
Abstract
Introduction:
Viloxazine extended-release (VLX-ER) is effective as monotherapy for attention-deficit/hyperactivity disorder (ADHD), and is often tried as an add-on treatment when psychostimulant therapy fails to provide an adequate treatment response. This phase 4, open-label study evaluated safety, tolerability, and efficacy of VLX-ER with optimized psychostimulants in pediatric participants with ADHD. Morning versus evening VLX-ER use was also evaluated.
Methods:
Children and adolescents (6–17 years) experiencing inadequate psychostimulant response (investigator-assessed ADHD Rating Scale-5 [ADHD-RS-5] score ≥24 and Clinical Global Impression-Severity of Illness [CGI-S] scores ≥3) during a 4-week screening period received flexibly-dosed VLX-ER, taken once daily in the morning (weeks 14) or evening (weeks 5–8), concomitantly with a psychostimulant. Safety (primary outcome) and efficacy were evaluated relative to baseline.
Results:
Fifty-six participants (26 children; 30 adolescents) enrolled, and 48 (85.7%) completed the study. Combination therapy was well tolerated, with only two participants (3.6%) withdrawing due to adverse events (AEs). The most commonly reported AEs were headache (17.9%), decreased appetite (12.5%), and upper respiratory tract infection (10.7%). Mean ± standard deviation investigator-assessed ADHD-RS-5 scores (baseline: 37.2 ± 8.4) improved progressively by −13.5 ± 9.7 points at week 4 and −18.2 ± 10.0 points at week 8 (p < 0.0001 each). Likewise, CGI-S scores (baseline: 4.4 ± 0.6) improved by −0.9 ± 0.9 at week 4 and −1.4 ± 1.1 at week 8 (p < 0.0001 each). Parent-assessed scales, including ratings of morning and evening ADHD behaviors and sleep disturbances, showed significant improvement relative to baseline regardless of morning (week 4) or evening (week 8) VLX-ER dosing.
Conclusion:
Combined treatment with VLX-ER and psychostimulant therapy showed acceptable safety and tolerability, with improvement in morning and evening ADHD behaviors and sleep disturbances relative to stimulant monotherapy. Timing of VLX-ER administration (morning or evening) did not appear to affect safety, drug response, or sleep improvement.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) affects 11.4% of children and adolescents in the United States (Danielson et al., 2024). When pediatric ADHD is not treated, inattentive and/or hyperactive/impulsive behaviors can negatively impact functional, occupational, academic, and social outcomes that may persist into adulthood (Hamed et al., 2015; Harpin et al., 2016; Shaw et al., 2012).
Psychostimulants have served as a primary ADHD treatment for several decades and are a recommended pharmacological treatment by clinical practice guidelines (Faraone et al., 2021; Schein et al., 2022a; UK NGC, 2018). Although psychostimulants are effective in mitigating ADHD behaviors for the majority of children with ADHD (Childress and Tran, 2016), 25%–38% may not experience adequate symptom response with psychostimulant monotherapy (Mattingly et al., 2013; McCracken et al., 2016). In one study of 779 children with ADHD primarily treated with stimulants, only 30.8% of participants achieved “complete symptom control” (Hodgkins et al., 2013).
Psychostimulants may also be limited by delays to affect onset following morning dosing as well as rebound effects at the end of the day (Cascade et al., 2008; Childress and Tran, 2016). Currently, no FDA-approved stimulants provide 24-hour ADHD symptom control, therefore, coverage for early morning or evening symptoms may be inadequate (Childress and Tran, 2016). Use of psychostimulants as a monotherapy may be limited by intolerable side effects (reduced appetite, headache, sleep disturbances, abdominal pain, and increased heart rate and blood pressure) or by potential misuse and abuse (Briars and Todd, 2016; Eiland and Gildon, 2024; Miech et al., 2023; U.S. Department of Health and Human Services SAMHSA, Center for Behavioral Health Statistics and Quality, 2020; Wolraich et al., 2019).
Nonstimulants can be used as ADHD monotherapy or combined with psychostimulants when either treatment alone does not provide adequate response (Briars and Todd, 2016; Schein et al., 2022b). Large, U.S.-based studies evaluating concomitant medication use in pediatric ADHD show psychostimulants and nonstimulants were used concomitantly in ∼10%–17% of children and ∼10% of adolescents with ADHD (Schein et al., 2022a; Zhou et al., 2020). However, few clinical studies have systematically evaluated their safety and efficacy in pediatric ADHD (Bahn and Seo, 2021).
Viloxazine Extended-Release [VLX-ER capsules; QELBREE®, (Supernus Pharmaceuticals Inc, 20225)] is an FDA-approved nonstimulant medication with demonstrated safety and efficacy in randomized, double-blind, placebo-controlled trials of pediatric and adult ADHD (Johnson et al., 2020; Nasser et al., 2022; Nasser et al., 2021a; Nasser et al., 2020; Nasser et al., 2021b; Nasser et al., 2021c). Follow-up long-term safety studies in children and adults demonstrated persistent efficacy over one or more years. Phase 1 studies in healthy adults of VLX-ER combined with methylphenidate or lisdexamfetamine dimesylate (Cutler A, 2023; Faison et al., 2021a; Faison et al., 2021b; Findling et al., 2023) showed preliminary safety of combined use. Based on substantial evidence of safety and efficacy for VLX-ER monotherapy, combined with a low likelihood of interaction with psychostimulants, this trial evaluated the safety and efficacy of polytherapy in children and adolescents with ADHD with suboptimal responses to current psychostimulant treatment regimens.
Materials and Methods
Study design and objectives
This phase 4, open-label, flexible-dose safety trial was conducted at 17 U.S. research centers [ClinicalTrials.gov identifier: NCT04786990 (2024)]. The primary objective was evaluation of the safety and tolerability of VLX-ER administered with methylphenidate or amphetamine to treat ADHD in children and adolescents (6–17 years). Secondary objectives were to evaluate the efficacy of combined VLX-ER and psychostimulant use, and the safety, tolerability, and efficacy of AM (morning) or PM (evening) administration of VLX-ER.
The protocol, protocol amendments, and informed consent form were reviewed and approved by a central institutional review board before study initiation, and each participant’s parent or legal guardian/representative provided written informed consent prior to enrollment; participants provided assent where appropriate. The study was performed in accordance with good clinical practice (GCP) guidelines as required by the Declaration of Helsinki, the International Conference on Harmonization guideline for GCP, and all applicable local and national legal guidelines.
Participants
Eligible participants were children and adolescents (6–17 years) with a primary diagnosis of ADHD per the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition [DSM-5 (APA, 2013)] criteria, confirmed by the Mini International Neuropsychiatric Interview for Children and Adolescents [MINI-KID (Sheehan et al., 1998)] at screening, and experiencing inadequate efficacy despite using a stable psychostimulant regimen (at least 5 days/week at an optimized dose) for at least 2 weeks. Inadequate efficacy was defined as having an ADHD Rating Scale-5 (ADHD-RS-5) (investigator assessed) (DuPaul et al., 2016) total score ≥24 and a Clinical Global Impression-Severity of Illness (CGI-S) (Guy, 1976) score ≥3 (mildly ill or worse) at both screening and baseline. Participants must have been willing to stay on this optimized stimulant dose for the entire study, be medically healthy based on screening tests, and functioning at an age-appropriate level intellectually. Unless sterile, sexually active males and females of childbearing potential agreed to use pre-specified, highly effective contraceptive methods throughout the clinical trial.
Individuals actively participating in another clinical trial or who participated 60 days prior to screening, had a history of VLX-ER hypersensitivity, a current diagnosis of a major psychiatric disorder (other than ADHD), or intellectual disability were excluded. In addition, those with a history of a disease/disorder, head trauma, or condition (e.g., encephalopathy) that would likely affect central nervous system functioning, interfere with study treatment, take priority over ADHD treatment, or could affect compliance and/or interpretation of results were excluded based on investigator judgment. Individuals were excluded if they had a history of suicidality, current suicidal ideation/behavior on the Columbia Suicide Severity Rating Scale (C-SSRS) (Posner et al., 2011), a history of substance use disorder or a positive urine screen for substances other than psychostimulants prescribed to treat ADHD, taking additional medication to treat ADHD (e.g., nonstimulants), or a history of physical, sexual, or emotional abuse (<1 year prior to screening). Other exclusion criteria were resting blood pressure or pulse rate >95th percentile, history of cardiac abnormalities, uncontrolled thyroid disorder, cardiovascular disease, cerebrovascular disease, abnormal screening laboratory or exam results that might interfere with safety, or use of any investigational or prohibited concomitant medications, including use of certain CYP1A2 substrates (e.g., theophylline, melatonin) within 28 days of baseline.
Intervention
The treatment period was 8 weeks of VLX-ER added to the participant’s current ADHD psychostimulant regimen (Fig. 1). Parents/guardians recorded ADHD medication use and timing in a daily paper or electronic diary. Psychostimulant dosage was to remain stable during the treatment period, but could be decreased once, if necessary. In children (6–11 years), the VLX-ER starting dose was 100 mg/day during week 1 and could be optimized to 100 mg/day/week within a range of 100–400 mg/day. In adolescents (12–17 years), the VLX-ER starting dose was 200 mg/day during week 1 and could be optimized by 100 or 200 mg/day/week within a range of 100–600 mg/day. Participants were instructed to take VLX-ER in the morning (AM) during weeks 1–4 and in the evening (PM) during weeks 5–8 (∼12 hours later than the AM dosing time).
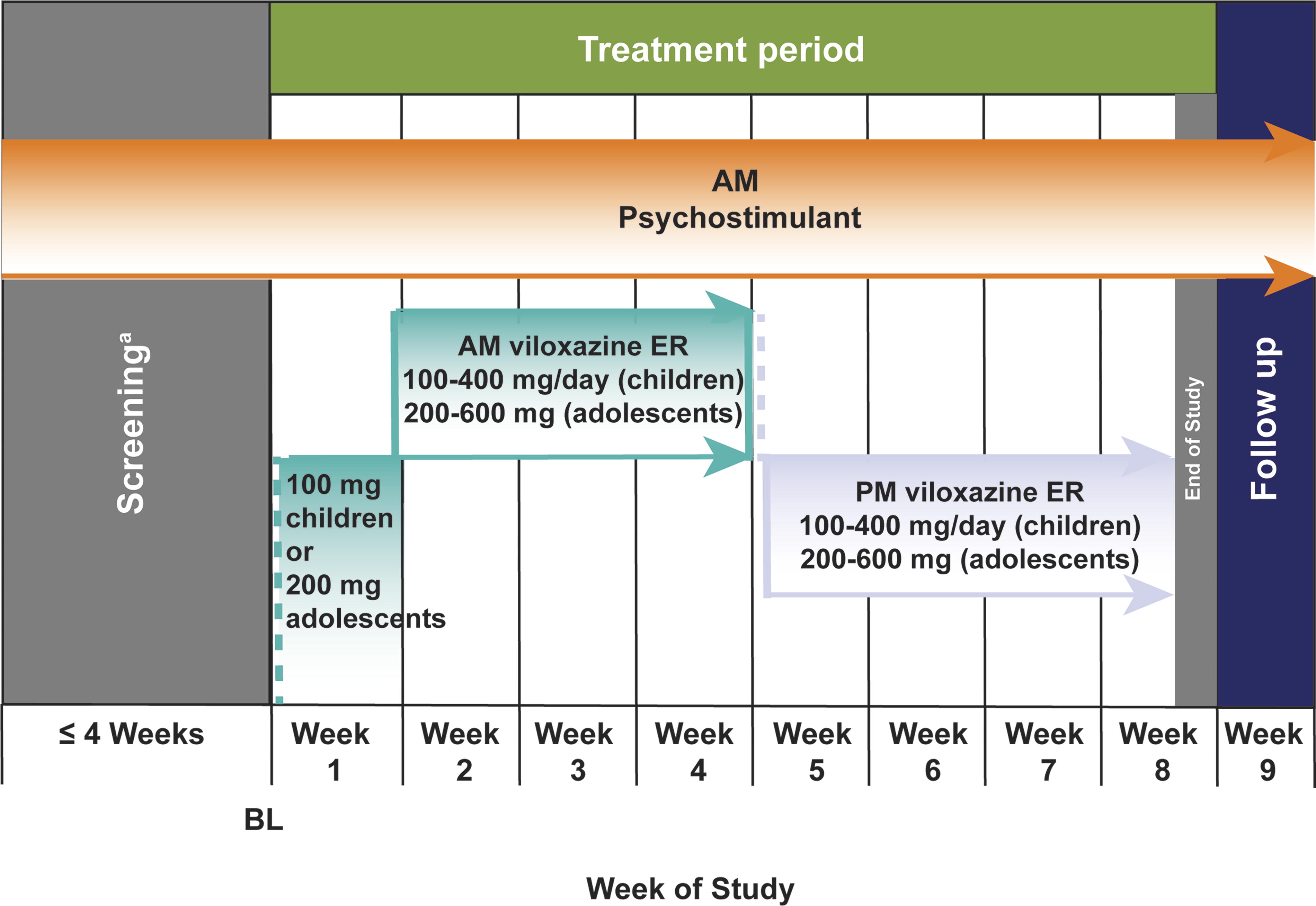
Study design. ER, extended-release.aScreening included a 7 day washout period immediately prior to baseline during which participants discontinued any prohibited concomitant medication(s) that could interfere with assessment of safety, efficacy, or tolerability per investigator judgement (see methods section for full list of prohibited medications).
Safety and efficacy endpoints
Safety endpoints (primary assessment) included adverse events (AEs), clinical safety laboratory tests, vital signs, weight, height, body mass index (BMI), electrocardiograms (ECGs), physical examination, and C-SSRS scores. Weekly caffeine intake was collected as VLX-ER may slow caffeine metabolism because it is a strong CYP-1A2 inhibitor. Except for physical examinations and urinalysis conducted at screening and week 8 (or early termination) and serum pregnancy tests (conducted only at screening), safety endpoints were assessed weekly.
Efficacy endpoints included the ADHD-RS-5 (investigator assessed) and the morning and evening versions of the ADHD-RS-5 (parent assessed) (DuPaul et al., 2016), CGI-S and CGI-Improvement (CGI-I) scales (Guy, 1976), the Weekly Parent Rating of Evening/Morning Behavior-Revised (WPREMB-R) (Faraone et al., 2018; Sutton et al., 2003) scale, and the Sleep Disturbance Scale for Children (SDSC) (Bruni et al., 1996; Mancini et al., 2019).
The child and adolescent versions of the ADHD-RS-5 are 18-item scales that directly correspond to the 18 ADHD symptoms listed in the DSM-5 (APA, 2013; DuPaul et al., 1998; DuPaul et al., 2016). Each item is rated from 0 (none) to 3 (severe) for a total score between 0 and 54. Scale items can be further separated into 9-item hyperactivity/impulsivity and inattention subscales. Morning and evening ADHD-RS-5 versions required the parent to rate scale items based on the child’s behavior in the morning hours and evening hours, respectively.
The WPREMB-R is a validated, 11-item scale assessing three morning and eight evening ADHD-related behaviors’ impact on activities of daily living (Faraone et al., 2018). Each item is rated from 0 (no difficulty) to 3 (a lot of difficulty), for a total score between 0 and 9 for morning behaviors and 0 and 24 for evening behaviors.
The CGI-S and CGI-I are 7-point scales that provide a standalone assessment of the clinician’s view of the participant’s ADHD symptom severity and improvement/worsening with treatment, respectively. The CGI-S is scored from 1 (normal/not at all ill) to 7 (extremely ill), and the CGI-I from 1 (very much improved) to 7 (very much worse) (relative to baseline).
The SDSC is a validated, 26-item, parent-completed rating of sleep disturbances over the past week across six dimensions. SDSC scores range from 26 (no sleep disturbances) to 130 (maximum). SDSC raw scores are converted to T-scores, where a T-score >70 (>95th percentile) is considered indicative of a clinically significant sleep problem.
ADHD-RS-5 and CGI scores were assessed by investigators at weekly visits. The WPREMB-P, SDSC, and morning and evening ADHD-RS-5 (assessed by parents and/or guardians) were evaluated at baseline, week 4, and week 8. Medication diaries were collected at each visit. A urine drug screen was performed at each visit to confirm psychostimulant use, and a confirmatory blood sample was collected to determine psychostimulant and VLX-ER concentrations.
Statistical analysis
Power or sample size calculations were not performed for this open-label study. To achieve a desired completion rate of ∼50 participants, assuming a 20% early discontinuation rate, study enrollment was planned for 60 participants.
All statistical analyses and data processing were performed using SAS version 9.4 or higher. Safety and efficacy endpoints were summarized overall and by period (weeks 1–4 and 5–8) based on observed values for all participants who received ≥1 VLX-ER dose (Safety Population). Safety outcomes were analyzed and reported descriptively. Efficacy outcomes were reported as change from baseline (CFB) except for CGI-I, which is already assessed relative to baseline and reported descriptively. Nominal p-values were calculated to assess treatment differences relative to baseline at each visit and for week 4 relative to week 8 using paired t-tests. The proportion of participants meeting predefined responder thresholds for ADHD-RS-5 (investigator assessed) (defined as ≥30% and ≥50% reductions from baseline) and CGI-S and CGI-I (defined as a score of 1 or 2) was also calculated. Response rates for these thresholds were reported by treatment week, and compared between week 4 and week 8.
Results
Baseline characteristics
The study occurred from July 27, 2021 to May 23, 2023 (first participant’s first dose to the last participant’s last visit). Of the 96 individuals screened, 56 received ≥1 VLX-ER dose (Safety Population), and 48 completed the 8-week study (Fig. 2). Participants had a mean age of 11.8 years, reflecting relatively balanced enrollment of children and adolescents, were mostly male (69.6%), White (80.4%), and not Hispanic or Latino (89.3%), with a mean ± standard deviation (SD) ADHD-RS-5 and CGI-S of 37.2 ± 8.4 and 4.4 ± 0.6, respectively, suggesting moderate to marked ADHD despite ongoing stimulant use (Table 1). The majority of participants took psychostimulants in the morning only. Seven participants took a scheduled afternoon dose of psychostimulants.
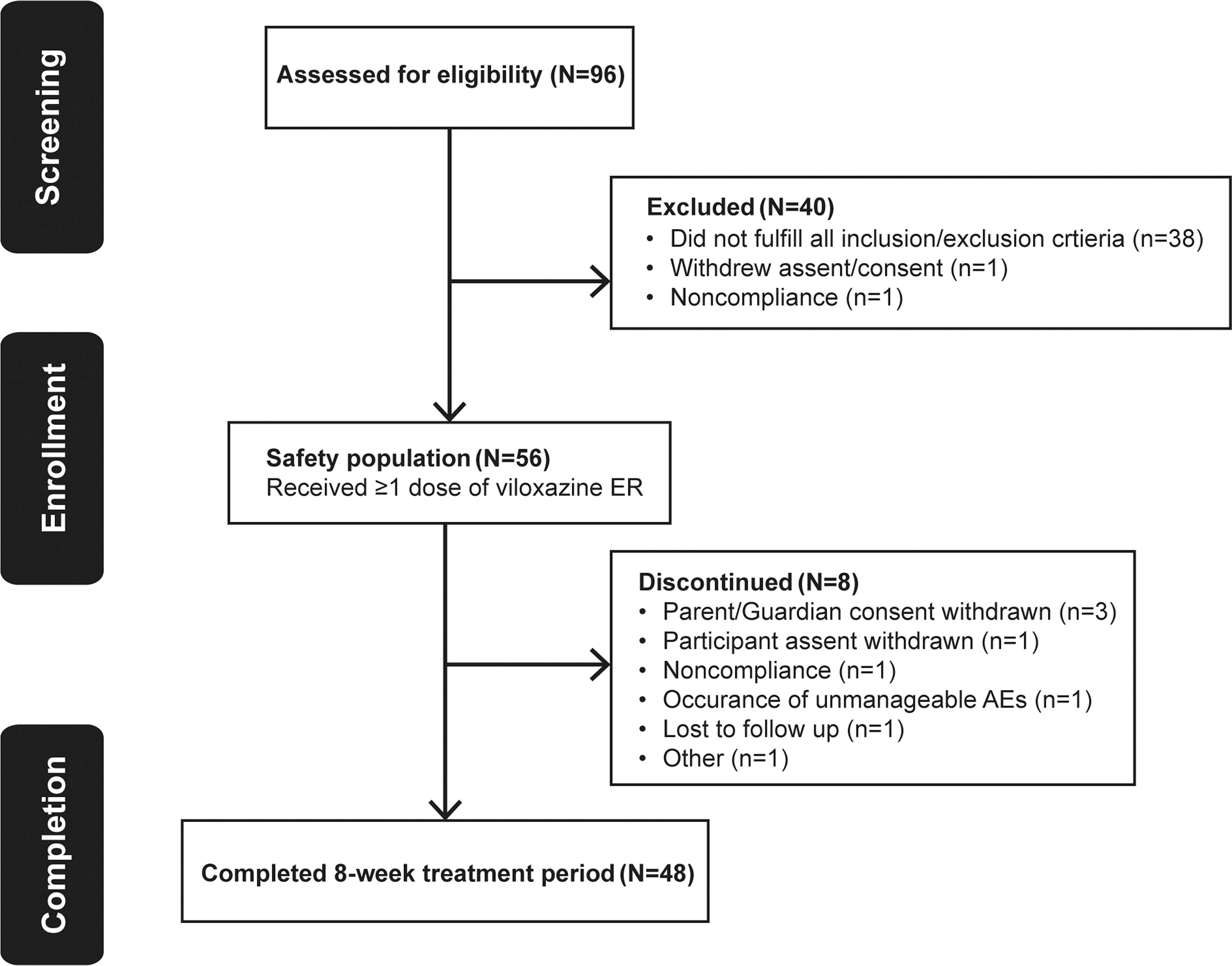
CONSORT flow diagram.
Baseline Demographic and Clinical Characteristics (Safety Population; n = 56)
Sample size for ADHD-RS-5 (parent assessed) IA/HI subscales: morning: n = 50, evening: n = 47.
ADHD, attention-deficit/hyperactivity disorder; ADHD-RS-5, ADHD Rating Scale-5; BMI, body mass index; CGI-S, Clinical Global Impressions-Severity; HI, hyperactivity/impulsivity; IA, inattention; SD, standard deviation; VLX-ER, viloxazine extended-release.
During the study, most dosing changes occurred between weeks 1 and 4 (morning VLX-ER dosing), with dosing distribution being similar at weeks 4 and 8 (Table 2). By week 8 (evening VLX-ER dosing), 38.1% of children and 40.7% of adolescents were using VLX-ER doses higher than their starting dose. Just over a third of participants (n = 21 [37.5%]) reported caffeine consumption during the baseline week, with fewer (8 [14.3%] to 14 [25.0%]) reporting caffeine use during subsequent weeks. For those who used caffeine during VLX-ER + psychostimulant treatment, overall weekly consumption was consistently low, without any observable pattern. Mean ± SD consumption ranged from 83 ± 56.7 mg/week (week 2) to 225 ± 232.0 mg/week (week 6).
Percent of Children and Adolescents for Each Viloxazine Extended-Release Daily Dosing Category at Weeks 4 and 8 (Safety Population [n = 56])
Safety outcomes
Of the 56 participants who received VLX-ER, 32 (57.1%) reported ≥1 AE (Table 3). The most commonly reported AEs (≥5% of participants) were headache, decreased appetite, upper respiratory tract infection, insomnia, nausea, diarrhea, fatigue, irritability, and vomiting. Of these, headache, decreased appetite, insomnia, nausea, fatigue, and irritability had a higher frequency of being assessed as treatment-related. Most AEs were first reported during weeks 1–4 (AM dosing) with fewer AEs (notably decreased appetite) having onset during weeks 5–8 (PM dosing). A post hoc analysis of AE presence during weeks 5–8 that included both AEs with onset during these weeks plus AEs that began in weeks 1–4 (AM dosing) and persisted beyond week 5 of PM dosing (Supplementary Table S1) demonstrated fewer differences in AE rates between AM and PM dosing, although appetite suppression appeared lower with PM dosing.
Summary of Safety Outcomes During Weeks 1–4 (AM Dosing), Weeks 5–8 (PM Dosing), and in the Study Overall (Safety Population; n = 56)
Adverse events were tabulated by time period of onset (weeks 1–4) or (weeks 5–8).
Participants were counted as having a treatment-related AE if they had any reported AE that was considered to be possibly related to VLX-ER treatment. If a relationship designation was missing, the AE was considered to be treatment related.
AESIs are defined as a seizure or AEs that might represent a seizure.
Participants were counted only once based on the greatest level of severity for any AE. If a severity designation was missing, it was considered to be severe.
One participant reported nausea and vomiting after stopping VLX-ER following study completion.
One participant reported diarrhea after stopping VLX-ER following study completion.
AE, adverse event; AESI, adverse event of special interest; SAE, serious adverse event; VLX-ER, viloxazine extended-release.
Except for one participant who experienced a severe case of influenza considered unrelated to VLX-ER treatment, all AEs were reported to be mild or moderate in severity and mostly abated or resolved without changes to the VLX-ER dose. No participant experienced a serious AE (SAE). AEs led to VLX-ER dose reduction for five participants and discontinuation for two participants; an adolescent male discontinued due to an AE of anxiety that started on study day 10 (morning VLX-ER dosing), and an adolescent female discontinued due to an AE of constricted affect that began on study day 43 (evening VLX-ER dosing) and resolved on study day 46. Each of these AEs was reported to be moderate in severity and possibly related to study treatment. For the five participants who reduced their dose, AEs appeared to resolve in four, for the fifth participant the AE (irritability) was considered unrelated to treatment, and resolved one day after the end of the study visit.
There were no clinically meaningful changes in laboratory parameters, with the exception that one participant had an elevated potassium level (6.9 mmol/L, normal range [NR] = 3.5–5.3 mmol/L), and a low calcium level (1.72 mmol/L, [NR] = 2.14–2.62 mmol/L) on day 57 that were reported as AEs. However, both parameters were considered unrelated, resolved on day 62, and did not lead to any VLX-ER dose changes.
Vital sign monitoring showed small increases from baseline to week 8 (n = 48) in mean ± SD sitting/standing blood pressure (systolic [3.3 ± 8.54/1.6 ± 6.82 mmHg]; diastolic [3.4 ± 8.63/1.5 ± 9.06 mmHg]) and pulse rate (6.3 ± 11.6/8.7 ± 14.9 beats/min); however, analysis from the baseline category (low, normal, or high) showed these values generally remained within normal ranges across study visits. For each measure, <15% of participants moved from a low or normal value at baseline to an elevated value at any visit, and only two participants (1 with tachycardia and one with increased diastolic blood pressure) had vital sign changes reported as AEs (both were assessed as mild and did not result in dose changes). Body weight and BMI remained relatively stable (week 8 CFB: −0.35 ± 1.85 kg and −0.25 kg/m2, respectively), and height increased by 0.51 ± 1.06 cm. One participant (age 12 years) had weight decrease reported as an AE that was assessed as mild and did not result in dose change.
ECG evaluations showed no clinically relevant changes, and there were no reported ECG-related AEs.
No participants reported suicidal ideation and behavior for any post-baseline visit.
Efficacy outcomes
The ADHD-RS-5 (investigator assessed) was significantly (p < 0.0001) improved by week 1 (vs. baseline), with further improvement at subsequent weeks (vs. baseline) (Fig. 3A, Supplementary Table S2). Improvement derived from significant reductions in both hyperactivity/impulsivity (HI) and inattention subscale scores (Supplementary Table S2). The 30% and 50% responder rates generally increased from week 1 through week 8 (Supplementary Table S3).
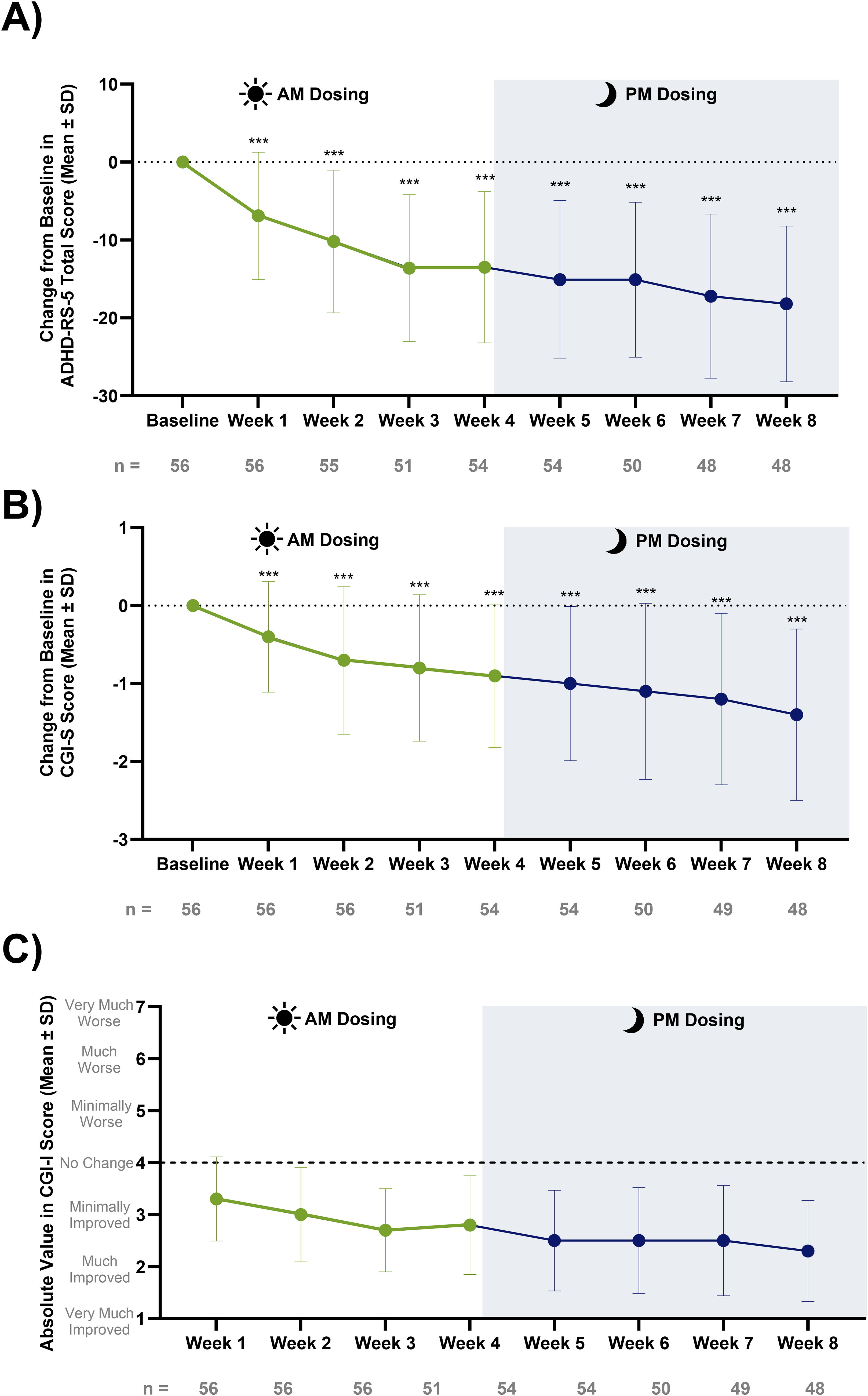
Improvements in the CGI-S and CGI-I mirrored that of the ADHD-RS-5, with significant improvement in CGI-S scores (vs. baseline) (p < 0.0001) starting from week 1 (Fig. 3B, C, Supplementary Table S2). CGI-S and CGI-I responder rates also generally increased in tandem from week 1 through week 8 (Supplementary Table S3).
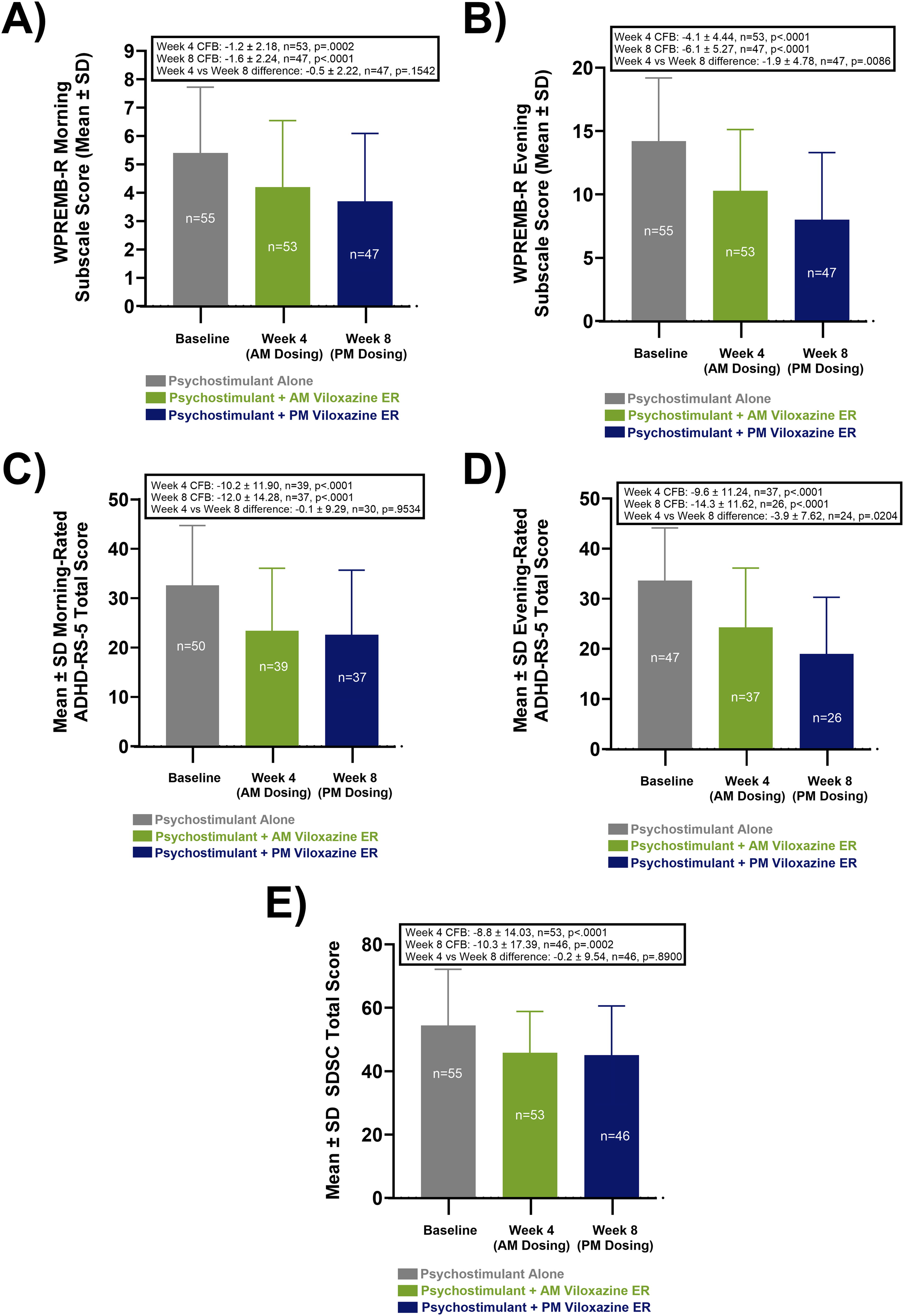
Parent-rated outcomes (WPREMB-R and ADHD-RS-5) showed similar incremental improvement at week 4 and week 8 that were nominally significant versus baseline in both morning- and evening-rated ADHD symptoms, and nominally larger at week 8 than week 4 for the WPREMB-R (Figs. 4A–4D, Supplementary Table S4). Changes in same-day ADHD-RS-5 assessments were statistically similar in the morning vs evening regardless of VLX-ER dose timing (week 4 [AM VLX-ER] CFB in morning rating: −10.2 ± 11.90 versus evening rating −9.6 ± 11.24, p = 0.56; week 8 [PM VLX-ER] CFB in morning rating: −12.0 ± 14.28 versus evening rating −14.3 ± 11.62, p = 0.51) (Fig. 4C, D, Supplementary Table S4). Similarly, ratings of sleep disturbance showed significant (p < 0.05) improvement relative to baseline under conditions of both morning administration of VLX-ER at week 4 and evening administration of VLX-ER at week 8 (Fig. 4E, Supplementary Table S2). Changes in SDSC subscales were significant versus baseline for all except the Sleep Breathing Disorder and Disorders of Arousal subscale scores, possibly due to low baselines for these two subscales. There were no significant differences between morning and evening VLX-ER administration (week 4 versus week 8) in any of the SDSC subscale scores.
Discussion
This open-label, flexible-dose study is the first to evaluate the safety, tolerability, and efficacy of VLX-ER, a nonstimulant, used concomitantly with psychostimulant medication to treat children and adolescents experiencing suboptimal ADHD response to stimulant alone. Participants experienced improvement across all ADHD symptom domains, and the combination showed good overall safety and tolerability, with no serious AEs and a low incidence (n = 2) of AEs resulting in study medication discontinuation. The most commonly reported treatment-related AEs (≥5%) were decreased appetite, insomnia, nausea, fatigue, and irritability, all of which were described as mild or moderate in severity. One participant had an elevation in heart rate, and another had an elevation in diastolic blood pressure sufficient to be characterized as an AE; however, each was categorized as mild in severity and did not result in dose change or treatment discontinuation. ECG monitoring showed no clinically relevant changes or signs of cardiac safety concerns. Although hampered to some extent by lack of two-way crossover, there were no obvious safety differences between AM dosing during weeks 1–4 and PM dosing during weeks 5–8, except that appetite suppression appeared lower with PM dosing, even when considering AEs with onset during AM dosing but persisting during PM dosing.
Notably, somnolence—the most commonly reported AE in VLX-ER pediatric ADHD monotherapy trials (16% incidence)—was virtually absent in this trial (n = 1, 1.8%). Conversely, insomnia—which had a 4% incidence in pediatric monotherapy trials-was somewhat more common in this trial (8.9%), though still well below the incidence of insomnia (∼23%) reported in the VLX-ER adult ADHD monotherapy trial and the incidence of insomnia in many pediatric psychostimulant monotherapy trials (children: 10%–23%–23%; adolescents: 12%–13%) (Arbor, 2020; Kollins et al., 2021; Shire, 2013, 2017; Supernus Pharmaceuticals Inc., 2025). Overall, these data suggest that concomitant psychostimulant use may have negated the potential sedative effects of VLX-ER in children and adolescents, while slightly increasing the potential for insomnia. The tendency for VLX-ER to cause insomnia in adults and somnolence in children has been surmised to stem from greater caffeine consumption in adults relative to children (VLX-ER prolongs the half-life of caffeine by inhibiting CYP-1A2).
Overall, fewer (14%−25%) children and adolescents used caffeine in this trial relative to the adult VLX-ER ADHD trial (>85%), and among those that did, caffeine use was typically lower (mean of ∼80–225 mg/week) than in the adult, double-blind trial (mean of ∼950–1100 mg/week for the VLX-ER group). Only two of the five pediatric participants in this trial who reported insomnia as an AE were using any caffeine at the time of AE occurrence. SDSC results show sleep quality improved following VLX-ER addition. Other safety outcomes and tolerability appeared similar to those in prior short-term VLX-ER pediatric ADHD monotherapy trials (Johnson et al., 2020; Nasser et al., 2021a, 2021b, 2021c).
In addition to being efficacious as monotherapy, nonstimulants are often used to augment psychostimulant response when psychostimulants alone do not optimally control ADHD symptoms (Wolraich et al., 2019). Our study supports this approach, with the combination of VLX-ER and psychostimulant showing incremental efficacy on all study outcome measures, including parent-rated and investigator-rated scales, subscales assessing inattentive and hyperactive/impulsive symptoms, ratings of morning and evening ADHD symptoms and ADHD-related behaviors, ratings of sleep disturbances, and global assessments of ADHD severity and improvement. Onset of symptom improvement was apparent by week 1, with mean changes in overall rating scales all showing nominal significance (relative to baseline) at weeks 4 and 8, which were generally larger in magnitude at week 8 than week 4. Improvement trajectory was relatively constant throughout the study (Fig. 3) despite the shift from morning to evening dosing, supporting VLX-ER efficacy throughout the 24-hour dosing cycle. The ability to take a medication at any time of day and expect consistent efficacy over the dosing cycle could be an advantage for patients, particularly if it avoids the need to await medication onset to facilitate morning functioning or prevents the need to use an immediate-release stimulant late in the day, when psychostimulant monotherapy wears off (Cascade et al., 2008) and where it may have a greater risk for disrupting sleep.
Despite that participants entering this study were presumably receiving some benefit from prior psychostimulant treatment, the magnitude of ADHD symptom response was consistent with that seen in the four VLX-ER phase 3 monotherapy trials in pediatric ADHD. Perhaps consistent with prior psychostimulant treatment, baseline ADHD-RS-5 scores in this trial were slightly lower (37.2 ± 8.35) than for the phase 3 trials (range: 39.9 ± 7.20 to 44.2 ± 6.81); yet the mean improvement in ADHD-RS-5 scores in this study (−18.2 ± 9.99) was at the high end of that seen with VLX-ER in the phase 3 trials (range: −16.0 ± 1.45 to −18.3 ± 1.36), suggesting a similar robust clinical response to that of monotherapy.
Our findings should be interpreted in light of several limitations. First, our sample size was relatively small and homogenous, and our study lacked a control group, rendering the findings preliminary in nature. Additionally, the lack of two-way crossover between morning-administered and evening-administered VLX-ER confounded the assessment of potential effects of dose timing on efficacy and safety outcomes by not allowing us to distinguish the degree to which differences in outcomes may have resulted from differences in time on treatment versus timing of dose administration. Also, only morning administration of psychostimulants was evaluated, therefore the results are not generalizable to children or adolescents who take psychostimulants at other times.
This study did not evaluate the degree to which symptom response may have been due to VLX-ER treatment alone versus the combined effect with psychostimulant. Future studies could attempt to taper psychostimulant following VLX-ER addition to determine the contribution of monotherapy versus combined therapy to overall response, especially given that research has suggested ∼40% of psychostimulant nonresponders will respond to nonstimulant treatment (and vice versa) (Newcorn et al., 2008). Trial enrollment was limited to pediatric ADHD; further study is needed to understand the effects of combined VLX-ER and psychostimulant in an adult ADHD population. Finally, the short-term nature of this study does not allow evaluation of long-term maintenance of response and safety outcomes.
Conclusions
This study provides preliminary evidence supporting the safety, tolerability, and efficacy of the combined use of VLX-ER and a psychostimulant to treat ADHD in children and adolescents experiencing suboptimal ADHD response to psychostimulant alone. Safety and efficacy outcomes were consistent with those of VLX-ER monotherapy, established in the pediatric clinical trial program. Additionally, consistent with presumptions of 24-hour symptom control, efficacy outcomes and overall tolerability appeared unaffected by VLX-ER dose timing, offering individuals the flexibility to take the medication based on personal preference.
Clinical Significance
This study provides preliminary safety and efficacy data to support the use of VLX-ER in combination with stimulants in pediatric patients with ADHD experiencing inadequate response to psychostimulant monotherapy. This trial also provides support for the safety and efficacy of VLX-ER when administered in the evening as opposed to the morning administration studied in phase 3 clinical trials. This added convenience could help individuals avoid taking a second stimulant dose later in the day and the associated risk for bothersome AEs such as insomnia.
Footnotes
Acknowledgments
The authors thank Greg Gargiulo of The Write Source, MSC, LLC, for medical writing assistance; Joseph T. Hull for protocol design, clinical trial management, oversight of site selection, initiation and training, data review, and production of the final study report; Rose Liberman for assistance with clinical trial conduct, study monitoring and closeout, review of top-line results, writing, review, and QC of the final clinical study report; Zulane Maldonado-Cruz for providing pharmacovigilance and medical monitoring during the clinical trial, review of safety endpoints, and critical editing and QC of the article; Gayathri Palety for providing pharmacovigilance and medical monitoring during the clinical trial and review of safety endpoints; Austin Eikenberry for article QC; and Jennifer Koch for article management and figure creation.
Authors’ Contributions
A.C.: Consulted on study design, investigator on trial, interpretation of data, critically reviewed drafts, and approval of final version of article. K.A.: Data curation, formal analysis, data validation, critically reviewed drafts, and approval of the final version of the article. G.C.: Critical review of the final/drafted clinical study report (CSR), ongoing data monitoring/review during trial, interpretation of data, data QA/QC for CSR, review of clinical protocol amendments and finalized versions, and critical review of the drafted/final article. J.E.: Interpretation of data, critically reviewed drafts, and approval of the final version of the article. K.H.: Study management, data collection, critically reviewed drafts, and approval of final version of the article. I.Y.: Medical and safety monitoring during the clinical trial, interpretation of safety data, review of safety endpoints, and review of final clinical study report. J.R.: Consulted on study design, interpretation of data, critically reviewed drafts, and approval of the final version of the article
Author Disclosure Statement
A.C. has received research support from, served as a consultant or speaker for, or served on an advisory board for Allergan, Aardvark, Axial, Axsome, Aytu, Takeda (Shire), Emalex, Akili, Arbor, Cingulate Therapeutics, Corium, Ironshore, Kempharm, Lumos, Neurovance, Noven, Otsuka, Pfizer, Purdue, Adlon, Sunovion, Tris, KemPharm, Supernus Pharmaceuticals, Inc. K.H., G.C., K.A., I.Y., J.E., and J.R. are employees of Supernus Pharmaceuticals, Inc.
Funding Information
This study was funded by Supernus Pharmaceuticals, Inc.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.