
Abstract
Background:
Alpha-particle-emitting radiotherapies are of great interest for the treatment of disseminated cancer. Actinium-225 (225Ac) produces four α-particles through its decay and is among the most attractive radionuclides for use in targeted radiotherapy applications. However, supply issues for this isotope have limited availability and increased cost for research and translation. Efforts have focused on accelerator-based methods that produce 225Ac in addition to long-lived 227Ac.
Objective:
The authors investigated the impact of 225Ac/227Ac material in the radiolabeling and radiopharmaceutical quality control evaluation of a DOTA chelate-conjugated peptide under good manufacturing practices. The authors use an automated module under identical conditions with either generator or accelerator-produced actinium radiolabeling.
Methods:
The authors have performed characterization of the radiolabeled products, including thin-layer chromatography, high-pressure liquid chromatography, gamma counting, and high-energy resolution gamma spectroscopy.
Results:
Peptide was radiolabeled and assessed at >95% radiochemical purity with high yields for generator produced 225Ac. The radiolabeling results produced material with subtle but detectable differences when using 225Ac/227Ac. Gamma spectroscopy was able to identify peptide initially labeled with 227Th, and at 100 d for quantification of 225Ac-bearing peptide.
Conclusion:
Peptides produced using 225Ac/227Ac material may be suitable for translation, but raise new issues that include processing times, logistics, and contaminant detection.
Introduction
Radionuclide therapy has been a pillar of nuclear medicine for over 80 years. Phosphorus-32 was first applied by Lawrence in 1938 for treatment of leukemia, 1 followed by other agents. Notably, the use of radioiodine for hyperthyroid conditions and cancer has become standard of care. 2 –4 Alpha-particle emitters have long been of interest for systemic applications in oncology and degenerative conditions. These charged helium nuclei deposit MeV of energy along a short path-length of only several cell diameters. 5 Alpha-particle-emitting radium isotopes localize to sites of bone turnover, and the treatment of ankylosing spondylitis using radium-224 (224Ra) had been used widely for decades. 6 The recent approval of radium-213 (213Bi) dichloride for metastatic prostate cancer has stimulated even further interest in the field. 7
Molecularly specific radioimmunotherapy involves conjugation of radionuclides to targeting vectors, often antibodies or peptides. The first clinical targeted α-particle-emitting radiotherapies developed by McDevitt et al. for bismuth-213 (213Bi) subsequently led to development of actinium-225 (225Ac)-conjugated antibody and small-molecule therapies. 8,9 The 225Ac isotope is considered nearly ideal for antibody-based targeted radiotherapy applications because the biological half-life of a full-length immunoglobulin and the physical half-life of the parent radionuclide (t 1/2 = 9.92 d) are well matched. 10,11
Increased translational efforts have revealed a supply-chain constraint for access to 225Ac. Production method, i.e., elution of a thorium-229 (229Th) generator–produces several times less than the estimated need for routine clinical practice. 12,13 Demand for the isotope has spurred several developmental efforts, including cyclotron, electron, or photon irradiation of radium-226, 14,15 and spallation of thorium targets by high-energy protons. 16,17 This latter accelerator-based approach can produce large quantities of actinium. This includes the desired 225Ac, along with an isotopic mixture of short-lived 224,226,228Ac (t 1/2 < 2 d) and long-lived 227Ac (t 1/2 = 21.8 year). Half-life, infrastructure requirements, and the cost and capabilities of physical separation methods preclude enrichment of the desired 225Ac. As such, the long-lived 227Ac is present in this drug formulation reagent.
227Ac decays predominantly to 227Th (t 1/2 = 18.72 d) followed by 223Ra (t 1/2 = 11.43 d), both of which are also alpha-particle emitters of interest for their therapeutic potential (Supplementary Fig. S1). 18 –21 This creates potential issues relating to radiochemical and dosimetric impact of this increasingly complex radionuclide mixture, which includes the concatenated decay progeny of both 225/227Ac. Studies to date have investigated the radiochemical impact of the presence of 227Ac in chelate radiolabeling. 16,22,23 At the end of bombardment, the radioisotopic impurity of 227Ac in the 225/227Ac material is reported to be low (0.2%–0.3%). 24 The ratio of isotopes changes rapidly with time as the 225Ac material decays during chromatographic separation from the target thorium and coproduced isotopes, shipment, and radiopharmaceutical preparation. 25
Early evaluation of the radiobiological impact of the impurity is underway in preclinical and modeling studies. 22,26 These and future studies are important as long-lived impurities in historical formulations of 224Ra may have contributed to incidences of treatment-induced malignancies, 27,28 however, evaluation of these data is ongoing. 29 As well, regulatory consideration regarding the handling and disposal of the chemical and biological wastes that are contaminated with this long-lived 227Ac material are a point of continuing discussion.
The authors' interest in the present study was to investigate the implications for quality control and clinical release processing for accelerator-produced actinium used to formulate a therapeutic agent; and the application of an automated synthesis system for such a radiopharmaceutical. They report and discuss the radionuclide and radioisotopic purity impact for this material following good manufacturing processes for labeling, including an automated radiolabeling module, and a variety of analytical methods.
Materials and Methods
Radionuclides
The 229Th generator-produced 225Ac and accelerator-produced 225/227Ac sources were supplied by the United States Department of Energy. The accelerator-produced material activity was 79.2 MBq (2.1 mCi) on 07/01/2021, 1 d following purification, and subsequently evaporated and shipped as dried nitrates (Supplementary Fig. S2). The certificate of analysis for this batch indicated an estimated 0.7 MBq (20 μCi) of 227Ac contaminant, as extrapolated from measurements of previously generated material. At receipt, the material in a Wheaton vial (3.0 mL) was measured using a well-style dose calibrator (Capintec CRC25R) set on precalibrated 225Ac setting number (#82) with 55 MBq (1.5 mCi) of starting material (at day 7 postradiomaterial purification, 07/06/2021). A representative certificate of analysis for generator produced material is included (Supplementary Fig. S3).
Radiolabeling
The DOTA-labeled peptide precursor was supplied by Modulation Therapeutics, Inc., (Fig. 1C). The labeling of the peptide was carried out under good manufacturing practice, within a shielded hot cell using a multifunctional automated radiosynthesis module (Trasis, AllinOne mini). The process was controlled remotely using Supervision (v.2.28 Rev. 2, Trasis). In brief, 46.6 MBq (1.27 mCi) of the source dissolved in 0.2 M HCl was loaded under vacuum in the initial vial for radiolabeling with the DOTA-conjugated precursor (200 μg) on day 5 postsource purification.
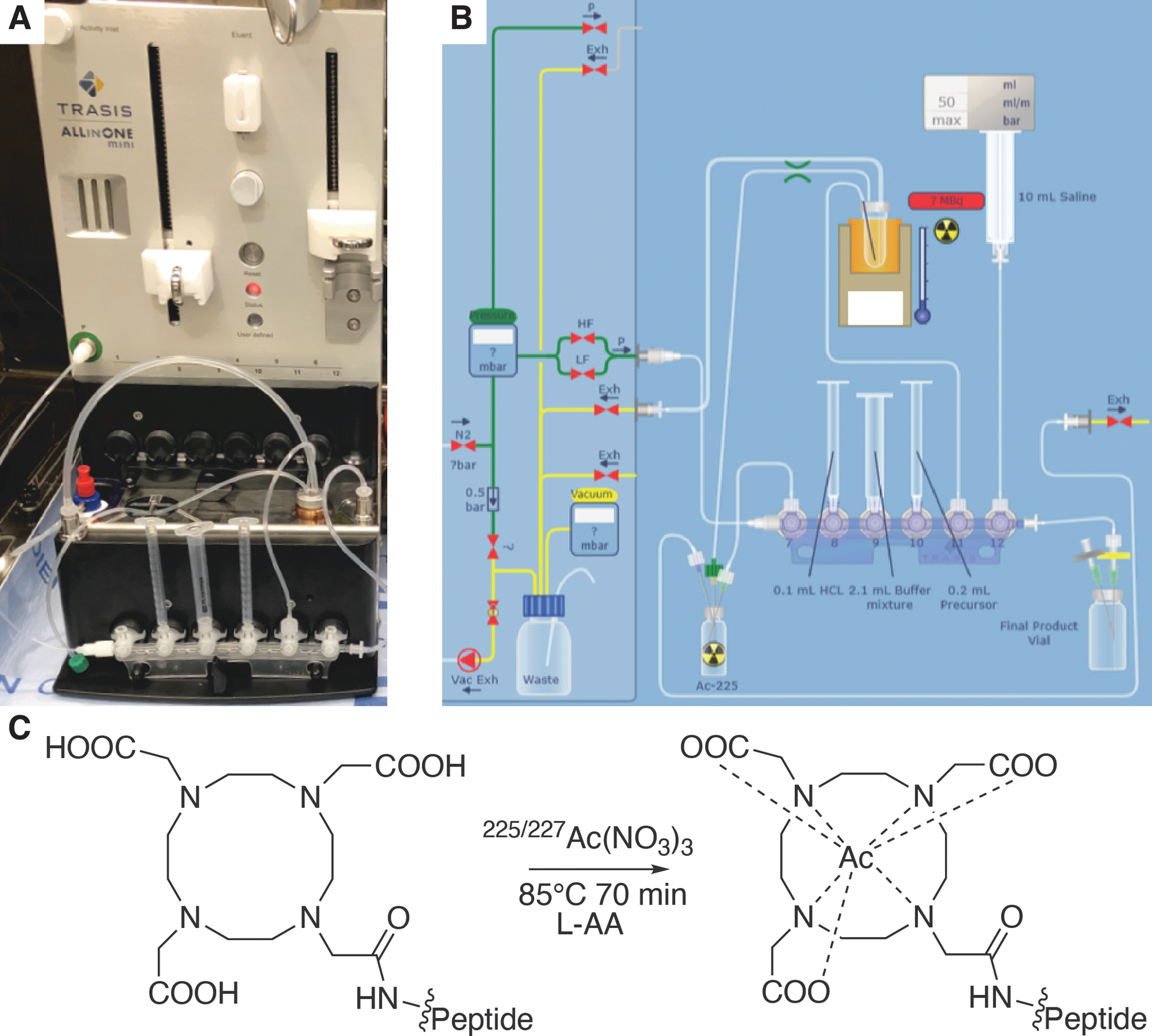
Automated Radiosynthesis Module and Conditions.
The source was transferred to the one-pot radiolabeling reactor cassette, in which the reaction occurred in Tris buffer (1 M, pH 7.2) at 85°C for 70 min in the presence of 20% v/v L-ascorbic acid at pH 6–8. The radiolabeled peptide was transferred in saline and passed through a 0.2 μm sterilizing filter, resulting in a final volume of 9.7 mL. The final radiolabeled sample was tested for sterility and presence of bacterial endotoxins.
Radio-instant thin-layer chromatography
A sample of the final drug product material (0.4 μL each) was spotted on glass-microfiber chromatography paper impregnated with a silica gel (115 mm length) and developed using 50 mM DTPA in ultrapure water. Acquisitions of 5 min were conducted at 225Ac/213Bi equilibrium (>4 h postmigration; typically 6–7 h) utilizing a radioTLC imaging system under default settings and analyzed using vendor software (AR-2000 and Winscan; Bioscan). Free radiometals migrated to the front and the bioactive-labeled material remained at the origin (0–50 mm).
Peptide purification and fractionation
Reverse phase high-pressure liquid chromatography (HPLC) was undertaken utilizing an Agilent system equipped with a HICHROM Vydac 218MS C18 (4.6 × 250 mm, 5 μm) column, running with a flow rate of 1 mL/min, and a UV detection set at 220 nm. Twenty microliters of the labeled peptide was injected for analytical chromatography and fractions were collected for radiotrace determination. The total separation was performed over 35 min and fractions were collected every 1 mL. A gradient mobile phase was used consisting of HPLC grade water (Solvent A) and acetonitrile (Solvent B) both with 0.1% trifluoracetic acid.
Gradient conditions were initially 100% (A), 0% (B); 0–20 min to reach 20% (A), 80% (B); 20–20.5 min was ramped to 100% (A), 0% (B); 20.5–35 min 100% (A), 0% (B). An elution time of 11.736 min was recorded from the radiolabeled product UV spectra, and 11.779 min was identified to be the unlabeled precursor.
Gamma counting
Collected fractions from the HPLC were measured using a COBRA-2 gamma counter (Packard) after the samples had reached an equilibrium state for 225Ac (>4 h), with an energy window channel setting of 15–800 keV. Acquisitions were recorded 1 min per sample, and the counts per minute were background corrected to determine radiochemical purity defined as the integration of collected fractions 10–14 mL (containing the peptide as determined by UV-trace) over the entire collection (35 mL). Two fractions contain detectable activity, noted as fr12 and fr13, and were submitted for further analyses.
Gamma spectroscopy
A high-purity germanium (HPGe) detector was used to acquire high energy resolution spectra for isotope identification. The cryocooled system (GEM-50195-S; Ortec) was used with samples placed 5 cm away from the detector enclosed in a 10 cm lead shield (HPLBS1; Ametek). The energy and efficiency calibrations were completed using a gamma ray source traceable to the National Institute of Standards and Technology (Eckert Ziegler Isotope Products) allowing preset calibration of each energy peak and quantification per isotope. The same sample-to-detector geometry was used to measure the HPLC collected fractions of interest and measured over 24 h. Counts were normalized to acquisition time for each gamma spectra, and the isotope quantification was conducted utilizing Gamma-Vision Software (version 8.0, Ametek) accounting for background, half-life, abundance, acquisition time correction, and normalized for each isotope recognized by the edited library.
Safety statement
Handling and evaluation of radiological materials require careful planning, infrastructure, and training. This is all the more important for high linear energy transfer emitters, as used in this work. All experiments were conducted by staff with certified Radiation Safety approvals and dose monitoring by the Washington University School of Medicine Department of Environmental Health and Safety, in approved areas. Samples were handled in either a hot cell or chemical fume hood, and characterization measurements were performed with sealed samples in appropriate and monitored areas. All samples used in this work have half-lives ≥10 d, requiring off-site storage and specialized disposal protocols handled by Radiation Safety.
Results
Accelerator produced 225/227Ac was received for radiolabeling using a preprogrammed automated radiosynthesis module (Trasis, AllinOne mini). In brief, the system consists of a volume and time-controlled heating and mixing platform for single-pot radiolabeling of the precursor DOTA-conjugated peptide. The advantages of an automated approach include a systematic and repeatable preparation method with reduced absorbed dose to the radiochemist. In addition, the device is located within an ISO7 hot cell, thereby reducing likelihood of radiocontamination to personnel, and biocontamination to the material. The module was programmed to fully automate the benchtop synthesis used to develop the radiopharmaceutical, see Figure 1. Here, the dry 227/225Ac-nitrate is dissolved in dilute acid, to which antioxidant and peptide is added and heated to 85°C for 70 min. The radiolabeled material was sterilized by filtration, resulting in 9.7 mL of 200 μg of peptide (48.47 MBq) for subsequent quality control analyses.
Initial radiochemical analyses were performed at the time of radiopharmaceutical production and at day 2 postlabeling (to model time elapsed required to reach and be administered to a patient). The authors first assessed the product using radio-iTLC. Radiolabeled peptide and control (uncomplexed) source radiomaterial were spotted and developed using 50 mM DTPA. The acquisition of the paper chromatographic strip by proportional counter was carried out at >6 h postdevelopment. This time is provided for 225Ac to reach secular equilibrium; and for migrated daughters, primarily 221Fr (t 1/2 = 4.9 min) and 213Bi (t 1/2 = 45.6 min), to decay. The radio-iTLC analysis indicated >99%% radiochemical purity, Figure 2A and B. The clinical release criterion for this radiopharmaceutical includes ≥95% RCP by radio-iTLC, which was met.
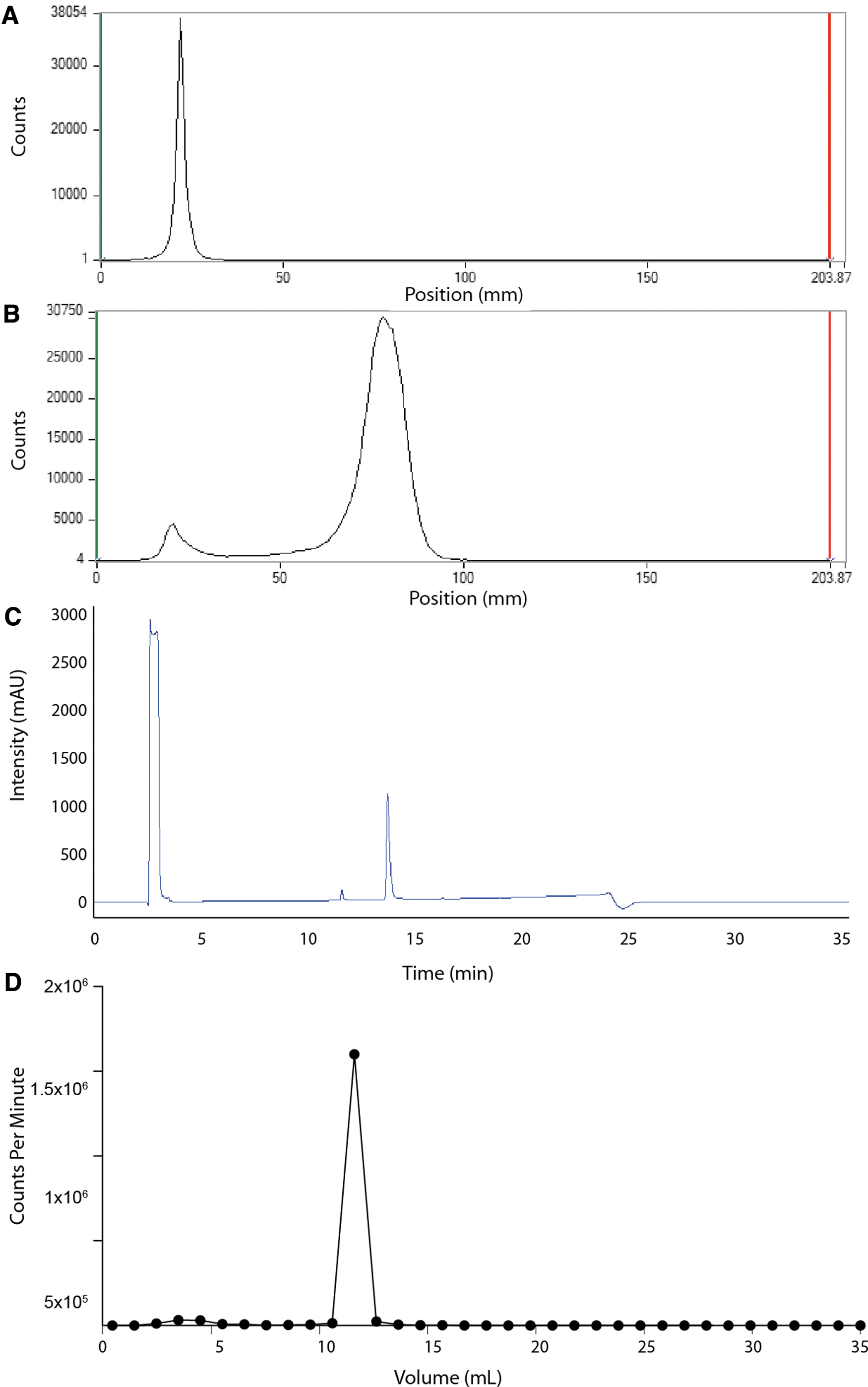
Radiochemical Purity Analysis.
Reverse-phase HPLC was also performed, demonstrating >90% chemical purity (Fig. 2C). The early (3–5 min) fractions correspond to the low retention of free metal ions (Fig. 2D). The collected peptide fractions were gamma counted, after reaching 225Ac secular equilibrium, and a 94.2% radiochemical identity was observed. This stands in contrast to the automated radiolabeling results from thorium-229 generator produced 225Ac >95% (n ≥ 5; Supplementary Fig. S4), analyzed by identical methods.
They next sought to investigate the radioisotopic content of the HPLC-separated peptide to interrogate this discrepancy. High-resolution gamma spectroscopy using cryocooled HPGe was acquired at multiple time points following radiolabeling to identify the peptide-complexed radionuclides. At the earliest analyzed time point, day 2 following radiolabeling, the abundant gamma emissions from 221Fr (218 keV) and 213Bi (440 keV) are prominent (Fig. 3A). The direct detection of 225Ac itself was also observed; and was quantifiable at 188 keV (this gamma ray line is isolated from daughter contributions). The activity determined from this 225Ac peak (99508 Bq) matched that of progeny 213Bi (98086 Bq) at equilibrium (Table 1). The HPGe method was in good agreement with the dose calibrator reading of the labeled radiopharmaceutical, after compensating for the proportion of activity injected by HPLC and that collected in fractions 12 and 13.
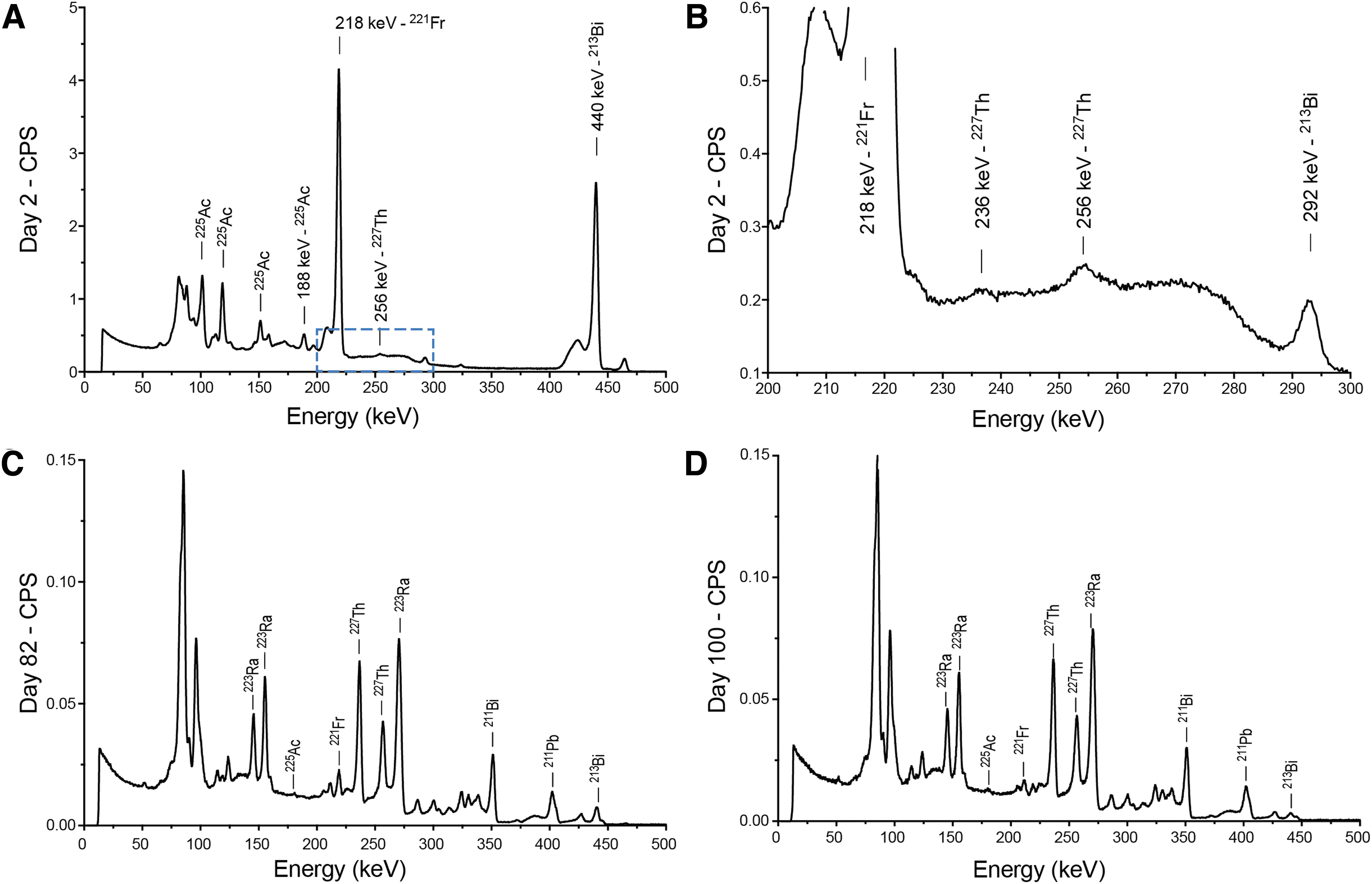
Gamma Spectroscopy of HPLC-Purified Peptide.
Activity (Bq) of Purified 227/22 5 Ac-Labeled Peptide Fractions Measured Utilizing Gamma Spectrometry Acquired at Day 2, 84, and 100 Postradiolabeling
The predicted activities are calculated for the half-life of each radionuclide from that measured at day 2. The characteristic gamma line for Ac-225 could not be determined (n.d.) after day 2.
Detailed observation also revealed the presence of 227Th in the purified radiolabeled peptide fraction at day 2. The amount measured here indicates that the 227Th present was labeled onto the conjugate at synthesis. In addition, 223Ra is not clearly detected at this early time point. This underlines successful chemical purification by HPLC of this nonchelated rare earth metal, and the direct DOTA-peptide labeling by both 225/227Ac and 227Th (Fig. 3B). The samples were reanalyzed for isotopic content at 82 and 100 d postradiolabeling (Fig. 3C, D).
Quantification of these isotopes' activities is presented in Table 1, for each of the displayed gamma spectrum measurements. Direct quantification of 225Ac was not successful at later time points due to interfering gamma and X-rays from the complex radionuclide mixture. The high-energy 440 keV of the 213Bi daughter could be cleanly assessed, and measured values at 82 and 100 d are within the error of the method as predicted from 225Ac-decay rate from the initial day 2 quantification. In contrast, the 227Th activity in the peptide fraction does not follow the predicted values for the half-life of this isotope, as it is fed by 227Ac decay.
The 256 keV line used for 227Th analysis has negligible interference from other emissions. Using the activity quantified at 100 d postradiolabeling (for which 227Th has reached >98% equilibrium with its parent), the authors calculated an initial 227Ac activity of 1514.2 Bq in the primary peptide fraction at the proposed day of injection. A further 68.1 Bq in the second largest fraction was measured, Table 2. The resulting isotopic purity of actinium-225 (defined as the percentage of 225Ac to all actinium isotopes present 30 ) of the isolated radiolabeled peptide is 98.46%.
Isotopic Purity at the Time of Patient Injection (Day 2 Postlabeling) of Purified Peptide
HPLC fraction 12 (11–12 min, the most concentrated fraction) and fraction 13 (12–13 min, the end of the collected peak) constitute the full radiolabeled 225/227Ac-peptide content. Ac-225 was measured by HPGe at day 2; Th-227 was evaluated at 100 d postradiolabeling, reaching equilibrium with Ac-227.
HPLC, high-pressure liquid chromatography; HPGe, high-purity germanium.
The time elapsed between final 225/227Ac purification from the accelerator bombarded material to the radiopharmaceutical labeling was ∼7 d. This provides time for ingrowth of 227Ac daughters. The authors have modeled the radionuclidic content using the Bateman equations, and displayed activity over time for all isotopes with a half-life greater than 9 d (Supplementary Fig. S5). The radionuclide content at the day of radiolabeling consists predominantly of 225Ac, as well as 227Ac and 227Th (which increases to the day of injection). The result is a radionuclidic purity for 225Ac of 97.8% at day 2 postlabeling, if they only consider therapeutic isotopes with a half-life greater than 9 d. These data demonstrate the importance of timing both between source purification and radiopharmaceutical administration, as well as controlling the molecular contents of the prepared material.
Discussion
Targeted radiotherapy is an emerging paradigm for the treatment of refractory and disseminated disease. Recent approvals by the European Medicines Agency and the US Food and Drug Administration (FDA), including iodine-131 (131I) meta-iodobenzylguanidine, radium-223 (223Ra) dichloride, and lutetium-177 (177Lu) octreotate, are poised to be the beginning of a trend that will likely see radiotherapies become commonplace in cancer care. These treatments can be considered inherently more complex than many nonionizing agents as issues of handling, time of administration, disposal, and overlapping medical, pharmaceutical, and nuclear regulations must be considered. These issues are relevant both in the preclinical research and development space as well as in the sphere of clinical translation.
Actinium-225 has been evaluated as an α-particle-emitting radionuclide for targeted radiotherapy applications for over 20 years, 8,30,31 with active clinical assessments ongoing. 31,32 The majority of the global supply for this work is 229Th-generator-produced material, limited to 20–40 GBq per year. 17,33,34 Meeting additional supply requirements has been a focus for alternative production methods from the United States Department of Energy (DOE) and others. Large-scale production by proton bombardment using thorium targets is a promising approach. 24,35 At this time, the DOE has submitted a provisional Drug Master File to FDA for evaluation of 225Ac produced through this process as an active ingredient in radiopharmaceutical drug manufacture.
Special consideration for the coproduced 21.7 year half-life 227Ac and its multiple α-particle-emitting progeny may be required. This includes the radiochemical impact as well as dosimetry and potential radiobiological effects of the contaminants; the handling of long-lived radionuclide materials and wastes; and the long-term contamination of radiochemical equipment and patient treatment spaces. 36
The complex mixture of radionuclides present in both starting material and in the final radiolabeled product when using 225/227Ac sources is a potential confounder in radiopharmaceutical quality control and release. Gamma ray spectroscopy can be used to detect isotopes from the 227Ac decay chain (Fig. 2), a technique which may not be accessible at all dispensing academic or tertiary medical centers. The analytical studies undertaken in this work further sought to define differences in peptide labeled with either 229Th generator-produced or accelerator-produced material. The conventional 229Th generator-produced 225Ac passed radiochemical purity quality control levels (defined as >95% as assessed by radio-iTLC) through multiple radiolabeling runs.
Using 225/227Ac starting material for radiolabeling under identical, automated conditions led to similarly high (>99%) yield by TLC, but a lower radiochemical purity of 94.2% by gamma counting. Further, their analysis of the radiopeptide at late time points (100 d postradiolabeling) enables an accurate assessment of the 227Ac content in the labeled radiopharmaceutical. Using this information in concert with the early (2 d postradiolabeling) gamma ray spectroscopy data, they can state that 227Th is initially chelated by the peptide. A limitation of this study is that these results are derived from a single run with the 225/227Ac starting material, which is produced at this time on an infrequent schedule.
However, the authors posit that this would not significantly alter their results as multiple batches of this material have had similar isotopic properties. Additional replicates would strengthen the basis of these observations and these results motivate further testing, and an assessment of the analytical methods leveraged to determine radiopharmaceutical purity and labeling efficiency.
At this time, no standardized methods of analysis or release for clinical use of 225Ac-constructs have been put forward. Techniques developed from clinical development efforts have relied predominantly on TLC of radioconjugate material from 229Th generator-produced sources. 8,32,37 Here, the francium-221 (221Fr) and 213Bi daughters of 225Ac presenting reasonable distinctive gamma rays (218 and 440 keV) readable using NaI detection or by gas ionization. The authors' analyses of the 225/227Ac material show that such systems are less well suited to deal with multiple α-emitting radionuclides with similar emitted energies. Accelerator-produced material present higher challenges of detection due to the multiple overlapping α-emitting daughters 227Th (236, 254 keV), 223Ra (145, 154, 270 keV), 211Pb (404 keV), and 211Bi (356 keV), limiting the capacity to clearly visualize and compute radiocontaminants.
In brief, the more complex mixture of isotopes present in the accelerator-produced material brings challenges for radiochemical analysis postlabeling. Radiopharmaceuticals produced using 225/227Ac sources may have to take this into account for pharmacy release criteria. Implied in this statement is that QC evaluation of the radiochemical and -nuclidic purity, along with the isotopic purity, may require systematic specific considerations for this material for each produced batch, including HPLC coupled with radiotracing and HPGe for release.
These data underline that purity considerations are driven by both the time of radiolabeling following the processing of the bombarded material, and the time between radiolabeling and administration. Experimentally derived expiration dates for the material may be required for radiopharmacy validation and release. Previous experience with radionuclide impurities can provide some guidance. Indium-111 (2.8 d) production results in a long-lived isomer, 114mIn (49.5 d). The contaminant produces a highly abundant high-energy beta particle that can significantly contribute to blood dose for leukocyte-labeled 111/114mIn. 38,39 As such, the material should be used as quickly as possible. The beta-particle-emitting lutetium-177 (6.65 d) produced via the 176Lu (n, γ) 177Lu reaction will contain 177mLu (160 d).
In the treatment setting, the detectable 177mLu may have only a minor contribution to imaging and therapy. 40,41 Literature, regulatory and manufacturer direction, also exists for the handling and consideration of impurities in yttrium-90, including strontium-90 and yttrium-88, and generator breakthrough for strontium-82,85/rubidium-82, germanium-68/gallium-68, and molybdenum-99/technetium-99m among others. 42,43
Standing-out against the historical considerations of impurities present in the radiopharmaceutical formulations above is the nature of the impurities for the 225/227Ac material. These include high-energy alpha- and beta-particle emitters, concatenated decay of isotopes with long half-lives. Radiopharmaceutical evaluation incorporating accurate information of the 225/227Ac, 227Th, and 223Ra and daughters in a formulation is thus necessary to fully evaluate the pharmacokinetic and dosimetric aspects of any drug. The authors have calculated the activity of 227Ac expected for a drug administration using the radiochemical yields and purity from the present work and administered activity levels of 225Ac in ongoing and proposed trials (Table 3). It should be noted that different radiolabeling chemistries have been used (a 2-step DOTA chelation at 65°C for the HuM195 antibody, and direct PSMA-617 chelation at 95°C) and different times of processing which will change the radionuclidic and isotopic purity of the final products. 31,32
Consideration of the localization, residency time, stability, and potential redistribution of the mixture of complexed and free radionuclides is likely to be distinct for each agent. Thus, the attendant radiotherapy and dosimetric impact could be evaluated on an individual compounding basis considering yield and timing for each patient. This may be considered onerous compared to current nuclear medicine therapy workflows; however, this is standard of practice for radiation oncology procedures for the deposition of ionizing radiation through either external beam or brachytherapy interventions. 43
In conclusion, this work observed and evaluated potential radiopharmacy quality control and release issues of 225/227Ac in a drug formulation. Standard measures from radio-iTLC and gamma counting of HPLC fractions were performed with generator- and accelerator-produced actinium-labeled peptide. Difference in results between radio-iTLC and gamma counting may be attributed to chemical and radiological properties of the complex radionuclide mixture. The preparation and injection times of therapies using this starting material will be important considerations in the context of radiopharmacy. These are in addition to the careful regulatory guidance required on the application, disposal, and licensing aspects, which will be necessary to a safe framework to both treat patients and to work with these long-lived contaminants.
Footnotes
Disclosure Statement
M.M., T.H., and D.M. are employees of Modulation Therapeutics Incorporated. T.J.W. and D.N.P. own intellectual property on the radiopharmaceutical. The remaining authors declare no conflicts of interest.
Funding Information
This work was supported, in part, by the National Institutes of Health R01CA229893, R01CA201035, and R01CA240711 and Modulation Therapeutics Incorporated.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.