
Abstract
Despite the consensus around the clinical potential of the α-emitting radionuclide astatine-211 (211At), there are only a limited number of research facilities that work with this nuclide. There are three main reasons for this: (1) Scarce availability of the nuclide. Despite a relatively large number of globally existing cyclotrons capable of producing 211At, few cyclotron facilities produce the nuclide on a regular basis. (2) Lack of a chemical infrastructure, that is, isolation of 211At from irradiated targets and the subsequent synthesis of an astatinated product. At present, the research groups that work with 211At depend on custom systems for recovering 211At from the irradiated targets. Setting up and implementing such custom units require long lead times to provide a proper working system. (3) The chemistry of 211At. Compared with radiometals there are no well-established and generally accepted synthesis methods for forming sufficiently stable bonds between 211At and the tumor-specific vector to allow for systemic applications. Herein we present an overview of the infrastructure of producing 211At radiopharmaceuticals, from target to radiolabeled product including chemical strategies to overcome hurdles for advancement into clinical trials with 211At.
Introduction
Targeted α therapy has for several decades attracted interest for therapy of disseminated cancer. The main rationale has been treatment of remaining minimal residual disease after primary treatments such as surgery, external radiotherapy, and/or chemotherapy. 1 –4 After the primary treatment, targeted α therapy has the potential to be a curative treatment for patients, given that proper care is taken when choosing the radioactive nuclide and treatment modality.
There are ∼400 (5-100%) α emitting radionuclides when including isotopes. However, among those only a few fulfill the criteria for nuclear medicine applications, that is, suitable half-life, absence of long-lived and/or toxic daughters, and feasible production of clinically relevant amounts. Narrowing the list to those with a half-life >1 h and no serial decay, which potentially will contribute to toxicity in vivo, only one nuclide remains, that is, astatine-211 (211At).
A few more radionuclides can be considered disregarding serial decay. These are thorium-227 (227Th), actinium-225 (225Ac), and radium-223 (223Ra). 5 –7 Including also nuclides with shorter half-lives, bismuth-213 (213Bi) and the lead-212/bismuth-212 (212Pb/212Bi) system can be added as potential choices. 8,9 All these nuclides have been used in clinical settings but Ra[223Ra]-dichloride (Ra[223Ra]Cl2; Xofigo®) is the only clinically approved α-emitting radionuclide. 10
However, when comparing the α-emitting radionuclides proposed for nuclear medicine applications in detail, 211At still emerges as one of the most promising nuclides. 211At can be produced relatively cost-effectively in reasonable yields with a medium energy cyclotron, comparable with the cost for cyclotron production of, for example, iodine-123 (123I). Despite the fairly straightforward production of 211At and a number of existing possible manufacturing sites, there are few facilities currently producing the nuclide. From this it can be concluded that the problem of 211At availability is not exclusively reflected by the possibility of radionuclide production. 211At needs to be converted into a chemically useful form after cyclotron production and it needs to be coupled to a carrier molecule. A chemical infrastructure must hence also be in place within a reasonable close proximity to the cyclotron production site.
To improve availability and motivate an increased production of the nuclide it is also essential to prove the potential of 211At-radiopharmaceuticals in clinical settings. So far there has been two minor clinical phase I trials completed with 211At-antibody radiopharmaceuticals, mainly financed by academic funding. 11,12 The results from these studies indicate therapy potential with no or very mild toxicity related to the treatment. To further prove the clinical potential of 211At, including progress into phase II/III studies, substantial funding is required in which both academy and industry need to share the economic risk. However, normally the industry needs evidence of the clinical potential before taking financial risk. So, in addition to the described practical hurdles of 211At production and availability, the funding of such research additionally suffers from this chicken and egg dilemma.
It is important to note that the scarce use of 211At is not reflected by difficulties in any of the radiopharmaceutical production steps. The production, isolation, and chemistry are well documented and feasible. However, a complete radiopharmaceutical infrastructure is less widely spread.
In this study we will, despite the hurdles, show a working outline to produce 211At including a chemical infrastructure to provide activities needed for patient treatments, using existing chemistry.
Astatine
Astatine, initially denominated “eka-iodine,” was first discovered in the United States in 1940 where it was produced at University of California, Berkeley through α-particle bombardment of natural bismuth. 13 The astatine isotope with the longest half-life is astatine-210 (210At) with a half-life of 8.1 h. 210At, however, decays by polonium-210 (210Po, an α-particle emitter with a half-life of 138 d), making it unsuitable for medical purposes. Because of the lack of stable or long-lived isotopes, many chemical and physical properties of astatine are still not known but derived from its neighboring halogens. 14,15
When comparing the physical properties of the α-emitting radionuclides proposed for nuclear medicine applications (211At, 227Th, 225Ac, 223Ra, 213Bi, 212Pb/212Bi) in detail, 211At emerges as perhaps the most promising alternative (Fig. 1). 211At decays with a half-life of 7.21 h in two branches: either by α-emission to bismuth-207 (207Bi) or by electron capture (EC) to polonium-211 (211Po). After the EC, 211Po promptly decays by α-emission to stable lead-207 (207Pb). This means that each decay will yield one α-particle. In addition, the EC branch to 211Po will yield characteristic polonium X-rays in the range of 70–90 keV. This enables simple quantification and the energy of the X-rays are in the range where it can be imaged with a γ camera. 16
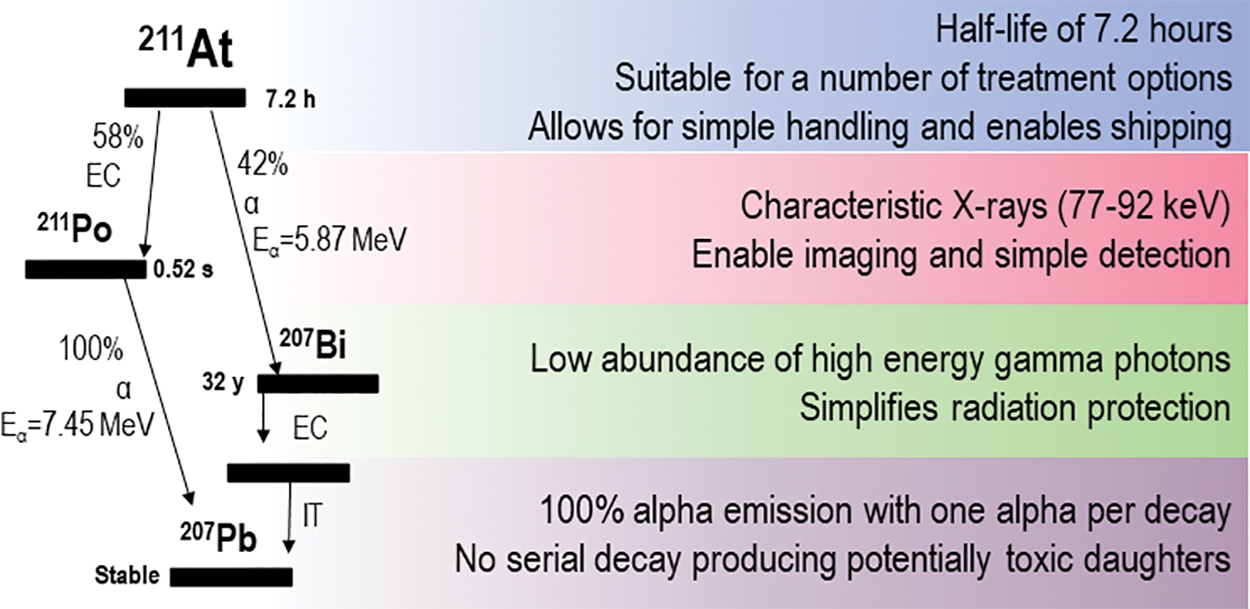
Physical properties of 211At. Color images are available online.
Astatine-209 (209At, half-life of 5.4 h) is another astatine isotope that decays in two branches either by α-emission to bismuth-205 (205Bi, 4%) or by positron emission (β+ decay) to polonium-209 (209Po, 96%). Due to high abundance of β+ it has been proposed as a theranostic pair to 211At. 17 However, as 209At is produced by high-energy proton spallation of actinide targets, 18 production of clinically relevant amounts can be foreseen to be problematic in a global perspective. In this sense, iodine-124 (124I) and 123I are more realistic theranostic pairs for 211At.
Production and the Chemistry Infrastructure of 211At
The overall infrastructure for producing 211At-radiopharmaceuticals can be divided into two main parts: (1) production, including targetry and cyclotron production of the nuclide followed by (2) the chemistry infrastructure, including isolation and work-up of 211At from the irradiated target and synthesis of the 211At-radiopharmaceutical. These steps are illustrated in Figure 2 and discussed in detail below.
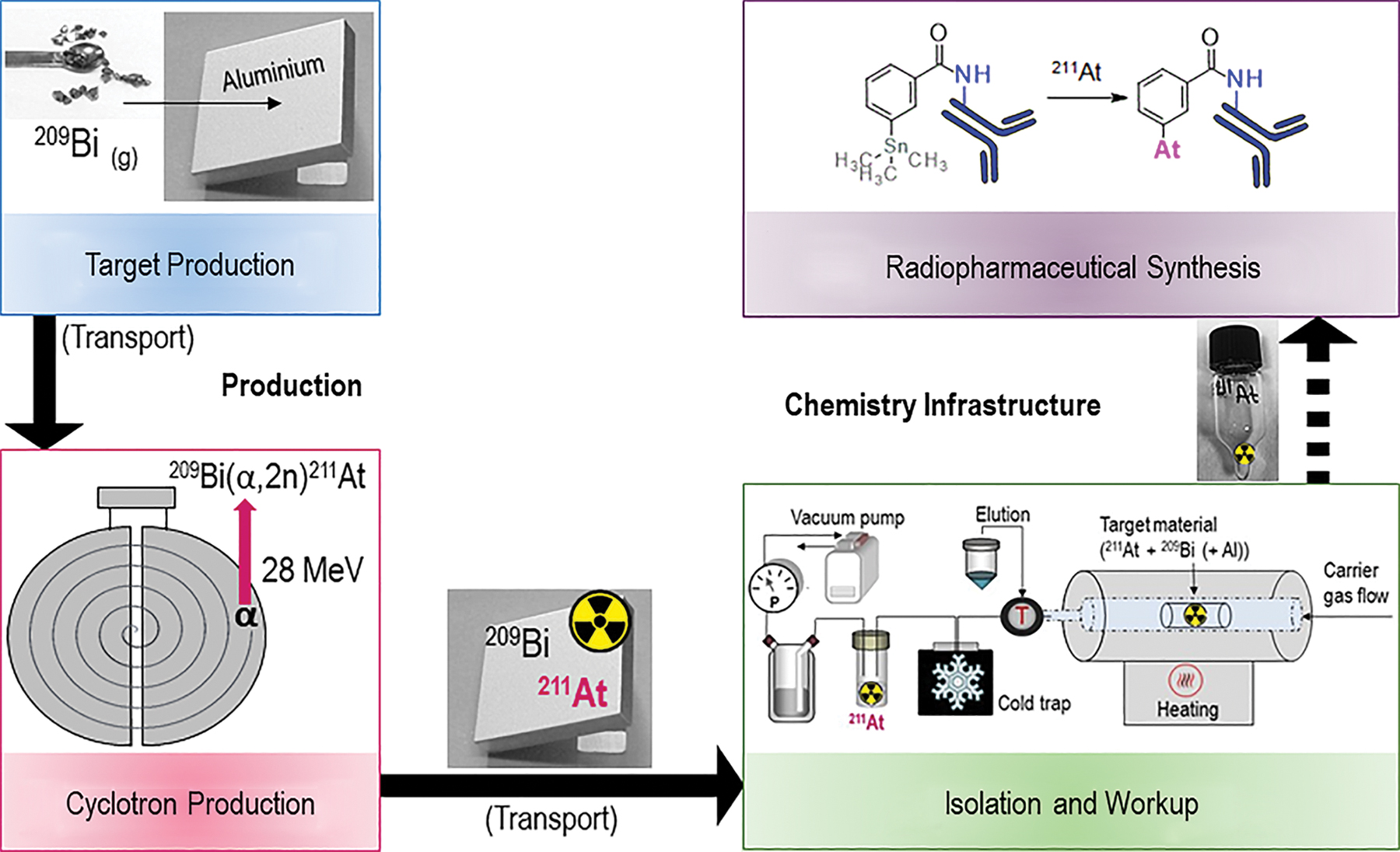
The chemical infrastructure for production of 211At radiopharmaceuticals. Color images are available online.
Targetry and cyclotron production of 211At
There are two possible routes for producing 211At, starting with a bismuth target: either indirectly through a 211Rn/211At generator in which 211Rn is produced by heavy ion irradiation of bismuth-209 (209Bi) through the nuclear reaction 209Bi( 7 Li,5n)211Rn, 18,19 or directly through cyclotron production utilizing the nuclear reaction 209Bi(α,2n)211At. 20 The advantage with the 211Rn/211At generator system is the longer half-life of 211Rn compared with 211At (14.6 h vs. 7.2 h). This will increase the distribution area of 211At from the production site with maintained astatine activity compared with the delivery of 211At alone. Although feasible, this route requires heavy ion high-energy beam irradiations and careful isolation and containment of the produced 211Rn, which is more technically demanding than the latter medium energy cyclotron production route. It is also possible to produce both 211At and 211Rn by high energy proton spallation of actinide targets but radiochemical yields from this production are to this date comparatively low. 21
Although 211At can be produced by heavy ion irradiation or spallation, the main production route has been and still is through the 209Bi(α,2n)211At reaction. The reason for this is the relative simplicity of production, the comparatively high yields, and significantly higher number of available facilities. A previous review by Zalutsky and Pruszynski related to these issues and the worldwide potential of producing 211At showed that there are a number of existing medium energy cyclotrons around the world with the capacity to produce 211At. 22 However, many of the cyclotrons mentioned in that review are old and some of them have been or are under decommissioning. In addition, a few new medium energy cyclotrons have recently been installed or are under installation and cyclotron models that can produce 211At are today available for purchase “off the shelf” from, for example, IBA, Belgium and Sumitomo, Japan. Because of this, an updated, nonexhaustive, worldwide list of cyclotrons capable of producing astatine is provided in Table 1. Astatine production on a regular basis is today mainly pursued in Copenhagen, Denmark, Nantes, France, several sites in Japan (Fukushima, Osaka, Takasaki, Chiba, and Wako Saitama) and at two facilities in the United States, Seattle, Washington, and Durham, North Carolina.
Facilities with Cyclotrons Capable of Producing 211At
The excitation function for the 209Bi(α,2n)211At reaction shows a maximum production yield using an α-beam of 31–35 MeV of the incident α particle. However, the energy should be kept at ∼28 MeV to avoid or minimize the competing reaction 209Bi(α,3n)210At. Coproduction of the 210At is unwanted because of its decay to the potentially toxic daughter 210Po. 23
There are several advantages by using natural bismuth as a target for producing 211At. As bismuth is readily abundant, there will not be any lack of target material, which enables a theoretical indefinite production of 211At. This can be compared with several other α-particle emitters such as 225Ac, 213Bi, and 223Ra, where nuclear legacy material has been initially used for production and where future targetry and/or production of clinical amounts is either limited in availability or complicated by the handling of the starting material. 24 –27
There are two main target set-ups for producing 211At from bismuth, either thin targets for internal cyclotron irradiations or thick targets for extracted beam irradiations. 20,28 For thick target preparation the bismuth can, for example, be electroplated or melted on the backing 29 or used as granulates that are melted during irradiation. 30 Thin targets are mainly prepared using physical vapor deposition of the bismuth on a backing material. 31 In any case, targetry for the production of 211At is a relatively straightforward and cost-effective process. However, one difficulty encountered with using bismuth as target material is its fairly low melting point (271.4°C). This in combination with a low melting and boiling point (Bp) of astatine (302°C and 337°C, respectively) means that an effective cooling of the targets and measures to enclose potentially evaporated astatine during the cyclotron irradiation are required. Therefore, the targets often have liquid cooling on the backside and front-side gas cooling. 32 In addition, melting of bismuth and evaporation of 211At during thin target irradiation can be prevented by application of a fine layer of aluminum on top of the bismuth layer. 33
The efficacy in cyclotron production of 211At is related to the helium ion source, the α-beam energy, the current of the beam, and the cooling of the target. These parameters need to be optimized, taking into account the described physical properties of bismuth and astatine.
As for today the most efficient and highest yields of 211At production reported come from Zalutsky et al. in Durham, NC. 32 They reported a maximum produced amount of 211At of 6.6 GBq at end of bombardment. Providing an efficient chemistry this would allow clinical radiopharmaceutical production for several patients in a single production run, in addition to transport within an area corresponding to several hours of decay from the production site.
Isolation and work-up of 211At from irradiated targets
After production 211At embedded in the bismuth layer of the target must be isolated and converted into a chemically useful form for further chemical processing, that is, labeling and production of astatinated molecules. The isolation from the activated target can be performed either by wet extraction or by dry distillation. 33,34 Using the wet extraction method, the irradiated target is partially or fully dissolved in strong acid, for example, nitric acid. 35 The 211At is then recovered using solvent extraction into an immiscible organic phase. 36 Before the solvent extraction step, it can be necessary to change the aqueous phase by distillation of the dissolution acid. In general the astatine also has to be back extracted from the organic phase or the solvent changed to achieve a chemically useful form for further labeling. 29 This sequential manual processing results in good yields but is time-consuming compared with the short half-life of the nuclide. 29,37 An alternative method to solvent extraction after target dissolution is astatine adsorption on a solid support followed by elution. 37
In dry distillation the irradiated target is heated well above the Bp of 211At (Bp = 337°C) releasing it from the bismuth (Bp = 1564°C), which remain as a solid/liquid. The evaporated 211At can then be condensed in a cold trap or captured through gas scrubbing utilizing different solvents. 38 Before the distillation it is advantageous to remove the activated bismuth layer from the aluminum backing of, for example, a thin target, as this allows the use of a small distillation unit that can fit in a hot cell or a glove box. Furthermore, reducing the volume of target material and preheating the distillation furnace will speed up the subsequent dry distillation, that is, the evaporation of 211At. In this way, dry distillation can be completed within 2 min. 33 Furthermore, if capturing the 211At in a cold trap (as a dry residue) it can be eluted in a small volume of a preferred solvent. In this way the overall preparation time, distillation, and work-up, can be limited to <20 min from the start of the distillation. 33
After distillation care should be taken when selecting the eluting solvent and to the following activity concentration. Chloroform has been found to be a suitable solvent for 211At. It readily solvates high activities of the nuclide and the resulting activity in the solvent is stable for at least a couple of hours. The high vapor pressure of chloroform enables evaporation of the solvent without loss of 211At activity, leaving the astatine as a dry residue. Although the 211At speciation in chloroform is not known, recent results suggest that radiolysis products of chloroform such as chlorine species may be important for the apparent stability of 211At in this solvent. 39 However, it should be noted that distribution ratios for 211At into chloroform from nitric acid are low, making this solvent unsuitable for astatine recovery using wet extraction. 15
Another option for eluting 211At from the cold trap is to use sodium hydroxide or an aqueous solvent containing a reducing agent to define an astatide state. 14 However, by introducing a reducing agent the subsequent halogen chemistry might be compromised. Yet another option is to use methanol. Contrary to chloroform, this may allow for direct labeling of protein conjugates but activity eluted in this solvent should be used immediately as 211At is not fully stable in methanol. Introducing an oxidizing agent, for example, a halogen-based oxidizing agent such as N-chlorosuccinimide, has been found to increase the stability of 211At in methanol. 40,41
In comparison, the dry-distillation method shows some advantages over manual wet-extraction methods. Besides generally faster isolation of 211At and hence also higher yields, distillation configurations with a cold trap enable elution of the 211At in small volumes ready for immediate use in the subsequent chemistry. 33
Independent of method, the isolation and work-up procedure should be performed as to minimize risk of internal contamination from the potentially volatile α-particle emitter, that is, in a sealed environment such as a negative pressure glove box or hot cell. Radiation protection for personnel from the decay of 211At in terms of external shielding, lead-glassed glove box window, or similar, is less important because of the low abundance of high-energy gammas [211At (687 keV) and 211Po (569.7 and 897.8 keV)] besides 211Po X-rays (70–90 keV), giving low external dose.
211At radiopharmaceutical synthesis
This section provides an overview of the different chemical approaches to produce astatinated radiopharmaceuticals. Much of the chemical development of 211At compounds is based on previous inorganic and organic chemistry of astatine and for more detailed information on this topic we like to refer to a comprehensive review by Guerard et al. 42
Astatine is the second heaviest element of the halogen group and it possesses many similar characteristics compared with its neighbor, iodine. In general, halogen properties can be used in astatine-labeling chemistry but there are also some obvious differences between astatine and iodine. For example, unlike iodine, astatine cannot be stably coupled to tyrosine residues of proteins. Instead of binding to tyrosine 211At has been found to form weak bonds with the sulfhydryl groups of cysteine. 43 This apparent instability of the direct 211At-protein bond compared with iodine can be ascribed to the fact that astatine, besides its halogen character, also displays some metallic properties. 44 The metalloid behavior of astatine has led to a few investigations in which its chelating possibilities were evaluated. 45,46 Although demonstrating the metallic properties, no chelate has so far resulted in a sufficiently stable bond with astatine. The development of radiopharmaceuticals with astatine has therefore, instead of using a metal-like chelate, mainly been directed to the formation of covalent bonds such as the aryl–astatine bond. Several routes, based on iodine chemistry, for synthesizing aromatic astatine compounds, have been investigated. These chemical reactions include halogen exchange, 47 diazonium salt reactions, 48 aryliodonium salt reactions, 49 and electrophilic substitution of metal-functionalized aromatic compounds. 50,51 Recently reactions involving boronic esters and boronic acids as leaving groups for aromatic astatination of biomolecules have also been explored. 52 Among the functionalized aromatic compounds, the most common strategy so far has been aryl–tin agents in which the 211At is introduced as an astatine–carbon bond in the aryl ring through an electrophilic destannylation reaction. 51,53,54
It should be noted that the bond strength of the astatine–carbon bond has been calculated to be weaker than the corresponding iodine–carbon bond. 55 The stability of the 211At-aryl carbon bond was recently evaluated by Teze et al. and the results showed that the apparent instability can be attributed to oxidative decomposition, which in vivo particularly occurs in lysosomes after cell internalization. 56 This also relates to the nonresidualizing properties of halogens and may explain the low in vivo stability of small 211At-labeled molecules, which after internalization processes in the kidney are decomposed, resulting in systemic reabsorption of free 211At. 57 To overcome the hurdle of exocytosis of 211At after internalization, a guanidinomethyl-containing reagent that increases intracellular retention of halogens (i.e., iodine and astatine) was developed by Zalutsky and colleagues. 58 The guanidine residue of the astatinated version of this reagent is postulated to sterically prevent cleavage of the 211At carbon bond after cell internalization. 57,59 Pyridine instead of aryl derivatives have also been used in protein halogenation in attempts to increase intracellular retention of 125I and 211At. 60,61
An overview of the different aromatic labeling strategies is given in Figure 3.
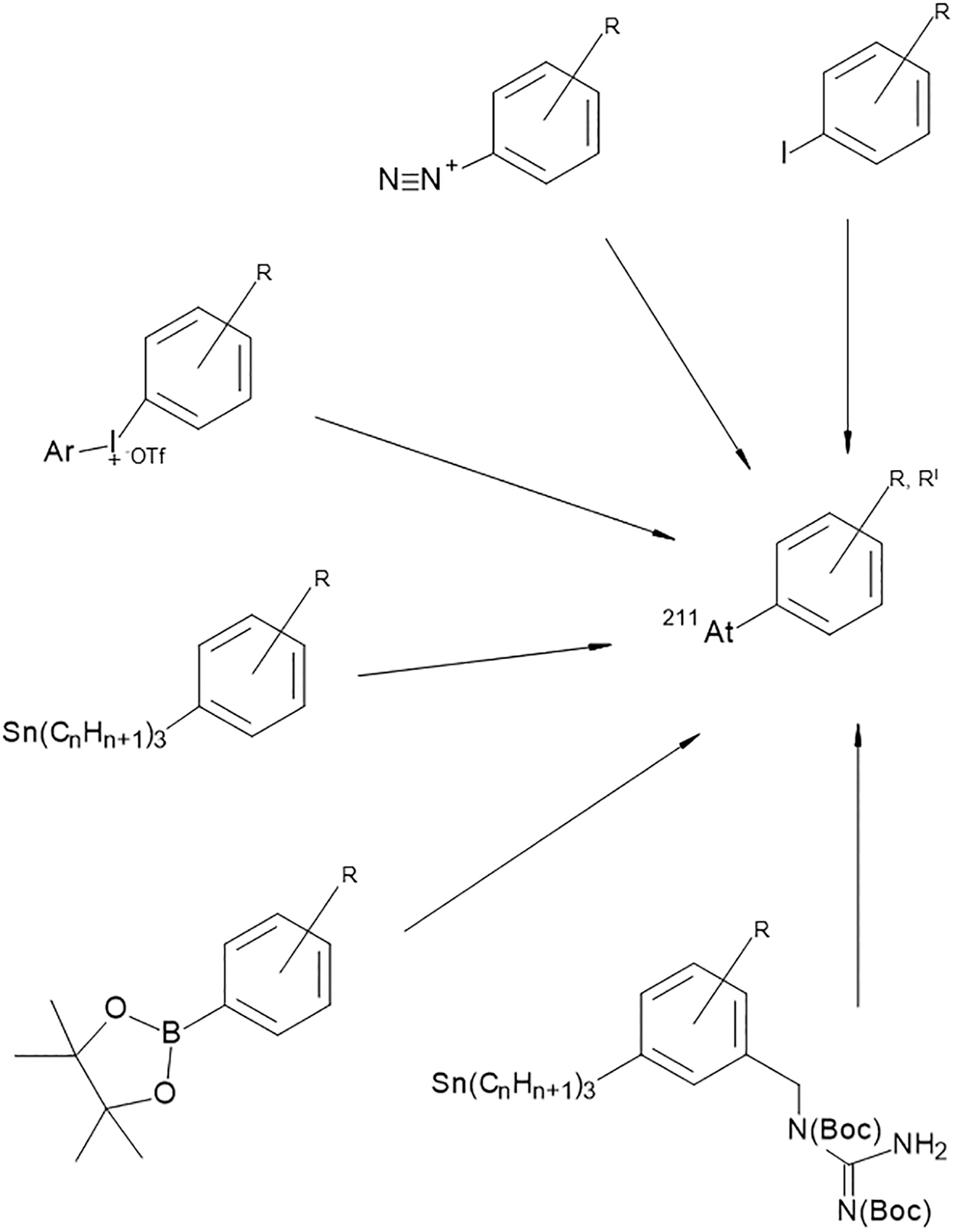
Examples of different aryl functional groups for 211At substitution reactions. From top: Iodine, isotope exchange; diazonium salt; aryliodonium salt; aryl tin; aryl boron pinnacol ester; aryl tin with guanidine moiety.
Despite the relative instability there are some recent reports on aromatic 211At compounds showing favorable in vivo distribution, for example, [211At]-meta astato benzylguanidine, 62 211At-labeled poly (ADP-ribose) polymerase (PARP) inhibitor, 63 and 211At-labeled sigma receptor ligands. 64,65 IgG antibodies have been the main vector for targeted α therapy using 211At and although labeled by aromatic reagents, the 211At antibodies show relative stability in vivo, particularly for intracompartmental applications. 11,12
Higher bond energies compared with the 211At-carbon bond have been shown between boron and astatine. 66 Based on this chemistry, Wilbur et al. developed and investigated several boron cage reagents, nido- and closo-carboranes, for radiohalogenation of biomolecules. 67,68 These reagents have also been implemented in protein labeling and were found to be more stable in vivo compared with the corresponding astatinated proteins labeled by aromatic reagents. 67,69,70
As previously mentioned, 211At cannot be labeled to tyrosine residues of proteins and an intermediate bifunctional reagent utilizing one of the above-detailed astatination strategies is therefore required. It commonly includes an N-succinimidyl ester, isothiocyanate, or a maleimide moiety, which can be coupled to amines or sulfhydryl groups of proteins and peptides. 54,71 –73 The conjugation of these bifunctional reagents to proteins can, depending on the nature of the reagent, be performed in two routes: either two radiochemical steps, astatine labeling to the bifunctional reagent followed by conjugation of the astatinated reagent to the protein, or one radiochemical step where preconjugation to the protein is performed before the astatine reaction (Fig. 4). By preconjugation the protein has been made susceptible for astatination, which is similar to that of labeling with metallic radionuclides where chelates are preconjugated to the protein. When applicable, the later route presents many advantages over the two-step approach, for example, higher radiochemical yields and higher specific activities can routinely be produced. 54,67 In addition, the radiochemical reaction can be completed within a few minutes consequently reducing the absorbed dose in the reaction volume. Including purification, the 211At radiopharmaceutical product can be prepared within 20 min. Using this type of labeling method in combination with recovery from dry distillation, the overall radiochemistry can be completed in <1 h.
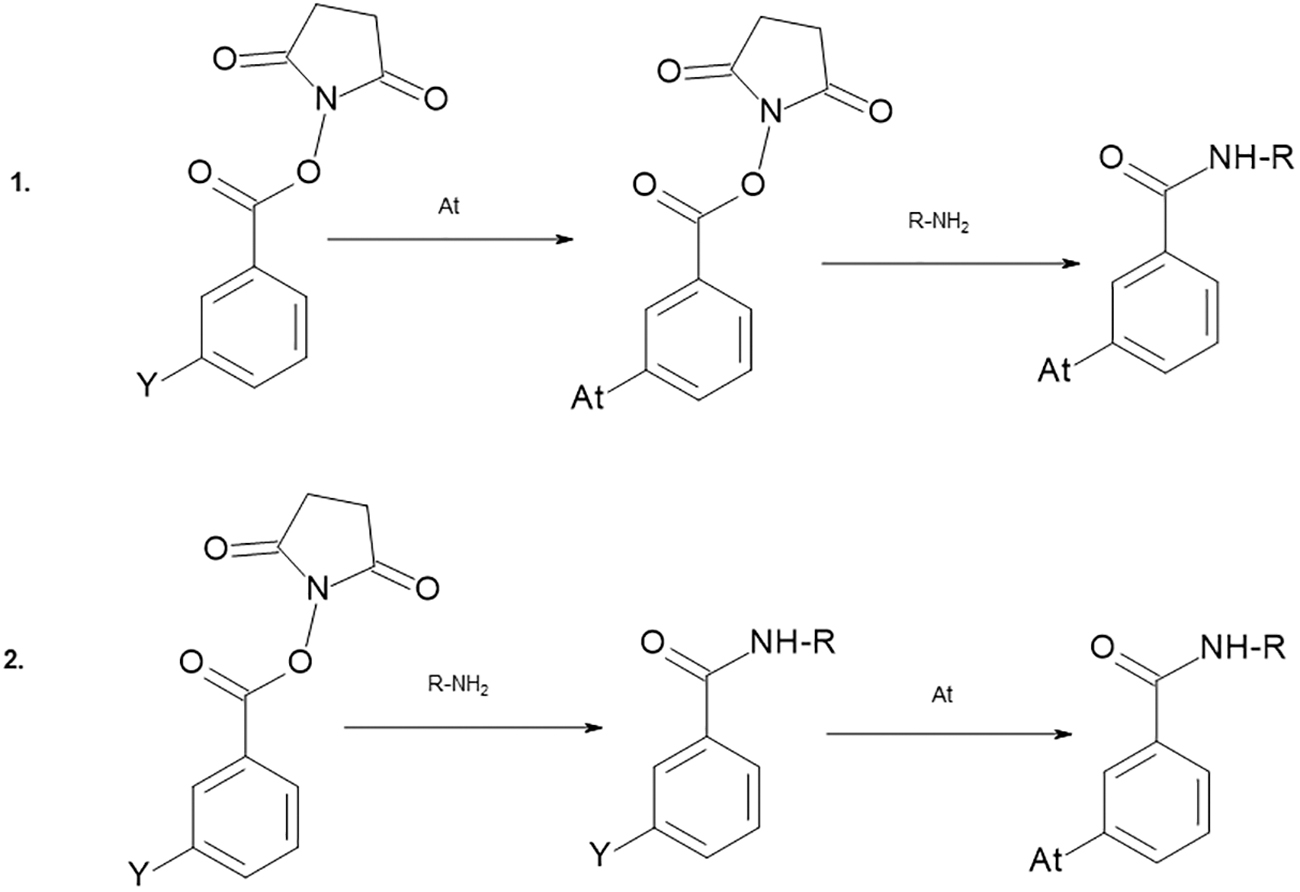
Methods for astatine labeling through a bifunctional reagent carrying an N-succinimidyl ester group for conjugation and a reactive group Y for astatination.
Automatic radiopharmaceutical production
The scarce availability of 211At is not only related to available cyclotrons and cyclotron production capacity. It is also related to the chemistry infrastructure including isolation/work-up after cyclotron production and the subsequent synthesis, which, because of its half-life, needs to be in place within a reasonably short distance to the cyclotron. At the moment all 211At research is based on custom systems for recovery after cyclotron production followed by manual synthesis methods. To meet the anticipated future demand for clinical trials and to increase basic and preclinical research with 211At, the chemistry infrastructure as a whole needs to be developed and significantly improved beyond these systems. One such improvement would be automatic pharmaceutical systems adapted to 211At research, that is, automatic systems for isolation and work-up of 211At after cyclotron irradiation followed by automatic synthesis of 211At radiopharmaceuticals. Automation would not only facilitate the synthesis of 211At compounds and radiopharmaceuticals but also increase the safety of handling 211At and increase reproducibility in pharmaceutical production by reducing the necessity of hands-on manual work and risk of human error.
A challenge for general automation lies in the difference in target configuration between different astatine-producing cyclotrons. The target configuration affects the design of the systems for recovery of 211At from irradiated bismuth targets and is one reason for the existence of different types of custom systems. One way to work around this problem would be to customize an automatic system for a specific target/cyclotron, providing access to astatine and astatinated compounds for several research groups centered around one cyclotron. Another way of circumventing the problem with differences in target configurations is to remove the activated bismuth layer from the backing. This provides a relatively similar starting point for automation independent of the original target design. As mentioned earlier, such a solution also allows for the use of a compact heating system in the recovery of 211At using dry distillation, meaning that the entire automatic system including recovery, work-up, and synthesis, more easily can fit into a glove box or hot cell.
There are three reports on automatic systems for astatine recovery: one platform for the complete synthesis from irradiated target to labeling of biomolecules using dry distillation, 74,75 and two automatic systems for converting 211At into a chemically useful form by wet extraction. 37,76 However, at present, there is no commercial automatic system available for isolation of 211At after cyclotron production and the production of 211At radiopharmaceuticals.
The automatic dry-distillation system utilizes a tube furnace and quartz glass setup for astatine evaporation where volatilized 211At is confined and moved within the system using a reduced pressure applied on the outlet of the system and a light flow of nitrogen on the inlet. The target material is removed from the backing before distillation and is inserted into a preheated distillation furnace. In this way the evaporation process is completed in <30 s. The evaporated 211At is condensed in a cold trap containing a system of capillary tubing and a valve placed between the furnace and cold trap to allow for elution of the activity throughout the capillary. After distillation the recovery of 211At from the activated bismuth is routinely 80%–90%. After recovery from the target the isolated 211At then enters the conjunct synthesis module where steps for synthesis are included such as reagent introduction and purification, which to a high extent depends on the radiopharmaceutical to be produced. The complete setup is controlled by preprogrammed software that can be combined with input from online activity monitoring of the 211Po X-rays from 211At decay. 75
An automatic wet extraction system for recovery of 211At from irradiated bismuth targets was recently reported by O'Hara (US 2018/0308599 A1). 76 In this system, the target is mounted in a target dissolution block in which parts of the target and produced 211At are dissolved by nitric acid. The nitric acid is then distilled and the activity redissolved in hydrochloric acid before being and loaded onto a resin column. The column is washed with an acidic wash solution followed by elution with an eluting solvent. The complete procedure from dissolution is controlled by software. Higher radiochemical yields of 211At isolated from the target and an overall simpler procedure is achieved with this method compared with previously reported manual wet extraction methods.
A similar automated wet extraction method was evaluated by Li et al. 37 The difference from the method reported by O'Hara is mainly the solid support, which in this case is based on tellurium. The irradiated target is dissolved by nitric acid and the nitric acid is converted to hydrochloric acid by addition of hydroxylamine hydrochloride. The solution is then loaded onto the tellurium column. The column is washed and the 211At is eluted with sodium hydroxide. High yields were obtained within a 90- to 100-min preparation time.
The advantage with this system is that it is somewhat less reliant on the target configurations of the cyclotrons compared with the described dry-distillation method.
Clinical Experience with 211At
At Fred Hutchinson Cancer Research Center (Seattle, WA), there is currently a clinical phase I/II study enrolling patients with acute myeloid leukemia and acute lymphoblastic leukemia for treatment with an anti-CD45 antibody, 211At-BC8-B10, before donor stem cell transplant. The study aims to determine side-effects and the best dose of 211At-BC8-B10. There are no results reported from this study yet (NCT03128034).
There are two completed phase I clinical studies using 211At-labeled antibodies: one as a postsurgery boost treatment for recurrent brain tumor and another with intraperitoneal treatment of ovarian cancer patients having relapsed after primary treatment. 11,12 Both these intracavitary studies showed mostly mild (grade I–II) side-effects across all dose levels from the treatment. In both studies, the antibodies were labeled using aromatic bifunctional agents with Stannyl-based according to Figure 3.
In the brain cancer study, performed at Duke University, Durham, North CA, 71–347 MBq 211At-labeled chimeric anti-tenascin monoclonal antibody 81C6 was given locally in the resection cavity followed by salvage chemotherapy. Results from 18 treated patients showed no dose-limiting toxicity. A few patients experienced low-grade neurotoxicity but no grade 3 toxicity was related to the treatment. The study showed that such a local intracavitary therapy in this dose range is safe and may result in a prolonged survival for the patients. 11
In the ovarian cancer trial performed in Gothenburg, Sweden, patients were treated intraperitoneally with 211At-MX35(Fab)2 antibody fragment, targeting the sodium-dependent phosphate transport protein 2B (NaPi2b). Nine women who had relapsed after standard primary treatment were, after treatment by salvage chemotherapy in complete, or near complete, remission, subsequently treated with 211At-MX35(Fab)2 in 1–2 L of peritoneal dialysis fluid (icodextrin 7.5%). By blocking the sodium/iodide transporter with potassium perchlorate in sodium iodide symporter-expressing organs such as the thyroid and stomach, a significant reduction in uptake of free 211At in the thyroid could be recorded. After the initial nine patients, an additional three patients were treated after a change in the protocol regarding preparation of the radiopharmaceutical. The synthesis route was simplified through preconjugation of the antibody fragments compared with previous two-step labeling, as given in Figure 4, enabling higher radiochemical yields and consequently higher activity preparations. The results from the 12 patients in total showed that there was little escape of activity from the peritoneal cavity during a 48-h time period, that is, most of the 211At activity decayed in the abdominal cavity. Dosimetric evaluation of organs at risk showed that this treatment can be delivered without causing any acute toxicities. Indeed, most toxicities recorded were low grade (I–II) and mainly related to the treatment procedure, with no observed hematological toxicity, thus no dose-limiting toxicity was reached. 77
Despite low risk of acute toxicity from 211At therapy, long-term stochastic risks may arise and should be considered, particularly for an upfront adjuvant therapy. 78 Weighing dose to various size microtumors 79 with recorded patient dosimetry 12 and calculated long-term stochastic risks, 78 an activity concentration of 200 MBq/L of 211At-labeled antibody may be a justifiable dose in future clinical intraperitoneal trials. 77
Discussion
Targeted α therapy is an emerging strategy for treatment of cancer, where the main rationale is treatment of small-scale disease, considering the high linear energy transfer and short path length of the α-particles. Therefore, α-particle therapy can be envisioned as an adjuvant treatment after standard primary treatments with, for example, surgery and chemotherapy. However, no such upfront targeted α-therapy setting for minimal residual cancer exists today. The only approved α-emitting radiopharmaceutical today is Xofigo, that is, [223Ra]RaCl2, which is used for palliative treatment of bone pain resulting from skeletal metastases in disseminated castrate-resistant prostate cancer. 80 Another α-emitting radionuclide that currently is intensively investigated for treatment of cancer is 225Ac. 81,82 Similar to 223Ra, 225Ac has frequently been clinically used on late-stage castrate-resistant prostate cancer. 83,84 Yet another radionuclide that has recently attracted interest for targeted α-therapy is 227Th, the rationale being an in situ generator of 223Ra. 85 At present, patients are enrolled in a phase I study for treatment of certain mesothelin-expressing cancers using 227Th (BAY2287411).
These radionuclides, 227Th, 225Ac, and 223Ra, are serially decaying radionuclides and moving treatment upfront to treat minimal residual metastases into a curable therapy would not be feasible. It is difficult to control the radioactive daughter distribution after decay of these radionuclides and therefore it may result in increased normal tissue toxicity and an increase in the risk for secondary cancer development. 86 –88
At present, 211At is the only radionuclide for targeted α therapy that can foreseeably safely meet the rationale of upfront treatment on minimal residual cancer. “Upfront” here means treating patients in complete remission after, for example, surgery and chemotherapy, before any sign of relapse. If the relapse frequency is <100%, this suggests that already-cured patients would be subjected to the targeted α therapy. 211At only releases one α particle per decay and does not result in any toxic α- or β−-emitting daughter nuclides. From toxicological and dosimetric perspectives, this is advantageous over the serially decaying nuclides described previously. However, one α-particle per decay means that less energy per nuclide can be deposited at or near the tumor site, compared with multiple α-particle-decaying radionuclides, and suggests that higher administered activities would be required.
211At has been applied in two clinical studies. 11,12 In Gothenburg, Sweden, 12 ovarian cancer patients were treated intraperitoneally with 211At-MX35(Fab)2 in a phase I study. Results indicate a well-tolerated treatment with no side-effects from the radiation. 77 The results proposed that calculated curative doses would be safe and adequate to administer to patients using existing labeling chemistry. 79 Based on the results from the phase I study, initiation of an upfront phase II/III trial to determine the effect of the treatment is motivated.
A phase II/III trial including a large number of patients will require improvement in the availability of 211At and a highly developed chemical infrastructure. This means that besides targetry and cyclotron production, an efficient system for converting 211At into a chemically useful form and subsequent chemical synthesis to produce the radiopharmaceutical are required.
A common way of facilitating chemical synthesis in nuclear medicine applications is automation utilizing radiopharmaceutical synthesizers, for example, in the production of [ 18 F] FDG or 68 Ga-DOTA-octreotate. In this way the synthesis will facilitate reproducible and repeatable production. An automatic approach would also be a possible solution to the chemistry infrastructure problem presently limiting 211At research. A system that automatically converts 211At into a chemically useful form after cyclotron production 37,75 and automatically produces the radiopharmaceutical 74 would meet the safety requirements in handling 211At and facilitating larger clinical trials and multicenter studies. Standardization and reliable quality production of 211At and a chemical infrastructure is a requirement for future routine hospital production of 211At radiopharmaceuticals. One may envision several different approaches for implementing the logistics of the supply of 211At and the subsequent synthesis of the 211At radiopharmaceutical. Besides optimization and improvement of the cyclotron production, a delivery chain must also be established. Target distribution from cyclotrons to surrounding treatment centers within a circa one half-life radius followed by automatic on-site production would most likely allow for the best use of the produced activity, limiting radiolysis of the product. In densely populated areas, or where larger hospital and producing cyclotrons are located in close proximity, target work-up and pharmaceutical synthesis at the cyclotron facility may also be an alternative.
An additional issue that needs to be addressed is the apparent in vivo instability of 211At compounds, particularly important for general systemic applications with this nuclide. Although relatively high stability of astatinated compounds is often observed in vitro, deastatination may occur in vivo, predominately when small targeting vectors that are metabolized by the kidneys are used. 56,89 The reabsorption of free 211At after sequestering of 211At molecules by the kidneys is believed to be related to intracellular oxidative decomposition after exocytosis and subsequent systemic recycling. 56,90
There are intense efforts made to improve the in vivo stability of 211At compounds to enable general nuclear medicine applications. 66,91,92 Many of the hampering features of 211At relate to its halogen characteristics. Like iodine, astatine is nonresidualizing after cell internalization and instability is to some extent related to intracellular decomposition and exocytosis of free 211At. Efforts to improve retention of 211At intracellularly have been made by utilizing different modifications of the aryl ring. 58,61 Despite the stability issue, relative stability of a few 211At compounds, proteins, or small molecules labeled by aromatic intermediates have been observed and may be explained by fast kidney clearance in relation to slow or noninternalization properties. 63 –65,93 In addition, 211At-labeled antibodies synthesized with existing well-established chemical methods show sufficient stability in intracavitary applications and allow for treatment in several different cavity modalities such as intraperitoneal, intrathecal, and in the blood compartment following, for example, leukemia or lymphoma.
Conclusion
In this review we show that despite existing challenges, there have been major steps taken in research for nuclear medicine applications with 211At, that is, the feasibility of producing 211At and the implementation of systems for target work-up and chemical synthesis. By providing a worldwide supply chain of 211At utilizing existing and new installations of cyclotrons and implementation of an efficient chemistry infrastructure, the main obstacles with this nuclide can be overcome and hence the demand concerning 211At radiopharmaceuticals for research and clinical trials could both be met and significantly enhanced.
Footnotes
Disclosure Statement
The authors E.A. and S.L. are the inventors of the Patent WO2015/195042 Automatic Process Platform for the Production of Astatine-211 [211At]-Radiopharmaceuticals.
Funding Information
This study was supported by the Swedish Research Council, the Swedish Cancer Society, the King Gustav V Jubilee Clinic Research Foundation, and grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALFGBG-435001).