
Abstract
Background:
BAY 1862864 is an α-particle emitting 227Th-labeled CD22-targeting antibody. This first-in-human dose-escalation phase I study evaluated BAY 1862864 in patients with CD22-positive relapsed/refractory B cell non-Hodgkin lymphoma (R/R-NHL).
Materials and Methods:
BAY 1862864 intravenous injections were administered at the starting 227Th radioactivity dose of 1.5 MBq (2 or 10 mg antibody), and the radioactivity dose escalated in ∼1.5 MBq increments (10 mg antibody) until the maximum tolerated dose (MTD) was reported. The primary objective was to determine the safety, tolerability, and MTD.
Results:
Twenty-one patients received BAY 1862864. Two dose-limiting toxicities (grade 3 febrile neutropenia and grade 4 thrombocytopenia) were reported in one patient in the 4.6 MBq (10 mg antibody) cohort. The MTD was not reached. Ten (48%) patients reported grade ≥3 treatment-emergent adverse events, with the most common being neutropenia, thrombocytopenia, and leukopenia, each occurring in 3 (14%) patients. Pharmacokinetics demonstrated the dose proportionality and stability of BAY 1862864 in the blood. The objective response rate (ORR) was 25% (5/21 patients) according to the LUGANO 2014 criteria, including 1 complete and 4 partial responses. The ORR was 11% (1/9) and 30% (3/10) in patients with relapsed high- and low-grade lymphomas, respectively.
Conclusions:
BAY 1862864 was safe and tolerated in patients with R/R-NHL.
Clinical Trial Registration numbers: NCT02581878 and EudraCT 2014-004140-36.
Introduction
Despite recent advances in cancer therapy, an urgent need for new therapies for patients with relapsed or refractory B cell non-Hodgkin lymphoma (R/R-NHL) remains. Prior studies established the sensitivity of patients with R/R-NHL to antigen-specific radionuclide-labeled antibodies and the sensitivity of those with early-stage follicular lymphoma (FL) to radiotherapy, which provided a rationale for the development of targeted alpha therapy (TAT) for the treatment of patients with NHL. 1 –4
TAT is a highly potent and selective anticancer treatment that delivers high-energy α radiation to tumor cells and the tumor microenvironment, leading to robust antitumor effects, while minimizing damage to the surrounding normal tissue. 5 α-particles have a high linear energy transfer, ranging from 50 to 230 keV/μm, which induces DNA double-strand breaks leading to cell cycle arrest and subsequent cell death. 5
227Th is an α-particle-emitting radionuclide with a half-life of 18.7 d, which decays through an α-particle emission to its first daughter radionuclide radium-223 (223Ra). 6 The physical half-life of 227Th is longer compared with other clinically used α-particle-emitting radionuclides 7 and is compatible with the biological half-life of an antibody in human plasma.
227Th can be efficiently complexed with the 3,2-hydroxypyridinone (3,2-HOPO) chelator conjugated to antibodies or other targeting moieties. 8 The longer physical half-life of 227Th, and low radiolysis and degradation rate of antibodies permit a pharmaceutical shelf life of 48 h for the 227Th-labeled antibodies. 7
CD22 belongs to the B cell-specific immunoglobulin superfamily and is expressed on the vast majority of R/R-NHL, but is absent on the surface of stem cells, B cell progenitors, and plasma cells. 9,10 CD22 functions as a negative regulator of B cell antigen receptor signaling. 11 The validity of the CD22-targeting approach has been shown in phase III studies of the recombinant anti-CD22 immunotoxin, moxetumomab pasudotox, in patients with CD22-positive hairy cell leukemia and the yttrium-90-labeled epratuzumab (anti-CD22 antibody) tetraxetan in patients with NHL. 12 –14
The 227Th-epratuzumab conjugate (BAY 1862864) is composed of an anti-CD22 humanized monoclonal antibody (epratuzumab; Immunomedics, Inc., NJ) conjugated to the 3,2-HOPO chelator at a chelator-antibody ratio of 0.8 and then labeled with 227Th at a nearly 100% yield. BAY 1862864 has demonstrated a cytotoxic effect at nanomolar concentrations when tested on CD22-positive cancer cell lines. In preclinical animal models, BAY 1862864 prolonged the overall survival of BAY 1862864-treated animals versus those who had received a control treatment and was well tolerated. 6
In this study, the authors report the safety, tolerability, pharmacokinetic data, and efficacy for the dose-escalation part of the first-in-human phase I study of BAY 1862864 in patients with R/R-NHL.
Materials and Methods
Study design and patients
This was an open-label, phase I study of BAY 1862864 in patients with CD22-positive R/R-NHL conducted in four centers (one in Sweden and three in the UK). The study was divided into a dose-escalation part, with a modified 3 + 3 design to determine the maximum tolerated dose (MTD), and an expansion part. Four treatment cycles, each lasting for 42 d, were planned, with additional injections allowed on the basis of a case-by-case benefit–risk assessment. Eligible patients were ≥18 years of age, had failed ≥1 prior chemotherapy or immunotherapy-based regimen, had relapsed or were refractory to the last treatment regimen, were ineligible for or failed high-dose therapy and autologous stem cell transplantation (HDT-ASCT), and had measurable disease. Patients were excluded if they had CD22-positive NHL with predominantly clinical leukemic presentation. Full inclusion and exclusion criteria are detailed in the Supplementary Data.
All patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, and applicable local laws and regulations. The protocol was reviewed and approved by the independent ethics committees at all participating sites. The study is registered in
Procedures and assessments
Validation of BAY 1862864 purity, identity, and stability are described in the Supplementary Data.
BAY 1862864 was administered intravenously as a slow bolus injection on day 1 of each 42 d treatment cycle. Patients were randomized 1:1 to receive either 2 or 10 mg of epratuzumab-chelator conjugate at a starting radioactivity dose of 1.5 MBq, based on nonclinical toxicology data. 15 The antibody dose was set to 10 mg for all the subsequent cohorts (the justification is given in the the Supplementary Data). The 227Th radioactivity dose was escalated in increments of ∼1.5 MBq, until the MTD was reported or a biologically active dose was determined.
The National Cancer Institute's Common Terminology Criteria for Adverse Events version 4.03 were used to grade toxicities and adverse events (AEs). The MTD was defined as the lower preceding dose level to the toxic dose. At the MTD, no more than one patient had to have experienced a dose-limiting toxicity (DLT) (defined in the Supplementary Data) out of six evaluable patients during the first 6 weeks of treatment (cycle 1). The evaluation of the QT/QTc interval was performed using the central triplicate 12-lead electrocardiography.
Pharmacokinetic evaluations and whole body measurements for 227Th and 223Ra were performed as described in the Supplementary Data.
Assessments of lymphoma lesions were carried out according to the 2014 Lugano classification criteria 16 using computed tomography (CT) and positron emission tomography (PET)-CT at screening, at the end of each cycle and at the end of study visit (12 weeks after the last injection).
Study objectives
The primary objective was to determine the safety, tolerability, and MTD of BAY 1862864, as assessed by the number of patients reporting DLTs during a period of up to 12 weeks after the first treatment. 17 The exploratory objectives included the evaluation of biodistribution of BAY 1862864, 18 pharmacokinetics, pharmacodynamics, immunogenicity, biomarkers, and tumor response to the treatment with BAY 1862864.
Statistical analyses
The choice of the number of patients per cohort was based on the standard phase I design for toxicity assessment.
All patients who had received at least one dose of BAY 1862864 were included in the safety analysis set (full analysis set). The incidence of treatment-emergent AEs (TEAEs) and drug-related TEAEs was determined as described in the Supplementary Data.
All patients evaluable for pharmacokinetics and pharmacodynamics, without major protocol deviations, were included in the respective analysis set. The efficacy population comprised all patients who had received ≥1 dose of BAY 1862864 and for whom any postbaseline efficacy data were available or who had clinical disease progression before the first postbaseline efficacy assessment (i.e., treatment failure).
The pharmacokinetic characteristics and the biomarker and efficacy data were summarized using descriptive statistics.
Statistical analyses were conducted using SAS, version 9.4 (SAS Institute, Inc., Cary, NC).
The study reports only data from the dose-escalation part.
Results
Patients
Between November 20, 2015, and November 11, 2019, 28 patients were assessed for eligibility. Seven patients did not meet the eligibility criteria, and 21 patients were enrolled and treated with different BAY 1862864 regimens (Fig. 1; full analysis set/safety analysis set). The majority (95% [20/21]) of patients had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1 (Table 1). Twenty-nine percent (6/21) of patients presented with diffuse large B cell lymphoma (DLBCL), 10% (2/21) with DLBCL originating from transformed FL, and 48% (10/21) with FL (Table 1). All patients (100%) had received ≥1 line of prior anticancer therapy, including rituximab (Table 1). Seventy-six percent (16/21) of patients had received prior chemotherapy and 29% (6/21) had received prior chemotherapy and immunotherapy (Table 1).
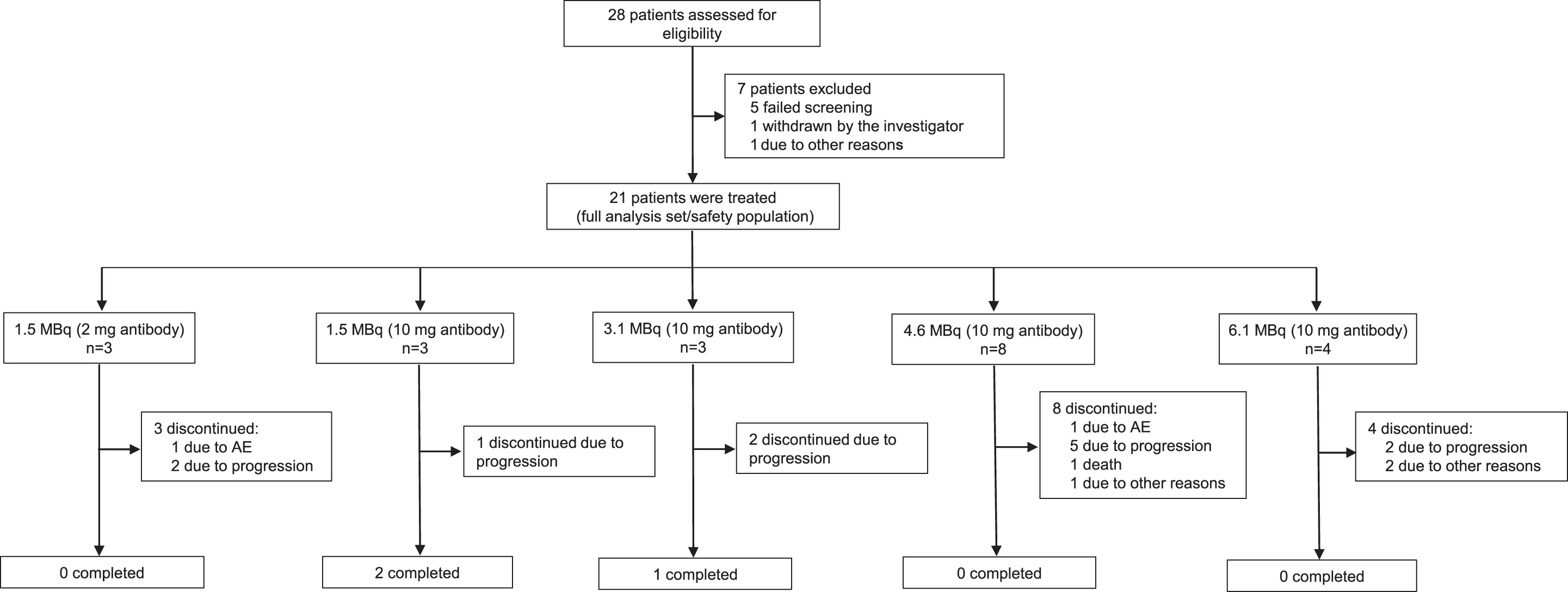
Patient disposition. AE, adverse event; MBq, megabecquerel.
Baseline Demographics and Disease Characteristics (Safety Analysis Set)
Data are n (%) unless otherwise stated.
Chemotherapy, chemotherapy/immunotherapy, high-dose chemotherapy/autologous transplant, monoclonal antibodies, other immunotherapy and other anticancer therapies.
Selected most common prior chemotherapies are shown.
DLBCL, diffuse large B cell lymphoma; ECOG PS, Eastern Cooperative Oncology Group performance status; FL, follicular lymphoma; FLIPI, FL international prognostic index; HCL, hairy cell leukemia; HDT-ASCT, high-dose therapy and autologous stem cell transplantation; IPI, international prognostic index; MZL, marginal zone lymphoma; NA, not available; PMBCL, primary mediastinal large B cell lymphoma.
Treatment
The median number of injections received for each cohort and the number of patients who had received one to five injections in each cohort are detailed in the Supplementary Table S1. Dose interruption or delay was reported in 10% (2/21) and treatment discontinuation in 5% (1/21) of patients due to an AE (Supplementary Table S1).
Safety profile
Any-grade TEAEs occurred in 95% (20/21) of patients, while grade ≥3 TEAEs were reported in 48% (10/21) of patients (Table 2). The most common grade ≥3 TEAEs were neutropenia (14% [3/21] of patients), thrombocytopenia (14% [3/21]), and leukopenia (14% [3/21]) (Table 2).
Treatment-Emergent Adverse Events in at Least 10% of Patients (Safety Analysis Set)
Data are n (%).
MBq, megabecquerel; TEAEs, treatment-emergent adverse events.
The most common drug-related any-grade TEAE was thrombocytopenia (48% [10/21] of patients) (Supplementary Table S2). Grade ≥3 drug-related TEAEs were reported in 24% (5/21) of patients, with the most common being neutropenia, thrombocytopenia, and leukopenia, each occurring in 10% (2/21) of patients (Supplementary Table S2).
Eighteen patients who had completed cycle 1 assessments were evaluable for DLTs. Three patients had not completed cycle 1 assessments due to progressive disease (PD). Overall, one patient experienced two DLTs, which were grade 3 febrile neutropenia and grade 4 thrombocytopenia, at the 4.6 MBq (10 mg antibody) radioactivity dose. No DLTs were reported for the other tested dose levels.
All patients with QT/QTc interval measurements had a change from the baseline in the Fredericia interval duration of ≤30 ms at all time points, except for one patient at cycle 1 day 1 (1 h postadministration of BAY 1862864) in the 1.5 MBq (2 mg antibody) cohort, who had a change from the baseline of >30–60 ms (Supplementary Table S3). Absolute values of the QTcF interval for patients with measurements show no QT interval prolongation (Supplementary Table S3). No liver toxicity was reported during the treatment period in any of the cohorts (data on file).
During the 12-month follow-up period, the most common any-grade AEs were neutropenia (19% [4/21]), thrombocytopenia (14% [3/21]), vomiting (10% [2/21]), and pyrexia (10% [2/21]), and the most common grade ≥3 AEs were thrombocytopenia (14% [3/21]) and neutropenia (10% [2/21]) (Supplementary Table S4). Potential drug-related spinal fractures were reported in two patients. No radiation-related toxicities, including benign and malignant neoplasms (except for a recurrence of a squamous cell carcinoma, resected >5 years ago, in a 76-year-old female with FL treated with a single 1.5 MBq [2 mg protein] dose), myelodysplastic syndromes, and acute myeloid leukemia, occurred during this period (Supplementary Table S4).
During the treatment and the follow-up period, median time to first grade 3 or 4 thrombocytopenia for 4 patients was 40 d (range 22–135). One patient with a prior history of HDT-ASCT in the 1.5 MBq (2 mg antibody) cohort, who received three injections, developed grade 3 thrombocytopenia 135 days after the first injection (Supplementary Table S5). The median time to recovery from a grade 3 or 4 thrombocytopenia nadir, which occurred in 2 patients during the treatment and the follow-up period, both in the 4.6 MBq (10 mg antibody) cohort, was 26 d (range 8–43) (Supplementary Table S5).
Grade 3 or 4 treatment-emergent serious AEs (TESAEs) were reported in 19% (4/21) of patients. 14% (3/21) of patients experienced at least one study drug-related TESAE (data on file). Three (14%) patients died during the study treatment period, but none of the deaths was deemed to be related to BAY 1862864. Two patients in the 4.6 MBq (10 mg antibody) cohort and one patient in the 6.1 MBq (10 mg antibody) cohort died due to disease progression.
Pharmacokinetics
Pharmacokinetic parameters for 227Th in blood and the total antibody in serum after a single dose administration in each treatment cohort are summarized in Table 3. Maximum 227Th blood concentrations increased proportionally over the radioactivity dose range of 1.5–6.1 MBq (Table 3). Changes in mean concentrations of 227Th in the blood and the total antibody in serum over time are shown in the Supplementary Figure S1A and B. The curve of 227Th concentrations in the blood over time paralleled that of the total antibody concentrations in serum across treatment cohorts in cycle 1 (Supplementary Fig. S1A, B).
Pharmacokinetic Parameters of 227Th and Total Antibody (Pharmacokinetics Analysis Set)
Data represent the arithmetic mean (percentage coefficient of variation), except for Tmax, which are the median (range).
AUC0–42 d, area under the concentration-time curve from 0 to 42 d; AUC0-TLAST, AUC from 0 to last concentration >LLOQ; Cmax, maximum observed drug concentration; CL, total clearance following IV dosing; IV, intravenous; LLOQ, lower limit of quantification; MBq, megabecquerel; MRTIV, mean residence time after an IV infusion; T1/2, half-life of terminal slope; Tmax, time to reach Cmax; TLAST, time of last drug concentration; VSS, apparent steady-state volume of distribution.
Across the different radioactivity dose levels, the median time to reach the maximum concentration (Tmax) of the total antibody in serum was between 0.14 h and 0.55 h, and the median time to reach the maximum activity of 227Th in the blood was between 0.15 h and ∼1 h (Table 3).
Using a high purity germanium (HPGe) detector, the whole body measurements for 227Th and 223Ra were performed in 3 patients treated with the 3.1 MBq (10 mg antibody), the 4.6 MBq (10 mg antibody), or the 6.1 MBq (10 mg antibody) radioactivity dose (1 patient in each dose level). 223Ra was observed during the elimination and decay of 227Th (Supplementary Fig. S1C–E). The 227Th and 223Ra measurements and those predicted based on physical decay are shown in the Supplementary Figure S1C–E. The noncompartmental analysis parameters for 227Th and 223Ra are detailed in the Supplementary Tables S6 and S7, respectively.
Pharmacodynamics
Treatment with BAY 1862864 resulted in the reduction of CD22-positive lymphocytes in the blood across all dose levels (Supplementary Table S8). No anti-BAY 1862864 antibodies were detected in any of the study samples analyzed from 21 patients (Supplementary Data, data on file).
Efficacy
The efficacy data are summarized in Table 4. A complete response (CR) was achieved in 5% (1/21) of patients and a partial response (PR) was reported in 19% (4/21) of patients using combined Lugano-CT and Lugano-PET-CT assessments according to the LUGANO criteria 2014 (Table 4). Fourteen percent (3/21) and 52% (11/21) of patients had stable disease (SD) and PD, respectively (Table 4). The best overall tumor response was not evaluable in 10% (2/21) of patients due to either discontinuation in cycle 1 or unavailability of post-treatment imaging data. A total objective response rate (ORR) (CR and PR) of 25% (5/21 patients) and disease control rate (DCR) (CR, PR, and SD) of 38% (8/21) were reported in the efficacy analysis data set (Table 4).
Efficacy of BAY 1862864 According to the Lugano 2014 Criteria a (Efficacy Analysis Set)
Data are n (%).
Lugano-CT and PET-CT combined.
CR, complete response; CT, computed tomography; DLBCL, diffuse large B cell lymphoma; FL, follicular lymphoma; HCL, hairy cell leukemia; MBq, megabecquerel; MZL, marginal zone lymphoma; NA, not available; PD, progressive disease; PET, positron emission tomography; PMBCL, primary mediastinal large B cell lymphoma; PR, partial response; SD, stable disease.
Efficacy data grouped according to indication showed an ORR of 11% (1/9) in patients with DLBLC, including one patient with primary mediastinal B cell lymphoma, an ORR of 30% (3/10) in patients with FL, and a PR in a patient with marginal zone lymphoma (Table 4).
Discussion
This first-in-human phase I study of BAY 1862864 in patients with R/R-NHL provides proof of the concept that a 227Th-radiolabeled anti-CD22 antibody is safe and may be beneficial for patients with R/R-NHL.
BAY 1862864 at a 227Th radioactivity dose level of up to 6.1 MBq (10 mg antibody) showed an acceptable toxicity profile in patients with R/R-NHL, and no dose-dependent or accumulative toxicity has been reported in this limited data set. Moreover, a repeated administration of BAY 1862864 at the 4.6 MBq (10 mg antibody) radioactivity dose at ∼6-week intervals was feasible, with the highest cumulative radioactivity dose reaching 13.8 MBq. Cytopenia, fatigue, and back pain were the most frequently reported any-grade TEAEs, while neutropenia, thrombocytopenia, and leukopenia were the key grade ≥3 toxicities. Based on nonclinical dosimetry studies (data on file), bone marrow toxicity was expected due to the estimated absorbed dose and the sensitivity of bone marrow to radiation.
Thrombocytopenia is a concern in patients with lymphoma treated with radioimmunotherapy. 19 Indeed, the marrow reserve is reduced due to the numerous prior therapies and treating physicians may be concerned about the risk of prolonged and severe thrombocytopenia and the attendant risk of hemorrhage.
Fourteen percent (3/21) of patients with R/R-NHL experienced grade ≥3 thrombocytopenia in this study, but the administration of increasing doses of BAY 1862864 did not result in a higher incidence of this complication and none of the patients treated at the highest 6.1 MBq (10 mg antibody) radioactivity dose level developed grade ≥3 thrombocytopenia. The median duration of thrombocytopenia was 8 d. Myelosuppression is also a concern in this patient group due to the fact that patients are often heavily pretreated before being considered for radioimmunotherapy.
The long-term follow-up safety data showed no adverse signal of delayed toxicity during the 12-month follow-up period. However, a relatively short long-term follow-up period, death due to disease progression, and termination of the follow-up period due to enrollment of patients with PD into other interventional studies precluded the evaluation of the long-term safety beyond 1 year. Three patients (14%) experienced fractures at least 6 months after the last treatment. A number of factors, such as advanced age, prior treatment with corticosteroids, chemotherapy, radiotherapy, and old spinal fractures, may have contributed to the fractures reported in this study.
Exposure to BAY 1862864 increased in a dose-proportional manner after administration of single radioactivity doses over a range of 1.5–6.1 MBq. Parallel time-concentration curves of 227Th and the total antibody across different treatment cohorts demonstrate the stability of BAY 1862864 in the blood.
The data collected using an HPGe radiation detector showed the median whole body half-life and the mean residence time (15.8 and 20.6 d, respectively) of 227Th to be in the same range as the physical half-life (18.7 d), which suggests that 227Th is eliminated from the body mainly through physical decay. In contrast, a comparison of the whole body activity of 223Ra measured with an HPGe detector and the predicted natural decay curves following administration of 3.1, 4.6, or 6.1 MBq (all at 10 mg antibody) dose, together with patient imaging data generated with the gamma camera, 18 suggest that 223Ra predominantly undergoes biological elimination, most likely through the gastrointestinal tract as reported in previous studies. 20,21
The efficacy data observed in this study indicate the first signs of potential clinical efficacy, with one CR being reported at the 3.1 MBq and four PRs at the 1.5, 3.1, and 4.6 MBq radioactivity dose levels. Importantly, patients with FL showed higher CR, PR, ORR, and DCR than patients with DLBCL, who had very aggressive disease and had either progressed or died following the treatment with a single BAY 1862864 dose in cycle 1.
Previous studies have demonstrated the efficacy of radioimmunotherapy in patients with NHL. Notably, patients with NHL without prior ASCT treated with the β-emitter yttrium-90-labeled epratuzumab tetraxetan achieved an ORR of 71%, including patients with bulky disease and poor prognosis, while patients with FL reported an ORR of 100%. 13 Radioimmunotherapy with yttrium-90-labeled ibritumomab tiuxetan in patients with follicular R/R-NHL showed an overall response rate of 80% versus 56% in the rituximab-treated control group, 22 and it was 74% in patients with rituximab-refractory follicular NHL. 2
In this study, the small sample size, disease heterogeneity at baseline, and the early termination of the trial due to reasons other than safety and efficacy of BAY 1862864 precluded the proper evaluation of a response rate.
Conclusions
The safety data reported in this study show that BAY 1862864 is safe and tolerated in patients with R/R-NHL.
Footnotes
Acknowledgments
Dr. Egle McDonald of Cancer Communications and Consultancy Ltd., Knutsford, UK, provided medical writing assistance, which was funded by Bayer.
Authors' Contributions
O.L. conceived and designed the study, acquired data, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. A.T.B. conceived and designed the study, developed methodology, acquired data, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. D.C. acquired data, analyzed and interpreted data, provided administrative, technical, and material support, supervised the study, approved the final version, and agreed to be accountable for all aspects of this work. C.H. conceived and designed the study, developed methodology, acquired data, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. E.L. conceived and designed the study, developed methodology, acquired data, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. A.C. conceived and designed the study, developed methodology, analyzed and interpreted data, drafted and revised the article, provided administrative, technical, and material support, supervised the study, approved the final version, and agreed to be accountable for all aspects of this work. J.P. analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. F.H. conceived and designed the study, developed methodology, acquired data, analyzed and interpreted data, drafted and revised the article, provided administrative, technical, and material support, supervised the study, approved the final version, and agreed to be accountable for all aspects of this work. F.B. conceived and designed the study, developed methodology, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. H.H. conceived and designed the study, developed methodology, analyzed and interpreted data, drafted and revised the article, supervised the study, approved the final version, and agreed to be accountable for all aspects of this work. L.-I.O. conceived and designed the study, developed methodology, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. I.S. conceived and designed the study, analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. C.M. analyzed and interpreted data, drafted and revised the article, approved the final version, and agreed to be accountable for all aspects of this work. O.L. (corresponding author): all coauthors have reviewed and approved the article before submission.
Data Sharing Statement
Availability of the data underlying this publication will be determined according to Bayer's commitment to the European Federation of Pharmaceutical Industries and Associations and the Pharmaceutical Research and Manufacturers of America “Principles for responsible clinical trial data sharing.” This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the US and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use
Disclosure Statement
O.L. declares no conflict of interests. A.T.B. declares no conflict of interests. D.C. received research funding from Amgen, Sanofi, Merrimack, AstraZeneca, Celgene, MedImmune, Bayer, 4SC, Clovis, Eli Lilly, Janssen, and Merck, and served on OVIBIO Scientific Advisory Board. C.H. declares no conflict of interests. E.L. declares no conflict of interests. A.C. is an employee of Bayer AG and a stockholder of Bayer, AstraZeneca, and Pfizer. J.P. is an employee and stockholder of Bayer AG. F.H. is an employee and stockholder of Bayer. F.B. is an employee and stockholder of Bayer AG. H.H. is an employee and stockholder of Bayer AG. L.-I.O. is an employee of Bayer AS. I.S. is an employee and stockholder of Bayer AG. C.M. declares no conflict of interest.
Funding Information
The trial was funded by Bayer.
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.