
Abstract
In patients with metastatic melanoma, sequential single-arm and randomized phase II trials with a therapeutic vaccine consisting of autologous dendritic cells (DCs) loaded with antigens from self-renewing, proliferating, irradiated autologous tumor cells (DC-TC) showed superior survival compared with similar patients immunized with irradiated tumor cells (TC). We wished to determine whether this difference was evident in cohorts who at the time of treatment had (1) no evidence of disease (NED) or (2) had detectable disease. Eligibility criteria and treatment schedules were the same for all three trials. Pooled data confirmed that overall survival (OS) was longer in 72 patients treated with DC-TC compared with 71 patients treated with TC (median OS 60 versus 22 months; 5-year OS 51% versus 32%, p=0.004). Treatment with DC-TC was associated with longer OS in both cohorts. Among 70 patients who were NED at the time that treatment was started, OS was better for DC-TC: 5-year OS 73% versus 43% (p=0.015). Among 73 patients who had detectable metastases, OS was better for DC-TC: median 38.8 months versus 14.7 months, 5-year OS 33% versus 20% (p=0.025). This approach is promising as an adjunct to other therapies in patients who have had metastatic melanoma.
Introduction
It has been suggested that autologous tumor cells (TCs) that are self-renewing and proliferating in tissue culture may be the best source of tumor-associated antigens (TAA) for active specific immunotherapy (ASI). 1 This source of TAA addresses issues of tumor heterogeneity, quantity of antigen, unique antigens that are patient specific, and current concepts of tumor stem cells, or tumor initiating cells. 2 For more than two decades, we have been investigating the use of autologous TC lines as the source of TAA in therapeutic vaccines for patients with metastatic cancer, especially metastatic melanoma. 3 –9 Such cell lines express various melanoma differentiation antigens 6 and certain antigens that may be specific for melanoma stem cells. 10 –13 Preclinical studies showed that immunity against cancer stem cells can be induced, 14 and that targeting a subpopulation of such cells can result in eradication of tumor. 15
Results of three successive phase II trials in patients with metastatic melanoma were previously reported. 4,5,9 Between 1990 and 2001, 74 metastatic melanoma patients were injected with irradiated TCs in conjunction with injections of granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon gamma (IFN-γ), and other adjuvants (trial #1). 4 The objective response rate (ORR) was 3/35 (9%) for patients with measurable or evaluable disease. Progression-free survival (PFS) was only 4.5 months, but median overall survival (OS) was 20.5 months and projected 5-year OS 29%, which was considered encouraging because contemporary patients with metastatic melanoma had a median OS of less than 1 year, and 5-year OS of only about 10%. 16 Survival was much better in patients who had no evidence of disease (NED) at the time that vaccine therapy was initiated, compared with those who had measurable or evaluable disease. Survival was similar whether patients received GM-CSF and/or IFN-γ as an adjuvant, but their survival was superior to that of patients who received other adjuvants or no adjuvant. 4 In a phase II trial in which patients were randomized to receive GM-CSF or IFN-γ as adjuvants, there was no difference in OS. 17
Between 2001 and 2007, 54 metastatic melanoma patients were injected with a dendritic cell (DC)-TC product consisting of autologous DCs that had been incubated with irradiated autologous TC, which were suspended in GM-CSF for injection (trial #2). 5 No objective responses were recorded for 15 patients with measurable disease per response evaluation criteria in solid tumors (RECIST). 18 The median PFS was again less than 5 months, but median OS was 5 years with a 54% projected 5-year survival rate at the time of publication. 5 The OS for patients treated with DC-TC was superior to OS for TC-treated patients in the previous trial. Patients with measurable disease had an inferior OS.
To establish whether vaccination with DC-TC (DC loaded with TAA ex vivo) was associated with better survival than injections of TC (relying on in vivo loading and expression of TAA by endogenous DC), a randomized phase II trial was conducted between 2007 and 2011 (trial #3). 9 Both the DC-TC and TC products were suspended in 500 μg of GM-CSF for injection. In this study, 24 patients were randomized to the TC arm, and 18 were randomized to the DC-TC arm. All patients were treated as per randomization with the imbalance serving as a consequence of early termination of the trial. Although determination of tumor response rate was not a major objective of this trial, one patient in the DC-TC arm had a delayed complete response of five sites of measurable disease, 7 and the response is known to have persisted beyond 4 years. 9 Survival was much better in the DC-TC arm with 2-year survival rates of 72% versus 31%.
Whether the apparent benefit of DC-TC over TC is evident regardless of tumor burden was not addressed in the randomized trial (#3) because of the small sample size (n=42). 9 It has been suggested that vaccines may be efficacious only in patients with a minimal tumor burden because of issues of immunosuppression, antigen heterogeneity, and the time needed to mount an effective immune response. 19 To address whether DC-TC was associated with longer survival than TC in patients with different degrees of tumor burden, we pooled data from all three trials, 4,5,9 which was felt to be reasonable since the key eligibility criteria for all three trials were the same, as were the treatment schedules. In all three trials, OS was one of the prospective endpoints, a secondary endpoint for the first two trials, and the primary endpoint for the randomized phase II trial.
Materials and Methods
Vaccine products and administration
The methods for producing the TC and DC-TC products were previously described. 4 –6,9 The protocol and operating procedures for manufacturing each patient-specific product were reviewed by the United States Food and Drug Administration in association with Biologics Branch Investigational New Drug (BB-IND) 5838 and BB-IND 8554. The methodology for establishing cell lines was the same for the three trials. The intent was to have each injection of either product potentially present antigens from about 10 million TCs. Each injection of TC product contained the antigens from about 10 million irradiated TCs. Each DC-TC product was produced by incubating 200–300 million DC with about 100 million irradiated TC and then divided into 10 aliquots. Therefore, the final cell numbers injected were greater for injections of DC-TC than TC.
For the three trials, the cell lines and vaccine products were manufactured in the Cell Biology Laboratory of the Hoag Cancer Center in Newport Beach, California, which operated between 1990 and 2011. The success rate for establishing cell lines was ∼50%, and it took a median of 4 months to establish a cell line. 6 In the three trials, cryopreserved vaccine products were thawed and administered subcutaneously (SC) per the same planned schedule of injections: once a week for 3 weeks, and then monthly for 5 months at weeks 8, 12, 16, 20, and 24. In the two trials in which TC were injected, individual doses ranged from 2 to 24 million cells (average 10 million). In the two trials in which DC-TC were injected, individual doses ranged from 5 to 35 million cells (average 14 million). There was substantial unintentional inter-patient variability in doses, but very little intra-patient variation among doses. The inter-patient variability in TC doses and DC-TC doses is the result of biologic differences among patients, including viability of TCs after irradiation and thawing, variations in numbers of peripheral blood mononuclear cells obtained during leukapheresis, and the number of DC emerging from culture in interleukin (IL)-4 and GM-CSF.
Patients
As per the Declaration of Helsinki, the three trials were conducted with the approval of appropriately constituted institutional review boards for the protection of human subjects. All patients gave written informed consent at the time of study entry. In the three trials, eligibility was limited to patients with metastatic melanoma for whom a cell line was established from tissue obtained at the time of surgical resection of a metastasis. 4,5,9 The same eligibility and ineligibility criteria were used in the three trials; none of the trials used extent or location of metastases to restrict participation. There was no restriction regarding prior or subsequent therapies, but concurrent anti-cancer therapy was not allowed. Patients with brain metastases were eligible if lesions had been resected or controlled with surgery, radiation, or stereotactic radiotherapy. Patients who were treated with ASI were enrolled or referred by their managing physician at a time when that physician felt the treatment was the most appropriate.
Subset populations
Subsets were defined by whether patients received the DC-TC or TC product, and by extent of disease at the time of enrollment for vaccine treatment. Extent of disease was defined as NED if the patients had no clinically or radiographically detectable metastatic melanoma as opposed to detectable disease, which included both measurable and non-measurable lesions. An additional population was defined by whether detectable disease was classified as measurable as per RECIST. 18
Statistics
Comparisons of proportions were made using the chi-square test. Averages were compared using the student's t-test. Hazard ratios for deaths were calculated using the proportional hazards assumption. Kaplan–Meier survival curves were compared using the Mantle–Haenszel log-rank test based on monthly events from initiation of treatment.
Results
There were 74, 54, and 42 patients who had enrolled in the three successive trials for a total of 170 patients. 4,5,9 Figure 1 shows the derivation of the four different patient subsets that were the focus of this analysis. Based on previous analyses that showed inferior OS for patients who did not receive GM-CSF or IFN-γ as an adjuvant and/or did not receive a minimum of the first three injections, 4 27 such patients were excluded from this analysis. In a randomized phase II trial of the Cancer Biotherapy Research Group (CBRG), 98 patients, including 51 with melanoma, were randomized to receive injections of TC along with injections of GM-CSF (n=49) or IFN-γ (n=49); there was no difference in survival or immune response. 17 However, in trial (#1), there was a dramatic difference in survival, 26% versus 0% 5-year OS for 53 patients who received GM-CSF and/or IFN-γ as an adjuvant compared with 21 patients who did not (p<0.001). 4 For this reason, in an effort to create a more closely matched group of patients, we excluded five TC patients who did not receive the first three weekly injections, and 20 TC patients who received neither GM-CSF nor IFN-γ as an adjuvant, which left us with 49 of the 74 patients. We also excluded 2 out of 24 TC patients from trial #3 who failed to receive the first three weekly injections. Thus, all 27 patients who were excluded came from the TC cohort: 7 were NED, and 20 had detectable disease. These excluded patients had a median survival of only 8.4 months, with 1-, 2-, and 3-year survival rates of only 44%, 26%, and 15%, respectively. Exclusion of the poor-prognosis 27 TC-treated patients left us with 49 TC patients from trial #1 and 22 TC patients from trial #3 for a total of 71 patients in the TC cohort for a comparison to the 72 DC-TC-treated patients.
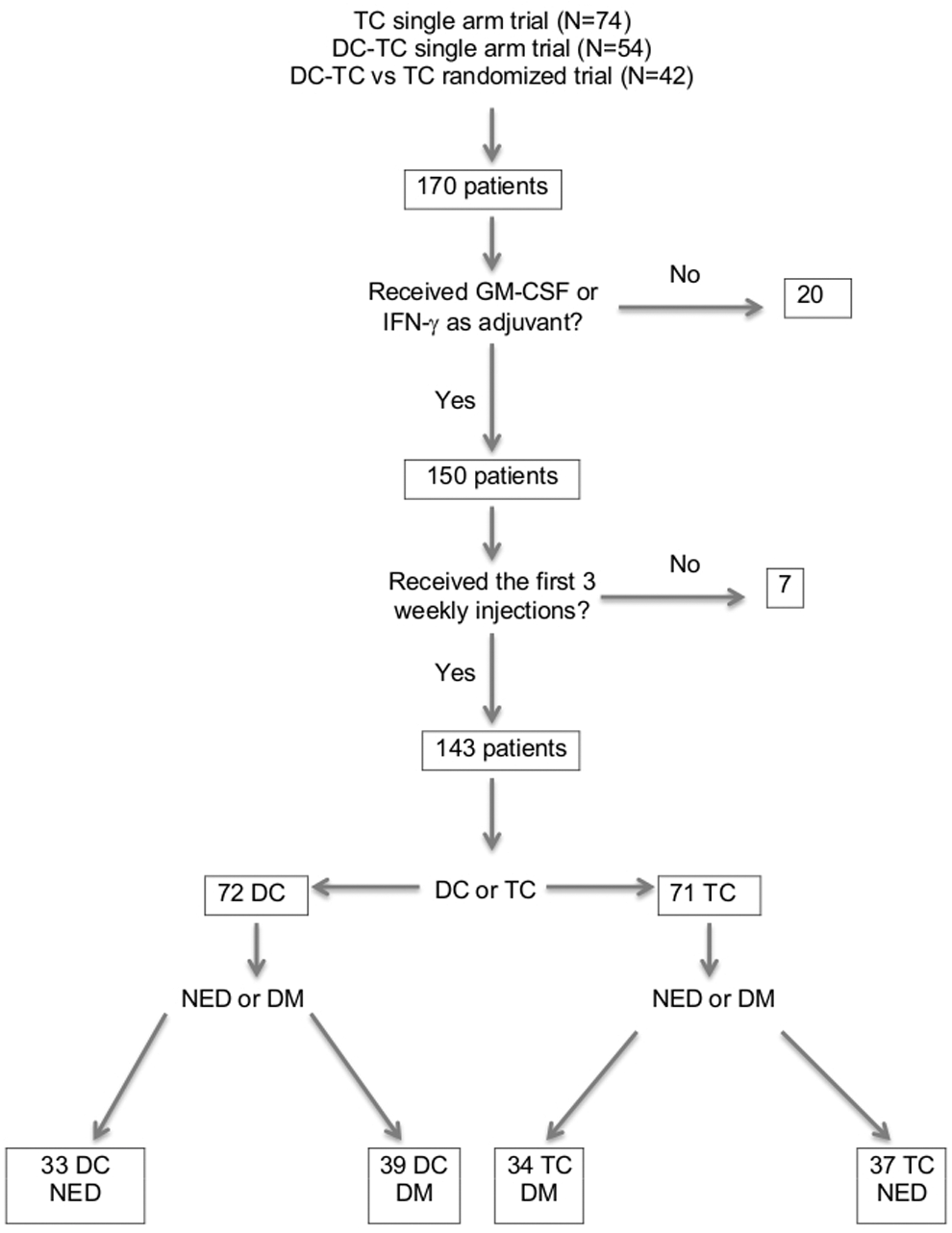
Pooled data and algorithm to derive subsets of patients by active specific immunotherapy (ASI) treatment with dendritic cell-tumor cell (DC-TC) vaccine or TC vaccine, and stratified by no evidence of disease (NED) or detectable metastases (DM).
Characteristics of the 72 DC-TC patients and 71 TC patients are shown in Table 1. There were no apparent differences between the cohorts in age, gender, performance status, most advanced stage of disease before vaccine therapy, or stage of disease at the time that vaccine therapy was initiated, although a somewhat higher proportion of DC-TC patients had M1c disease, and a smaller proportion were NED at the time of treatment. The DC-TC patients received a higher average number of injections, but the reason that patients received fewer than the eight planned injections was progressive disease in all except for one DC-TC patient who had known cardiovascular disease and died of a cerebrovascular event after three injections. All DC-TC patients received GM-CSF as an adjuvant compared with 62% of TC patients, but as noted earlier, a randomized trial revealed no survival differences for patients treated with GM-CSF compared with IFN-γ as an adjuvant with the vaccine. 17 There were no differences among survival curves on the basis of quartile divisions by the number of cells injected for either the TC or DC-TC subsets of patients. Baseline lactate dehydrogenase (LDH) serum levels were not required or recorded for patients enrolled in trial #1 conducted during 1990–2001. Only 4 out of 24 TC patients from trial #3 had an elevated LDH, but 2 were excluded from this analysis because they did not receive the first three weekly injections. In contrast, LDH was elevated in 25 out of 54 DC-TC patients in trial #2, and in 7 out of 18 DC-TC patients in trial #3. Thus, a total of 32 out of 72 (44%) of DC-TC patients had an elevated LDH at the time of study entry. In trial #3, 4 out of 24 (17%) TC patients and 7 out of 18 (39%) DC-TC patients had an elevated LDH.
M, distant metastases; M1a, skin and/or soft tissue; M1b, lung±skin and/or soft tissue; M1c, visceral or elevated LDH with M1a or M1b; ECOG PS, Eastern Cooperative Oncology Group performance status; SD, standard deviation; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon gamma; DC, dendritic cell; TC, tumor cell; LDH, lactate dehydrogenase.
Figure 2 displays the survival curves for pooled DC-TC versus pooled TC. Survival was longer in the patients who received DC-TC with a median survival of 60 months versus 22 months, 2-year survival of 71% versus 49%, and 5-year survival of 51% versus 32% (p=0.004). Trial #3 had also shown a 2-year survival advantage for DC-TC (72% vs. 31%, p=0.007). 9
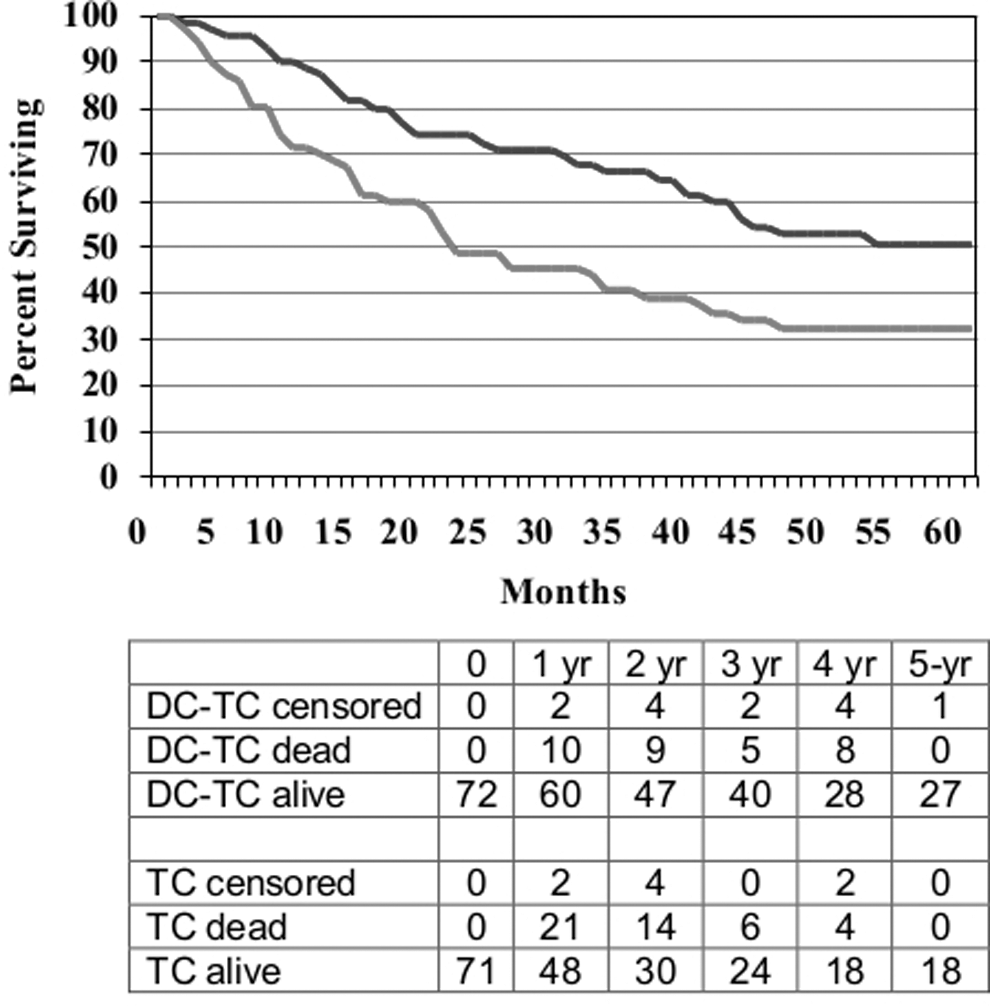
Overall survival (OS) for patients with metastatic melanoma who received either DC-TC (n=72) or TC (n=71), HR=0.57, p=0.004.
The difference in survival for patients based on whether or not they were NED at the time of treatment is shown in Figure 3. As expected, survival was much better for those who were NED at the time that ASI was initiated with a median survival not reached at 5 years versus 25.5 months, and 5-year survival of 56% versus 27% (p=0.001).
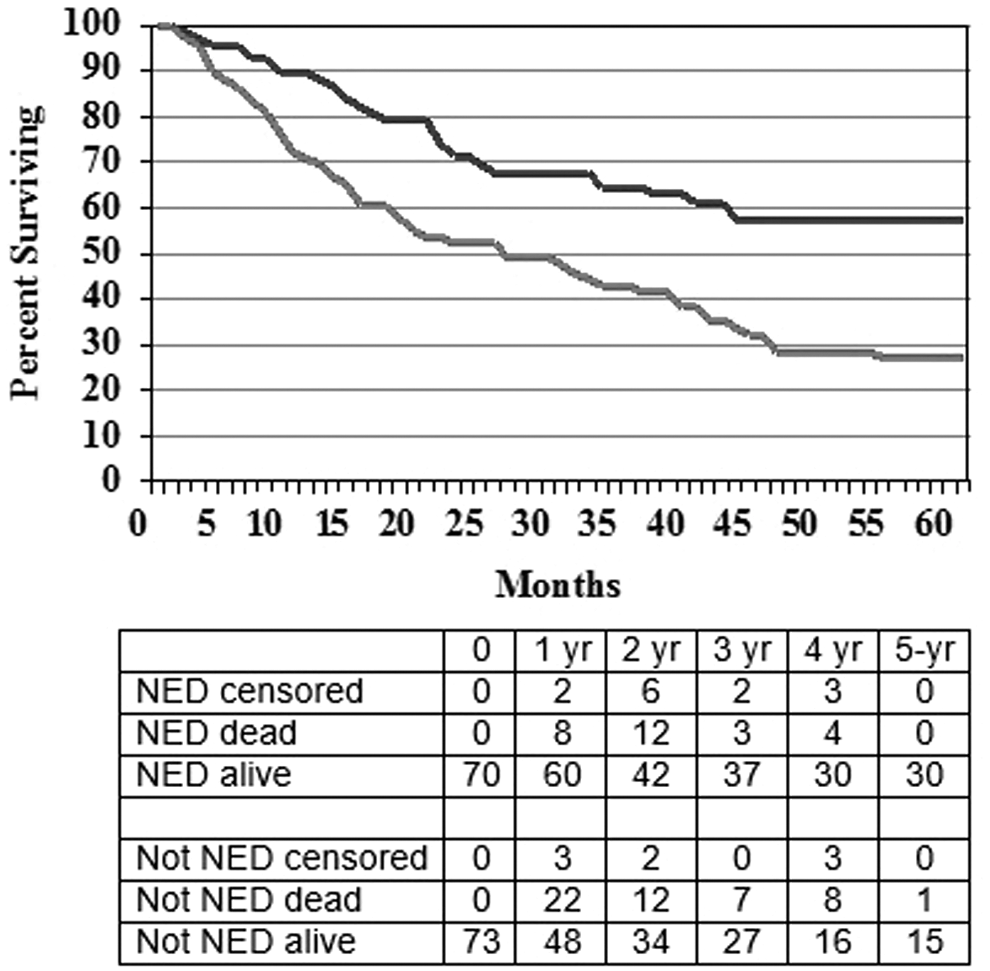
OS for patients with metastatic melanoma who at the time of treatment had NED (n=70) compared with those who had detectable disease (n=73), p=0.001.
The DC-TC versus TC comparison for the NED subset of patients is shown in Figure 4, and for the non-NED subset in Figure 5. Median follow up was more than 5 years for all cohorts, and no patients were lost to follow up. For the 70 patients who were NED when ASI was initiated, survival was better for those who received DC-TC with median OS not reached versus 32.2 months and 5-year survival rates of 73% versus 43% (p=0.015, HR=0.39). In the subset of non-NED patients enrolled in the randomized trial, there was a similar benefit for 8 patients treated with DC-TC compared with 11 treated with TC (p=0.005). For the 73 patients who were not NED at the start of treatment, that is, they had measurable disease or detectable disease that was considered non-measurable or equivocal, survival was also better for those who received DC-TC with median survival of 38.8 months versus 14.7 months and 5-year OS 33% versus 20% (p=0.025, HR=0.66). In the subset of NED patients enrolled in trial #3, limited data showed a similar benefit for 10 patients treated with DC-TC compared with 13 treated with TC (p=0.11).
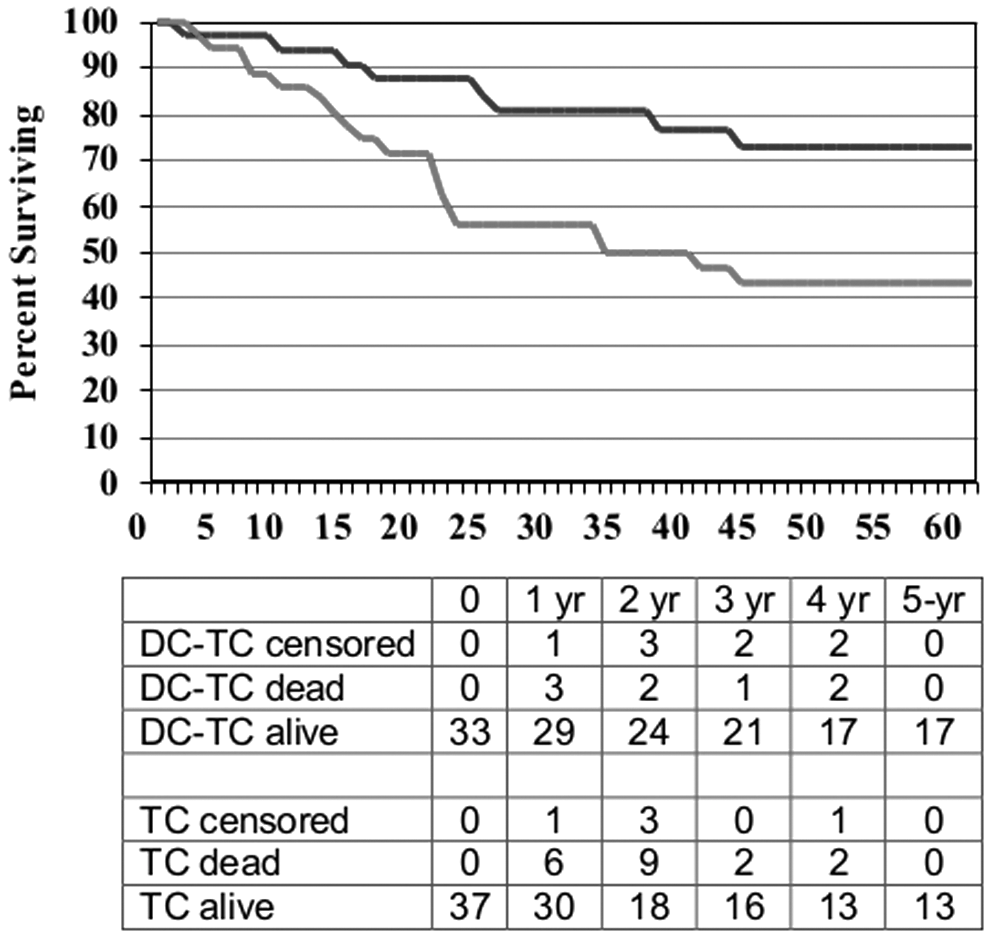
Survival curves for patients who had evidence of disease at the start of ASI by treatment product: DC-TC (n=33) versus TC (n=37), HR=0.39, p=0.015.
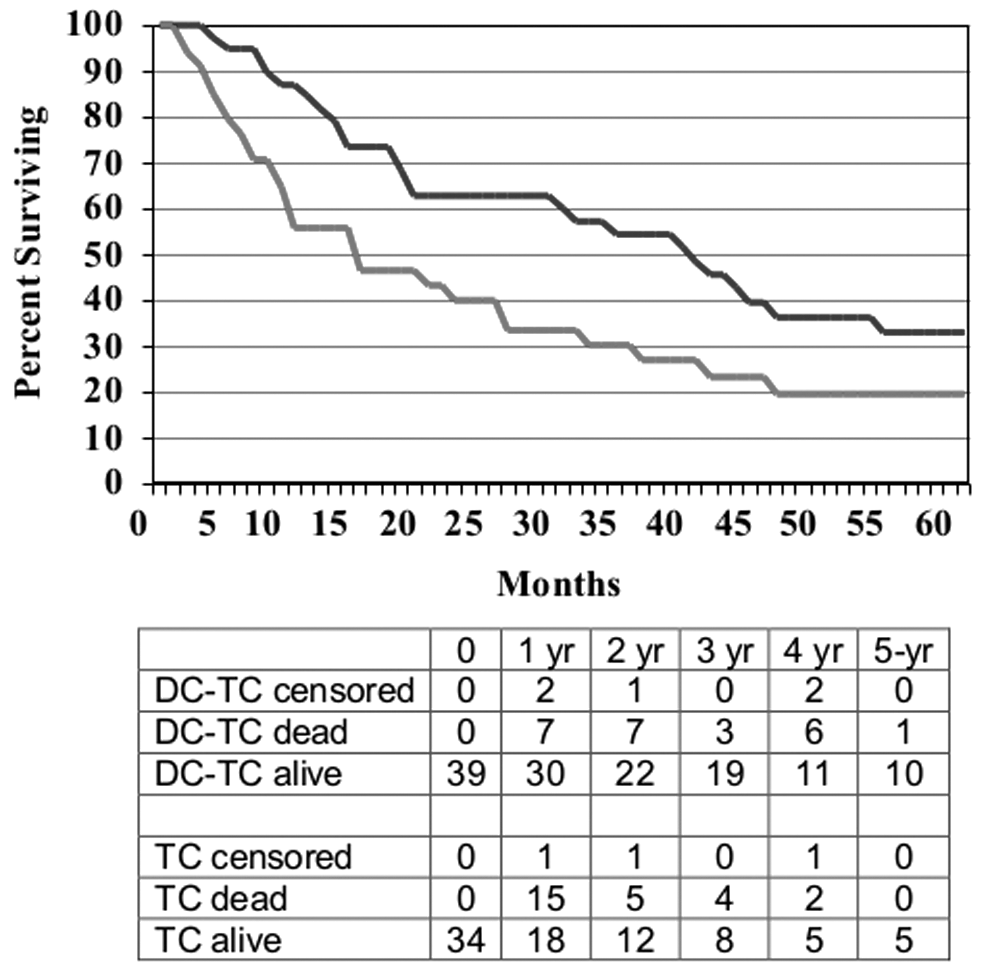
Survival curves for patients who had detectable metastatic melanoma at the start of ASI by treatment product: DC-TC (n=39) versus TC (n=34), HR=0.66, p=0.025.
The question of benefit for patients with measurable disease was more problematic, because tumors were characterized as evaluable/measurable by those other than RECIST in trial #1, while RECIST were used in trials #2 and #3, which included the DC-TC products. 5,9 There were 15 DC-TC patients in trial #2 who had measurable disease by RECIST; in trial #3, there were 8 DC-TC patients and 9 TC patients who had measurable disease by RECIST at the time that treatment was started. The 23 DC-TC patients with measurable disease had a median survival of 18.8 months, a 2-year survival of 46%, and 5-year OS of 21% compared with a median of 6.5 months and 5-year OS of 11% for the 9 TC patients with measurable disease (p=0.039, HR=0.80).
Discussion
The pooled data from these trials suggest that the DC-TC product is associated with longer OS than the TC product in patients with metastatic melanoma, both in the subset of patients who were NED at the time that ASI was initiated and in patients who were not-NED, that is, they had detectable disease, including measurable, non-measurable, and equivocal disease. The results for these 143 patients is consistent with the results observed in small subsets in trial #3. 9 Importantly, the pooled data suggest that there is benefit in both patients who have been rendered free of disease, typically by surgical resection of metastatic sites, and in patients who had recurrent or persistent detectable metastatic disease at the time of ASI. It is less clear whether there is a benefit in patients who have measurable disease, but the data in patients whose tumor burden was defined using RECIST (trials #2 and #3) suggest there is benefit in this subset of patients as well. There are insufficient data to address whether there is a benefit across the various subsets of measurable distant metastatic disease, that is, soft tissue or lymph node metastases (M1a), lung metastases with or without soft tissue or lymph node metastases (M1b), or other visceral sites of disease or any metastatic sites plus an elevated LDH (M1c).
The major limitation of this study is that it relies on pooled data and sequential trials rather than concurrent trials or randomized trials. However, the three trials had the same eligibility/ineligibility criteria, and variables that were previously found to be associated with worse survival (not receiving the first three injections and/or not receiving GM-CSF and/or IFN-γ) were used to exclude 27 TC-treated patients who had a worse outcome. The analysis of these larger numbers of patients yielded results that are consistent with what was observed in the small subsets of patients randomized in trial #3.
Both ASI products used in these trials utilized TAA derived from autologous, proliferating, self-renewing TCs rather than allogeneic cell lines or peptide antigens. Technological advances have made it possible to prove that individual tumors contain tens to thousands of non-synonymous mutations that may be perceived as foreign by the host immune system. 20 It has also been shown that such mutations result in antigens that are unique to individual patients and that can be targeted with millions of T cells administered with IL2 to produce therapeutic benefit in patients whose immune systems have been ablated with chemotherapy and radiation. 21 The use of proliferating cells may facilitate exposure to antigens that are cell-cycle specific, and the use of self-renewing cells may facilitate exposure to antigens that are associated with tumor initiation. 2
Historically, the greatest limitation for this patient-specific autologous cell line approach was the difficulty in establishing autologous TC lines, and the length of time needed for such success. During the conduct of the three trials pooled for this report, the cumulative success rate for establishing melanoma TC lines was 300 out of 617 (49%) and a median of 4 months to establish. Recent advances in tumor stem cell media formulation appear to have greatly increased the probability of successfully establishing a cell line and the rapidity of establishing a cell line. Enzyme-digested, cryopreserved tumors that were unable to be grown using previous methodology have been successfully grown within a few weeks with specific stem cell media (Andrew Cornforth, pers. comm.). These improvements in manufacturing should greatly increase the ability to conduct clinical trials with such products and enhance commercial feasibility. 22
An important issue is what might be the appropriate clinical niche for such a therapeutic vaccine. The distribution of patients in trials #2 and #3 (n=96) was ∼45% NED, 35% measurable, and 20% equivocal or detectable, but non-measurable as per RECIST. This ASI should be viewed as potentially additive or synergistic with any anti-melanoma therapy that can reduce tumor burden, but it is unlikely to be associated with long-term survival. Such modalities include surgical resection of metastatic melanoma, radiation therapy of limited disease, IL2, inhibitors of enzymes associated with BRAF mutations, monoclonal antibody checkpoint inhibitors, and, occasionally, chemotherapy.
Five-year OS rates after surgical resection of melanoma metastases range from 20% to 30% depending on location, whether or not there is more than one metastatic site resected, and other aspects of patient selection. 23,24 More recently, patients rendered surgically free of metastatic disease and then treated with vaccines consisting of peptides, Bacille-Calmette-Guerin (BCG), or BCG with allogeneic cell lines have had 5-year OS rates of 40%–45%. 25,26 In our pooled data, the 5-year OS rates were 43% for TC and 73% for DC-TC. There is no standard adjuvant therapy for patients who have undergone such resections, although GM-CSF has been used in this setting. 27
Enhancing long-term endogenous immune anti-tumor effects and overall long-term survival rates is increasingly the goal of immunotherapy. 28 Vaccine approaches that enhance immune recognition of TAA is one such strategy. The anti-cancer benefit associated with IL2, ipilimumab, and antibodies to programmed death receptors (PD1) or their ligands (PDL1) is dependent on recognition of TAA by the patient's immune system. The 5-year OS for metastatic melanoma patients who are healthy enough to receive treatment with high-dose IL2 is about 15%. 29,30 It was recently reported that in IL2-treated melanoma patients, 5-year survival was 39% for patients treated with IL2 and TC or DC-TC, compared with 13% for those who received only IL2. 31 Ipilimumab is associated with 5-year OS of about 20% as initial therapy, and in previously treated patients. 32
The anti-PD1 antibodies nivolumab and pembrolizumab will likely be associated with 5-year survival rates that are greater than 30%. 33,34 Studies in B16 melanoma tumor-bearing mice suggest that the benefits of anti-checkpoint antibodies combined with anti-tumor vaccines are greater than either product alone. 35,36 Theoretically, whether administered before, concurrent with, or after IL2, ipilimumab, nivolumab, pembrolizumab, or related immunotherapies, this ASI product may add to the survival benefit associated with such therapies because of the potential to induce or enhance long-term anti-tumor immunity to TAA that was previously not recognized or only weakly recognized.
The combination of BRAF and MEK enzyme inhibitors has produced rapid ORRs in a high percentage of metastatic melanoma patients with PFS about 9–10 months, and fewer secondary cutaneous malignancies are reported when an MEK inhibitor is combined with a BRAF inhibitor. 37 –39 However, because of the toxicities associated with these agents, 40,41 and the relatively rapid emergence of resistant disease, these agents have had little, if any, impact on 5-year survival rates. There remains an unmet need for a minimally toxic adjunctive therapy that can enhance or maintain the therapeutic benefit derived from the enzyme inhibitors.
Footnotes
Acknowledgments
This work was supported by the Cancer Biotherapy Research Group (CBRG) and Hoag Hospital Foundation. The authors would like to acknowledge the laboratory contributions in the manufacturing of these patient-specific products by Shankar Nayak (1990–1997), Patric Schiltz (1995–2006), Senthamil Selvan (1999–2008), and Andrew Cornforth (1995–2002, 2007–2011). They would also like to thank biostatisticians Curtiss Church and David Jackson for their assistance, respectively, with calculations of log-rank tests and hazard ratios.
Disclosure Statement
Dr. R.O.D. served as Chief Medical Officer for California Stem Cell, Inc. (CSC) Irvine, CA, which in 2011 acquired the intellectual property associated with the autologous vaccines described in this article. In May 2014, Caladrius Biosciences, Inc. (at the time named NeoStem, Inc.), acquired CSC. Dr. R.O.D. is now an employee of Caladrius Biosciences, Inc., and owns stock in that company. Dr. R.O.D. and/or other immediate family members have stock in other companies that make anti-melanoma products, including Bristol-Myers Squibb, Glaxo-Smith-Kline, and Pfizer. Dr. R.O.D. serves as a consultant to Prometheus, Inc., San Diego, CA. and participates in a speakers' bureau for Genentech. Dr. E.F.M. participates as a member of the Genentech Speakers Bureau. Dr. L.S.S. is an advisor to Bristol-Meyers-Squibb and participates in a speakers' bureau for Genentech. All other authors declared no conflicts.