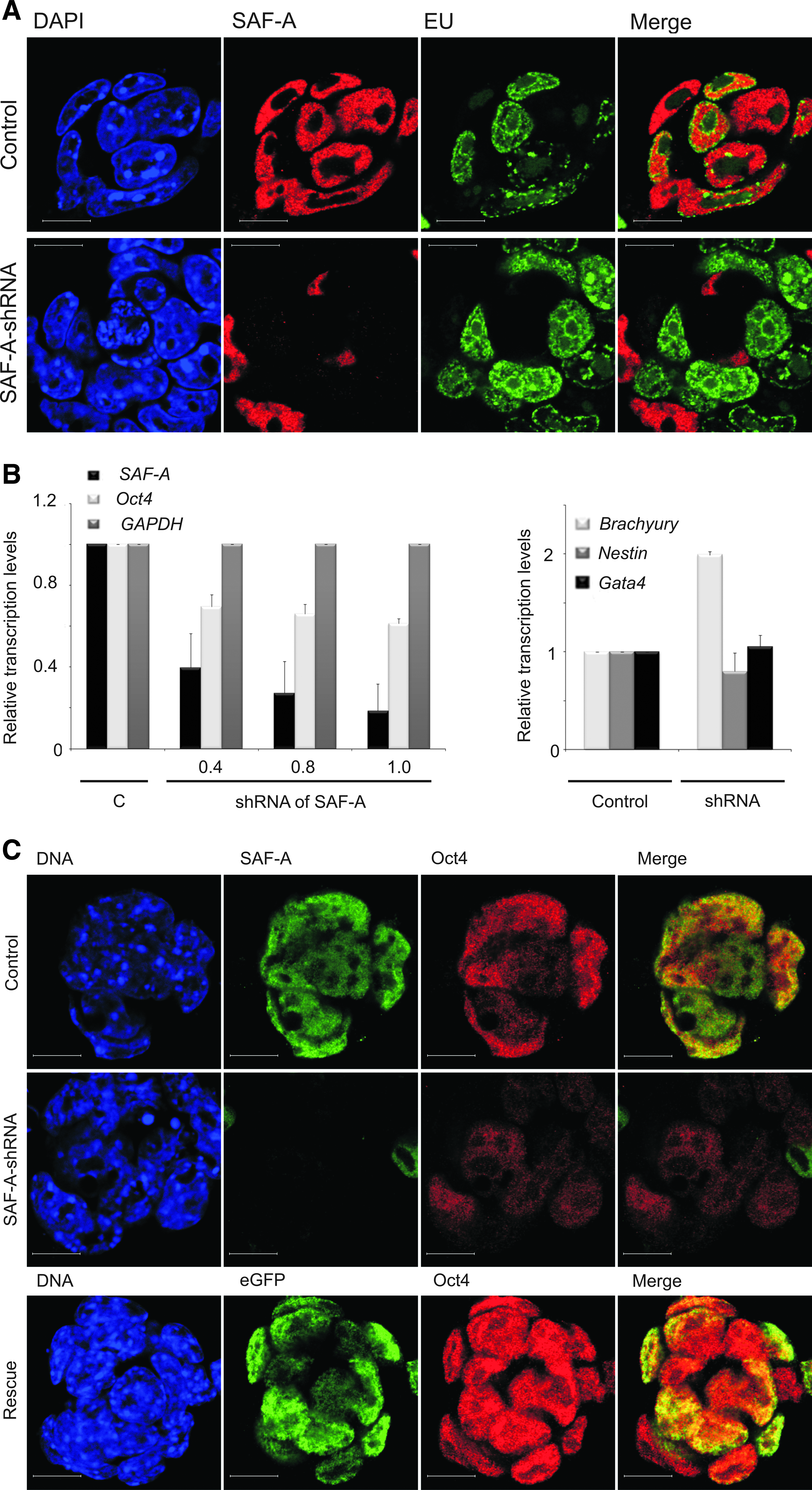
SAF-A is involved in Oct4 transcription regulation. (
Our RNAi experiments have established a link between expression of SAF-A and maintenance of mES cells via Oct4 expression. As mentioned above, we have observed consistent gene silencing results using multiple shRNA constructs as well as siRNA in mES cells. The next issue addressed was if ectopic expression of human SAF-A (hSAF-A) can rescue the knock-down phenotype of mES cells. In these experiments we used two different mouse-specific shRNA constructs that do not affect expression of hSAF-A. Immunofluorescence analysis using confocal microscopy was employed to analyze changes in Oct4 protein levels in the presence or absence of ectopic eGFP-hSAF-A. As expected, shRNA mediated knock down of endogenous SAF-A (green) decreases Oct4 (red) levels (Fig. 2C, middle panel). Remarkably, this Oct4 downregulation was observed even though the culture media was supplemented with LIF. Overexpression of full-length eGFP-hSAF-A in the knock-down cells results in increased Oct4 levels (Fig. 2C, lower panel) indicating rescue of the knock-down phenotype. These experiments support our hypothesis that SAF-A plays a role in positive regulation of Oct4.
Endogenous SAF-A physically interacts with RNA pol II independently of CTD phosphorylation and mRNA
Recent reports that SAF-A may interact with the CTD of RNA pol II (Kim and Nikodem, 1999; Kukalev et al., 2005) intrigued us to examine a possible interaction between SAF-A and RNA pol II in mES cells. Visualization of endogenous protein–protein interactions can be achieved by state-of-the-art in situ proximity ligation assay (PLA) (Soderberg et al., 2006). The in situ PLA is based on recognition of proteins by pairs of antibodies raised in different species. A red signal is generated only when the distance between two epitopes is shorter than 40 nm by using specie specific secondary antibodies covalently coupled to one oligonucleotide each that can become ligated with bridging DNA sequences and used as template in rolling circle amplification. The amplified DNA is visualized by annealing of complementary labeled DNA strands. Technical and biological controls in our system show that the in situ PLA is highly sensitive and very specific for interactions between endogenous proteins (Johansson et al., 2010) (Supplementary Figs. 3 and 7). By performing in situ PLA followed by confocal microscopy, a considerable number of SAF-A:RNA pol II complexes can be seen in mES cell nuclei (Fig. 3A and Supplementary Fig. 4). To explore this discovery in more detail, extracts prepared from mES cells were subjected to coimmunoprecipitation (co-IP) with either anti-SAF-A or anti-RNA pol II followed by Western blotting. Consistent with the in situ PLA, and colocalization studies (Supplementary Fig. 5), the RNA pol II was immunoprecipitated with anti-SAF-A and vice versa (Fig. 3B, -RNAse). Importantly, the addition of RNase A to immunoprecipitation did not affect these results, which implies that SAF-A interacts with RNA pol II independently of mRNA (Fig. 3B, +RNAase).
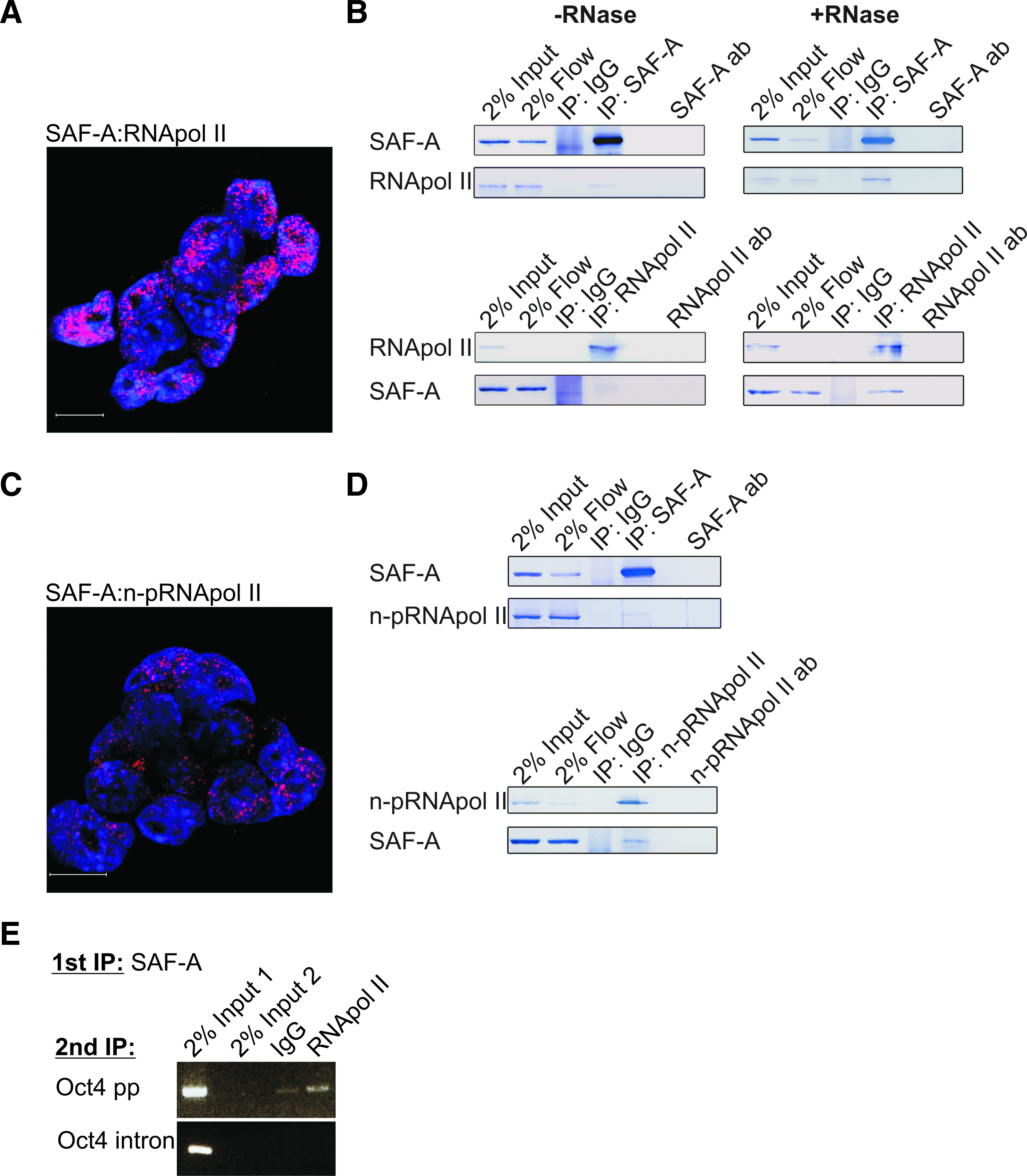
Endogenous SAF-A physically interacts with RNA pol II independently of CTD phosphorylation and mRNA. (
We next investigated if SAF-A interacts specifically with the nonphosphorylated form of the RNA pol II CTD, which is specific for initiation of transcription (Lu et al., 1991). In situ PLA using a combination of anti-SAF-A and an antibody that is specific for the nonphosphorylated form of the RNA pol II CTD (Fig. 3C and Supplementary Fig. 6) as well as co-IP (Fig.e 3D) show that SAF-A also interacts with the nonphosphorylated RNA pol II CTD.
To address whether SAF-A:RNA pol II complex is attached to the Oct4 proximal promoter region we performed sequential ChIP (Re-ChIP). Chromatin immunoprecipitations with SAF-A followed by RNA pol II specific antibodies were performed to pull down SAF-A:RNA pol II complexes binding chromatin in mES cells. The SAF-A:RNA pol II binding to the Oct4 proximal promoter and intron regions was examined by PCR. The PCR analysis showed an enrichment of Oct4 proximal promoter in the SAF-A:RNA pol II precipitations relative to IgG precipitations. On the contrary, the Oct4 intron region was not precipitated (Fig. 3E).
In conclusion, the above data show that SAF-A:RNA pol II complex is attached to the Oct4 promoter and that endogeneous SAF-A physically interacts with RNA pol II independently of CTD phosphorylation and mRNA, and suggest that SAF-A might be involved in transcription initiation of Oct4.
Endogenous SAF-A interacts with endogenous Oct4 and Sox2
In addition to the proximal promoter, the distal enhancer which is located in conserved region 4 (CR4) is important for Oct4 gene expression in the ICM and ES cells. Previous reports have indicated that Oct4 is subject to self-regulation. It has been shown to form a heterodimer with Sox2, which binds to the distal enhancer to regulate Oct4 transcription (Chew et al., 2005; Okumura-Nakanishi et al., 2005). Chromatin configuration reduces the distance between the proximal and distal enhancer dramatically, and facilitates crosstalk between these regions. We therefore asked if endogenous SAF-A interacts also with endogenous Oct4 and/or Sox2. To test this hypothesis we employed in situ PLA, using anti-SAF-A in combination with monoclonal anti-Oct4 or anti-Sox2. Interaction between Oct4 and Sox2 was used as a positive biological control, whereas lack of interaction between Sox2 and Ncl was used as negative biological control (Supplementary Fig. 7). These experiments revealed that endogenous SAF-A interacts with or binds in close proximity to endogenous Oct4 (Fig. 4A, upper panel and Table 1) as well as endogenous Sox2 (Fig. 4B, upper panel and Table 1). Interestingly, withdrawal of LIF for two days resulted in loss of SAF-A:Oct4 (Fig. 4A, lower panel, and Table 1) and SAF-A:Sox2 (Fig. 4B, lower panel, and Table 1) complexes. Thus, complex formation between endogenous SAF-A:Oct4 and SAF-A:Sox2 is dependent on LIF signaling.
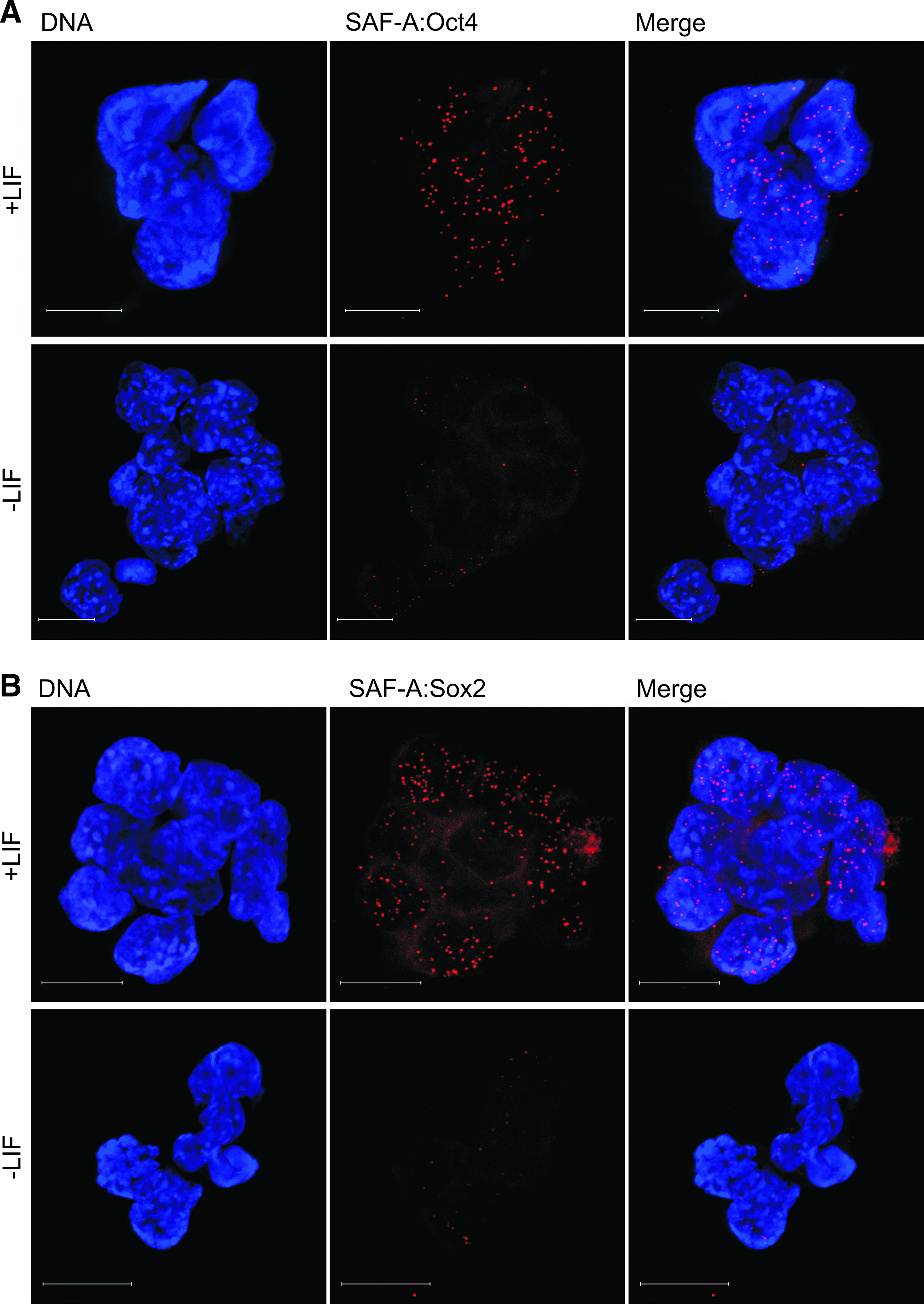
SAF-A is found in complex with Oct4 and Sox2 in an LIF signaling-dependent manner. In situ PLA detection by immunofluorescent confocal microscopy using antibodies against SAF-A in combination with antibodies against (
The number of red spots, representing individual endogenous protein–protein complexes detected by in situ proximity ligation assay (PLA) in each cell, was counted.
The number of complexes between SAF-A and transcription factors Oct4 and Sox2 decrease as ES cells are induced to differentiate by LIF withdrawal [for 2 days (2d)], as does the number of SAF-A:STAT3 complexes, which links LIF signaling and Oct4 expression. In contrast, the number of complexes between SAF-A and RNA pol II remains constant as ES cells are induced to differentiate by LIF withdrawal. This is also true for the number of complexes between SAF-A and HATs CBP and PCAF, which are used as controls. Data are mean ± SD (n ≥ 10).
Differentiation of mES cells by LIF withdrawal does not decrease SAF-A levels
In an attempt to understand how LIF/STAT3 signaling influences SAF-A's transcription regulation of Oct4, we first monitored SAF-A and Oct4 mRNA levels by RT-Q-PCR and SAF-A and Oct4 protein levels by immunofluorescent confocal microscopy, in undifferentiated and by LIF withdrawal differentiation-induced mES cells over a period of days (1–4 days). When cultured in medium without LIF the mES cells differentiate slowly, and differentiation is asynchronous. This results in a morphologically mixed cell population. RT-Q-PCR reveals that Oct4 mRNA levels gradually decrease throughout the period of differentiation (Fig. 5A). However, in contrast, and contrary to our expectation, we find that SAF-A mRNA levels initially increase, only to return to original levels at day 3 (Fig. 5A). Oct4 and SAF-A protein levels exhibit patterns similar to the corresponding mRNA levels as judged by immunofluorescent confocal microscopy (Fig. 5B). Oct4 protein levels gradually decrease throughout the period of differentiation, whereas SAF-A protein levels initially increase and then return to original levels at day 3. Taken together, these observations suggest that differentiation of mES cells is accompanied by a peak in SAF-A expression, resulting in higher SAF-A protein levels. Thus, Oct4 downregulation cannot be ascribed simply to a decrease in SAF-A protein levels.
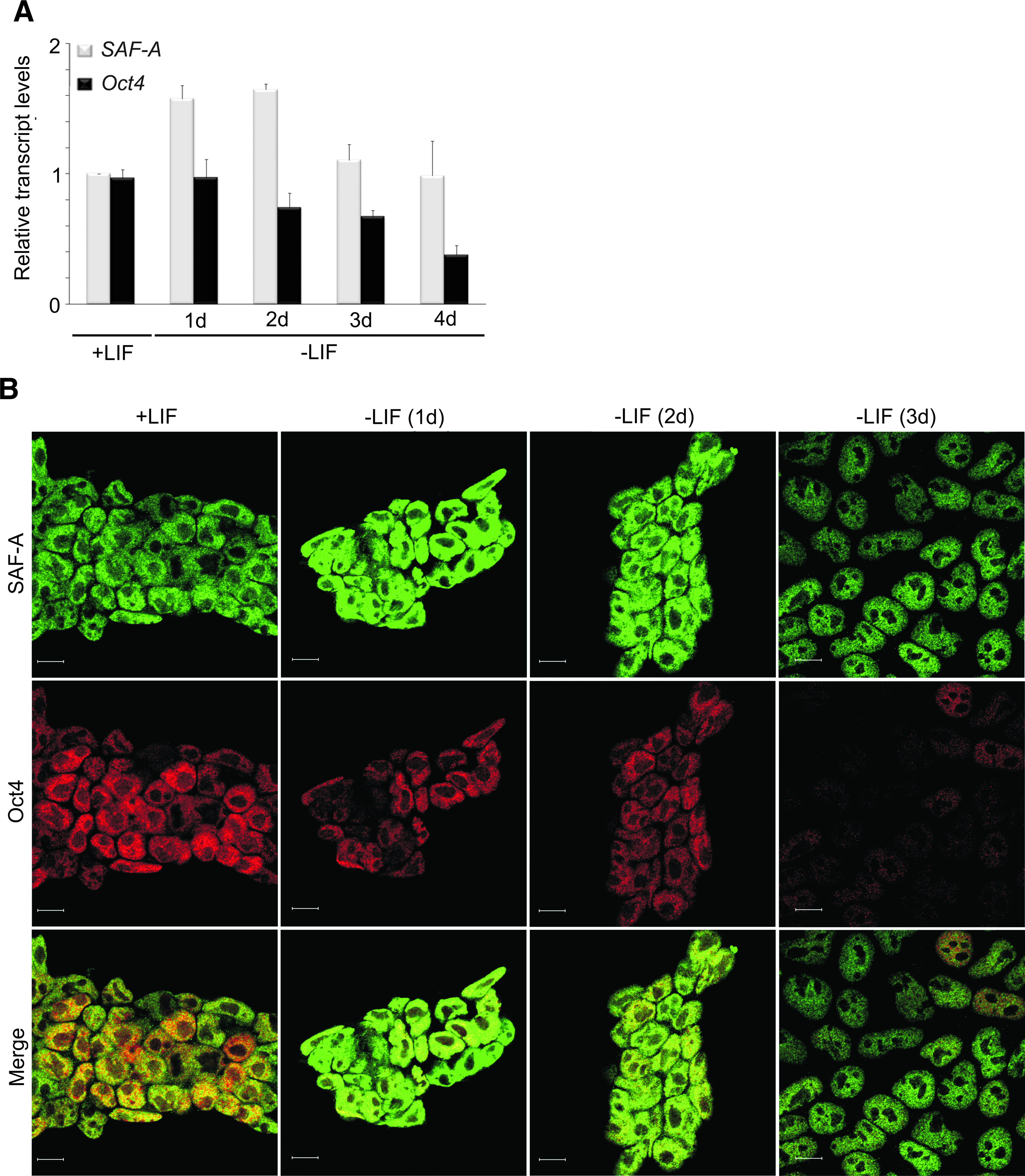
Differentiation of mES cells by LIF withdrawal does not decrease SAF-A levels. (
STAT3 is in complex with SAF-A in mES cells and fewer complexes are detected after LIF withdrawal
The finding that Oct4 downregulation is not due to decreased SAF-A levels, in combination with the previous finding that LIF can maintain Oct4 expression via STAT3 (Niwa et al., 1998), prompted us to ask how LIF signaling via STAT3 incorporates a role for SAF-A in Oct4 transcription regulation. In situ PLA using anti-SAF-A in combination with monoclonal anti-STAT3 was used to reveal interactions between endogenous SAF-A and endogeneous STAT3 in mES cells (Fig. 6, upper panel, and Table 1). Withdrawal of LIF for 2 days results in decreased number of complexes (Fig. 6, lower panel, and Table 1). The observation that SAF-A also interacts with STAT3 might provide a link in a chain of interactions ranging from extrinsic stimuli by LIF on the cell surface, to an intrinsic response in the nucleus, which possibly result in Oct4 expression.
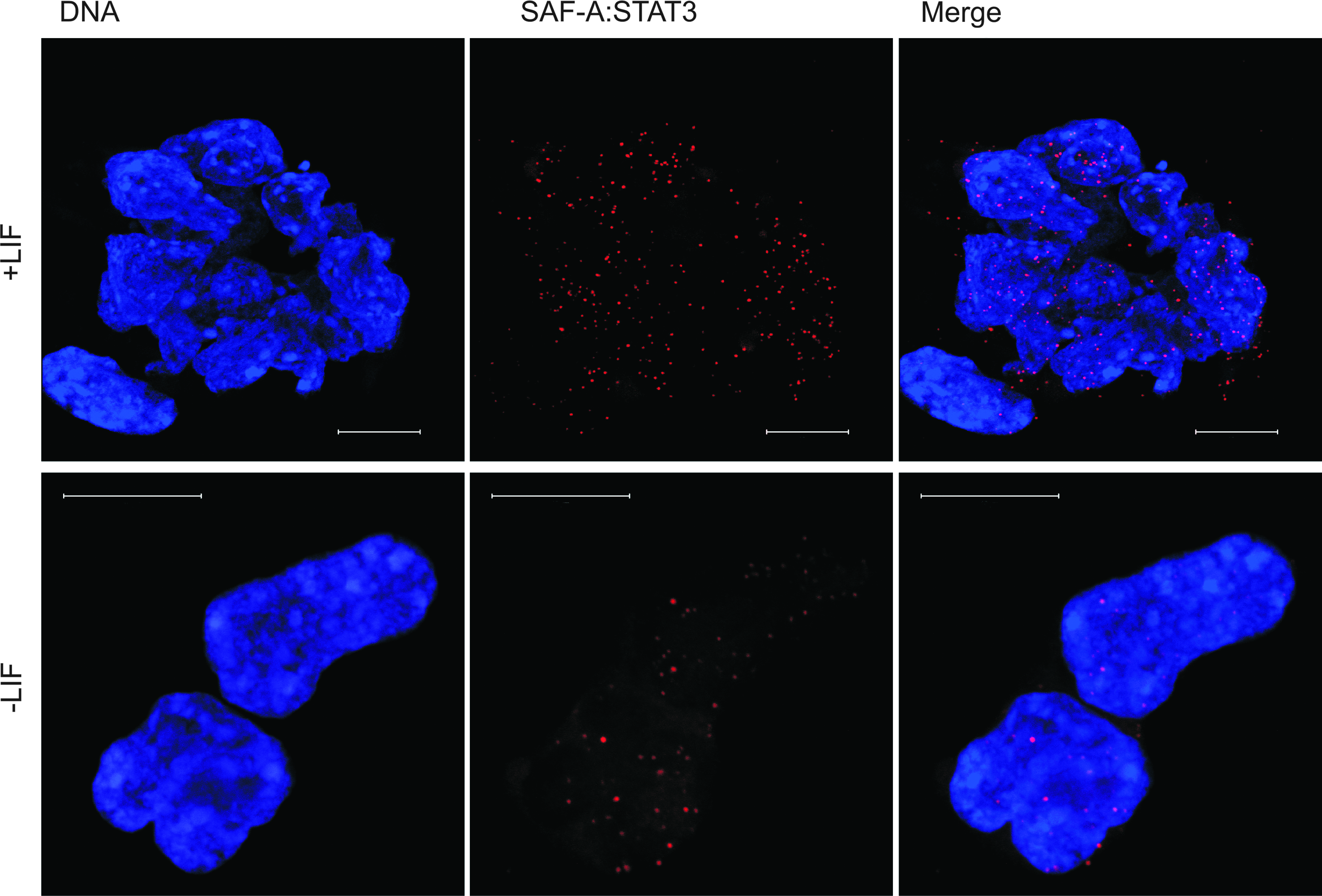
SAF-A is found in complex with STAT3 and complexes are seen upon LIF withdrawal. In situ PLA detection by immunofluorescent confocal microscopy by using antibody against SAF-A in combination with antibody against STAT3 in mES cells. Induction of differentiation by LIF withdrawal (−LIF) for 48 h results in decreased number of SAF-A:STAT3 complexes. Each red dot represents close interactions between endogenous SAF-A and endogenous STAT3. DNA was counterstained with Hoechst 33342 (blue). Scale bars represent 10 μm. (See color version of this figure at www.liebertonline.com).
Discussion
One critical unresolved issue in the field of ES cell biology is the molecular details regarding regulation of Oct4 expression. Here we provide evidence for SAF-A as a key regulator of Oct4 expression in ES cells upon LIF signaling. SAF-A is a nuclear protein that contains five distinct predicted structural domains (Fig. 7A), one of which is the SAP [SAF-A/B, Acinus, and protein inhibitor of activated STATs (PIAS)] domain, a putative DNA binding domain, found in diverse nuclear proteins, for example, in developmental pluripotency-associated gene 2 and 4 (Dppa2 and Dppa4) reported to be regulated by Oct4 and Sox2 (Boyer et al., 2005; Chakravarthy et al., 2008; Maldonado-Saldivia et al., 2007). Alternative splicing yields two isoforms of human SAF-A, which are identical with respect to in vitro nucleic acid binding capabilities (Fackelmayer and Richter, 1994). The single 800 aa SAF-A variant found in mouse has a calculated molecular mass of 87,918 Da, and is 97.25% identical to human SAF-A isoform B. Such exceptionally high cross-species sequence identity of SAF-A indeed suggests that it has essential functions, and the fact that no viable SAF-A gene knockout has been reported supports this suggestion. Moreover, a hypomorphic mutation in a noncoding region of the SAF-A gene, which yields a mere twofold reduction of SAF-A levels, results in postimplantation lethality at E6.5 (Roshon and Ruley, 2005). This further indicates that maintenance of precise SAF-A levels is critical for cellular differentiation and embryonic development.
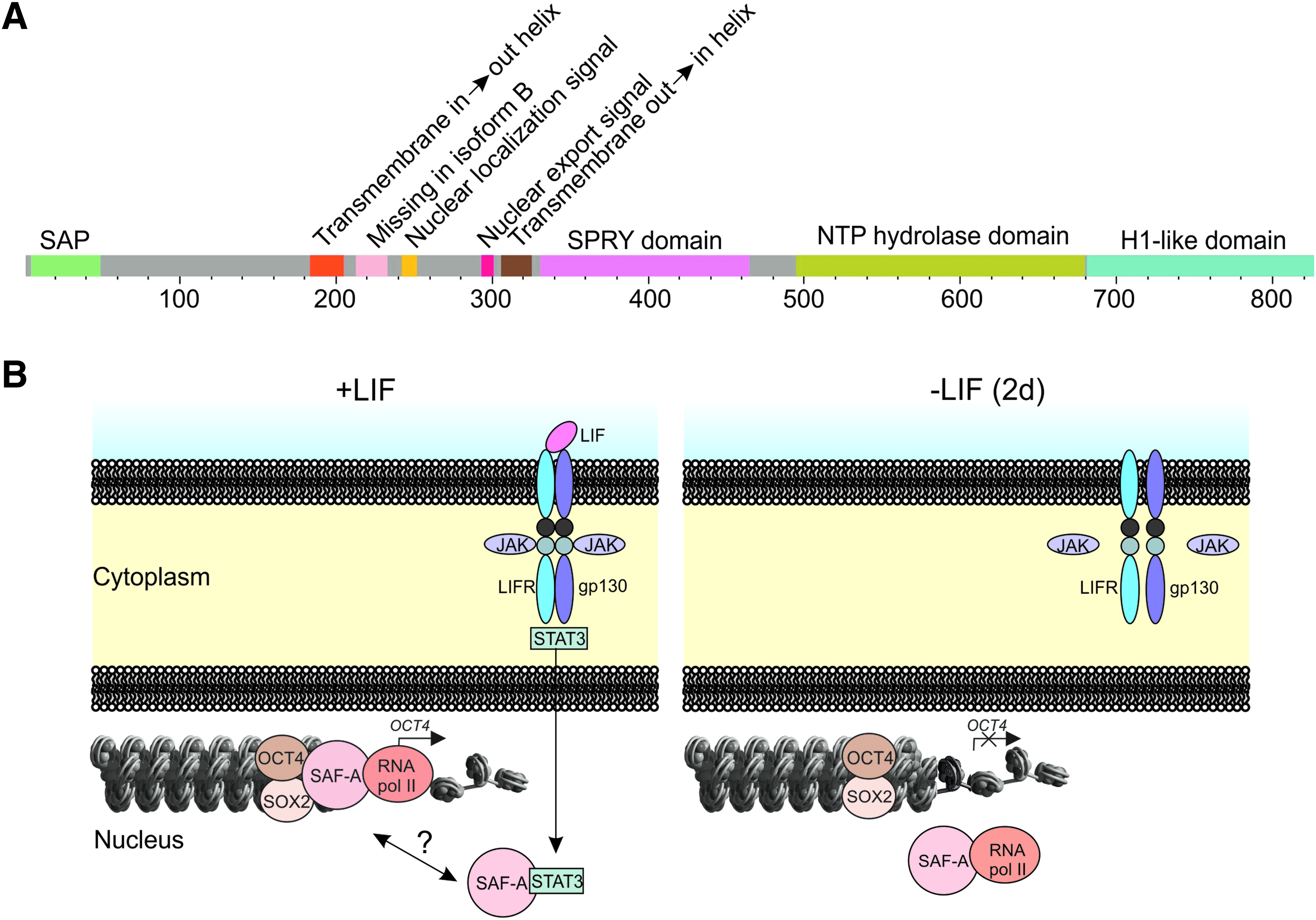
SAF-A features and tentative model for SAF-A mediated transcription initiation of Oct4 in ES cells. (
SAF-A has been demonstrated to bind RNA (Dreyfuss et al., 1993) and single- and double-stranded DNA (Gohring and Fackelmayer, 1997; Gohring et al., 1997) to establish transcriptionally active chromatin loops. Previous reports have indicated that SAF-A interacts with gene promoters and its interaction is controlled by external factors for example circadian rhythm (Ahmad and Lingrel, 2005; Gao et al., 2005; Onishi et al., 2008; Zhao et al., 2009). Here we find that SAF-A preferentially binds the Oct4 proximal promoter, that the interaction is broken rapidly upon lineage commitment induced by removal of LIF, and support these findings by ChIP analysis in mES cells. Previous reports have demonstrated that upon differentiation the Oct4 promoter becomes methylated; however, methylation is detected much later than our observed detachment of SAF-A. Considering that SAF-A level does not decrease after LIF-removal but rather increase we conclude that the promoter release is not controlled directly by the level of SAF-A. It is possible that in wt ES cells a modification of SAF-A, rather than its mere presence/absence, may be a critical parameter for SAF-A's change in promoter binding. This could cause a conformational change in the SAF-A protein. We also demonstrate by functional assays that SAF-A indeed regulates Oct4 transcription: RNAi mediated knock down of SAF-A in mES cells maintained under pluripotency conditions, that is, in the presence of LIF, considerably decreases Oct4 levels, remarkably to the same Oct4 level that is observed during early differentiation induced by LIF deprivation. In addition, huge cell loss and morphological changes of the ES cells were detected upon SAF-A knock down. RNAi rescue experiments of ES cells by human SAF-A show reexpression of Oct4, interestingly, in many cells to a higher level than wild type. These results provide evidence that SAF-A is required for Oct4 transcription in ES cells in the presence of LIF signaling.
The SAF-A NTP hydrolase domain has previously been suggested to interact with RNA pol II in HeLa cells (Kim and Nikodem, 1999; Kukalev et al., 2005). Using in situ PLA and co-IP we confirm that SAF-A interacts with RNA pol II in ES cells. Importantly, we also demonstrate that endogeneous SAF-A interacts with endogenous RNA pol II independently of CTD phosphorylation and independently of the presence of RNA in ES cells. In addition, we found that SAF-A:RNA pol II complex is attached to the Oct4 proximal promoter region when Oct4 is actively transcribed in pluripotent mES cells. Besides the previous suggestion that SAF-A interacts with RNA pol II during transcription elongation (Kukalev et al., 2005), our data thus indicate that SAF-A plays important roles already during Oct4 transcription initiation considering that nonphosphorylated CTD of RNA pol II (npCTD-RNA pol II) has previously been reported to be involved in transcription initiation (Lu et al., 1991). Our results suggest two distinct but not mutually exclusive roles for SAF-A in ES cells: the complex between SAF-A and RNA pol II with a phosphorylated CTD (pCTD-RNA pol II) reflects transcription elongation events, and the complex between SAF-A and npCTD-RNA pol II reflects transcription initiation events. Moreover, there are only two Oct4 alleles per cell. Provided Oct4 is subject to biallelic expression in ES cells, a complex that is completely unique for the Oct4 promoter would generate no more than two dots by in situ PLA per nucleus. The average numbers of detected SAF-A complexes involved in transcription initiation and elongation are substantially higher. SAF-A thus controls not only Oct4, but a larger set of genes in ES cells. In support of this, we have detected by ChIP that SAF-A associates with Nanog promoter and the association of SAF-A with Klf2 promoter has previously been reported (Ahmad and Lingrel, 2005). Moreover, SAF-A has a role in global transcription in ES cells as our RNAi-EU incorporation results indicate. In conclusion, our data suggest that SAF-A controls a network of genes, including the pluripotency genes Oct4, Nanog, and Klf2, at the level of transcription initiation and elongation.
Research on iPS cells has demonstrated that Oct4 expression can be initiated by the introduction of a limited number of genes. So far always including the Oct4 gene itself, and in most cases also the Sox2 gene. Thus, Oct4 is involved in transcription initiation of its own gene, perhaps as a heterodimer with Sox2 (Chew et al., 2005; Okumura-Nakanishi et al., 2005). The finding that SAF-A is in complex with both Oct4 and Sox2 might suggest that SAF-A serves to bring together factors required for Oct4 expression in ES cells and load them on the promoter together with RNA pol II. In accordance with this suggestion, the number of complexes that SAF-A forms with Oct4 decreases approximately by 70% upon induction of differentiation by LIF withdrawal (Table 1) for 2 days, although Oct4 levels are not significantly reduced during this time period. The number of complexes between SAF-A:Sox2 is decreased to the same extent as well, suggesting that any of these lost interactions can be considered as a candidate for controlling SAF-A dissociation from the Oct4 proximal promoter. It should be mentioned that it is not a new concept that RNA pol II interacting factors regulate Oct4 expression by the interaction with also Oct4 itself. Recently, it has been reported by two independent groups that RNA pol II association factor, Paf1, affects Oct4 expression (Ding et al., 2009; Ponnusamy et al., 2009). To this, we now add an additional RNA pol II binding factor that interacts sequence specifically with the Oct4 proximal promoter at the initiation of transcription start site dependent on extrinsic LIF signaling.
LIF has been shown to signal via STAT3; however, we and others have failed to detect direct STAT3 binding to the Oct4 promoter (Kidder et al., 2008). Our findings that SAF-A is in complex with STAT3 and that the number of complexes decreases upon LIF withdrawal could explain how Oct4 expression is maintained by LIF signaling, although SAF-A:STAT3 complexes are only reduced approximately 50% after 2 days of LIF withdrawal.
Our discoveries allow us to propose a model for Oct4 transcription initiation (Fig. 7B). This explains how signaling by extrinsic LIF proceeds via STAT3 (Matsuda et al., 1999; Niwa et al., 1998), which translocates into the nucleus and could interact with SAF-A. SAF-A then binds the Oct4 promoter due to STAT3 or some unknown factors. The transcription factors Oct4 and Sox2 associate with SAF-A. Because Oct4 and Sox2 have been found to interact preferentially with the enhancer region of the Oct4 promoter (Chew et al., 2005; Okumura-Nakanishi et al., 2005) it is likely that the complex with SAF-A, Oct4 and Sox2 links enhancer with the proximal promoter regions. SAF-A next recruits npCTD-RNA pol II to the transcription start site to initiate Oct4 transcription, and continues to interact with RNA pol II following CTD phosphorylation to process nascent mRNA as transcription elongation proceeds. Upon early initiation of differentiation mediated by LIF withdrawal for 2 days (2d), SAF-A no longer binds the Oct4 promoter and Oct4 transcription is blocked. SAF-A and RNA pol II were found to be in complex even after LIF withdrawal. It should be mentioned that we have not completely ruled out the order in which detected interactions occur.
Understanding determinants of pluripotency at the molecular level may facilitate the generation of patient-specific cells for drug discovery and cell therapies. This report identifies SAF-A as a regulator of the pluripotency factor Oct4 in ES cells by direct interaction with the Oct4 proximal promoter.
Footnotes
Acknowledgments
We acknowledge the Swegene Centre for Cellular Imaging at the University of Gothenburg for use of imaging equipment, and the Proteomics Core Facility at the University of Gothenburg for peptide sequencing. This work was supported by the Åke Wiberg Foundation, the Swedish Research Council, the Swedish Cancer Society, the Clas Groschinsky Memory Foundation, and the Magnus Bergvall Research Foundation.
Author Disclosure Statement
The authors declare no conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.