
Abstract
Introduction:
The effective management of irritability is a key need in young people with autism spectrum disorder (ASD). We evaluated the efficacy and safety of brexpiprazole in children and adolescents with irritability associated with ASD.
Methods:
This was an 8-week, phase 3, randomized, double-blind, placebo-controlled trial (NCT04174365) and 26-week, open-label extension (OLE, NCT04258839) of brexpiprazole (0.25–3 mg/day based on body weight) in children and adolescents (5–17 years) with a diagnosis of ASD, score ≥18 on the Aberrant Behavior Checklist–Irritability (ABC-I) subscale, and score ≥4 on the Clinical Global Impressions–Severity scale.
Results:
Of the 119 randomized participants (brexpiprazole = 60, placebo = 59), 104 completed double-blind treatment, and 95 enrolled and 70 completed the OLE. Similar reductions in mean ABC-I subscale score were seen in both groups (least-squares mean ± standard error reduction from double-blind baseline of −10.1 ± 1.3 with brexpiprazole vs −8.9 ± 1.3 with placebo). Thus, the primary endpoint did not show a significant treatment effect (LS-mean [95% confidence interval] treatment difference: –1.22 [–4.49, 2.05], p = 0.46) and the hierarchical efficacy analysis ended at this point. At the end of the OLE, participants had a mean ± SD reduction from open-label baseline of −6.1 ± 8.2 in ABC-I subscale score. During double-blind treatment, 51.7% participants treated with brexpiprazole had ≥1 treatment-emergent adverse event (TEAE) versus 35.1% with placebo; no severe or serious TEAEs were reported. The only potentially treatment-related TEAE that occurred in ≥5% of participants was somnolence (12.1% for brexpiprazole vs 5.3% for placebo). During the OLE, the most commonly reported TEAE was increased weight (n = 13, 13.7%).
Conclusions:
Treatment with brexpiprazole did not demonstrate significant efficacy versus placebo for the treatment of irritability associated with ASD. The safety profile was consistent with that observed in adult and adolescent patients with schizophrenia.
Introduction
The umbrella term “irritability” describes proneness to anger that may reach a pathological extent (Vidal-Ribas et al., 2016). It is estimated that approximately 20% of people living with autism spectrum disorder (ASD) exhibit symptoms of irritability, with higher rates in people with sensory differences, and comorbid anxiety, depression, symptoms of attention-deficit/hyperactivity disorder (ADHD), and/or oppositional defiant disorder (Alatrash et al., 2024). High levels of irritability in children and adolescents with ASD often manifest as behavioral problems such as oppositional behavior, aggression, temper tantrums, self-injurious behaviors, and severe noncompliance (Mazefsky et al., 2019). There is general consensus in national and international guidelines that pharmacologic interventions can help young people with ASD to benefit from educational and other interventions and to remain in less restrictive environments through the management of severe and challenging behaviors (Fuentes et al., 2021; Howes et al., 2018; Volkmar et al., 2014).
Alterations in several neurotransmitter systems including dopaminergic, serotonergic, and noradrenergic neurotransmission have been implicated in the underlying pathophysiology of ASD and its behavioral manifestations (Beversdorf, 2020; Marotta et al., 2020). Based on their efficacy on the Aberrant Behavior Checklist (Aman et al., 1985)–Irritability (ABC-I) subscale, two second-generation antipsychotics, risperidone and aripiprazole, are approved by the U.S. Food and Drug Administration (FDA) for agitation and irritability in autism. Haloperidol is approved in the European Union for persistent, severe aggression in children and adolescents with autism, when other treatments have failed or are not tolerated.
Brexpiprazole is an atypical antipsychotic with broad pharmacological activity across noradrenergic, serotonergic, and dopaminergic neurotransmitter systems implicated in psychiatric disorders (Maeda et al., 2014; Oosterhof et al., 2015). In the pediatric population, brexpiprazole is approved by the FDA for the treatment of schizophrenia in children aged 13 years and older (FDA, 2025) , and recent safety data from an open-label, long-term maintenance trial of brexpiprazole (1–4 mg) in adolescents (aged 13–17 years old) with schizophrenia support a favorable pediatric safety profile that is generally consistent with that observed in adult patients (Atkinson et al., 2024). Collectively, this evidence suggests that brexpiprazole should be investigated as a treatment for irritability in ASD.
The primary objective of the double-blind, randomized, controlled, open-label extension trials was to evaluate the efficacy, safety, and tolerability of brexpiprazole in children and adolescents with irritability associated with ASD, aged 5–17 years.
Methods
Study conduct
This was a phase 3, multicenter, randomized, double-blind, placebo-controlled, 8-week trial (NCT04174365) of flexibly dosed brexpiprazole (0.25–3 mg/day based on body weight) followed by a 6-month open-label extension study (NCT04258839) conducted at 29 specialist centers across the United States (27 sites participated in the extension study). All sites received training for study assessments. The double-blind study started on October 30, 2019, and the open-label extension study completed on March 16, 2023. Institutional review boards at the participating sites approved the protocols, and the trials were conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice Guidelines. Separate electronic or written informed consent was obtained for each study from a guardian or legal representative for all participants prior to assessment of eligibility.
Participants
Participants aged 5–17 years, weighing ≥15 kg, with a diagnosis of ASD according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria, supported by the Autism Diagnostic Interview Revised (ADI-R), a score ≥18 on the ABC-I subscale, and a score ≥4 (moderate severity) on the Clinical Global Impressions–Severity (CGI-S) scale were eligible for participation in the double-blind study. The open-label extension was open to those participants who completed the double-blind phase and who, in the investigator’s opinion, could benefit from treatment. Participants who turned 18 years old during the double-blind study were permitted in the open-label extension study. Participants had to have a mental age of ≥2 years, as determined by the investigator based on school participation, social history, or medical records.
Key exclusion criteria for the double-blind study were a current primary DSM-5 diagnosis of bipolar I or II disorder, schizophrenia, schizoaffective disorder, and major depressive episode, as determined using the Kiddie-Schedule for Affective Disorders and Schizophrenia–Present and Lifetime version (Kaufman et al., 1997) or posttraumatic stress disorder. Participants with comorbid ADHD that is the primary diagnosis or is not adequately treated or stable, a diagnosis of Fragile X or Rett’s disorder, epilepsy, a history or current evidence of other unstable medical conditions, or prior use of long-acting injectable antipsychotics were also excluded. Participants at significant risk of committing violent acts, serious self-harm, or suicide based on history or routine psychiatric status examination were also excluded. Use of concomitant psychotropics including other antipsychotics (washout of ≥7 days for oral risperidone and ≥14 days for oral aripiprazole), antidepressants, mood stabilizers, benzodiazepines (except diazepam, lorazepam, oxazepam, and clonazepam when used as rescue therapy), and stimulants (unless used for the treatment of ADHD) was prohibited during the study.
Study design
Following a screening period of up to 28 days, participants who continued to meet entrance criteria at the baseline visit were randomized, 1:1 using a computer-generated code (stratified by site), to receive 8 weeks of double-blind treatment with brexpiprazole or matched placebo, supplied in child-resistant blister cards. Titration to target dose began at the baseline visit and was based on body weight (participants weighing <50 kg received a target dose range of 1–1.5 mg, and those with a body weight ≥50 kg received a target dose range of 1.5–3 mg) and investigator’s decision in order to reach a desired therapeutic effect (at day 15). Participants weighing <50 kg received 0.25 mg once daily (QD) for days 1–3, increasing to 0.5 mg QD at days 4–7 and 1 mg QD at days 8–15; at day 15 the dose could be maintained at 1 mg QD or increased to 1.5 mg QD for the rest of the study. Participants weighing ≥50 kg received 0.5 mg QD for days 1–3, increasing to 1.5 mg QD at days 4–7 and 2 mg QD at days 8–15; at day 15 the dose could either be reduced to 1.5 mg QD, maintained at 2 mg QD, or increased to 3 mg QD for the rest of the study.
The double-blind trial was originally designed with in clinic (screening, baseline, and weeks 2 and 8) and virtual visits (weeks 1, 3, 4, and 6), with the primary and key secondary efficacy endpoints assessed at each post-screening visit. The schedule was modified due to the COVID-19 pandemic to allow virtual visits at week 2 and to allow planned virtual visits to be performed either in clinic or virtual based on the investigator and family’s decision. For the open-label extension study, the last visit of the double-blind trial could serve as the baseline so long as it was conducted in clinic. Open-label visits were then performed virtually at weeks 1, 2, 3, 4, 8, 14, 18, and 22, with the end-of-treatment visit at week 26 performed in clinic. The first dose of open-label brexpiprazole (dose based on body weight) was taken the following day after the last dose of double-blind medication so that treatment continued without interruption. Participants previously on placebo were titrated in the same way as in the double-blind study.
The primary efficacy endpoint was the change from double-blind baseline to week 8 in ABC-I, and the key secondary endpoint was the CGI-S. Safety was assessed at each visit through reporting of treatment-emergent adverse events (TEAEs), vital signs, and the Columbia-Suicide Severity Rating Scale (C-SSRS) child version. Physical and laboratory measures, body mass index (BMI), and extrapyramidal symptoms (Simpson Angus Scale [SAS], Abnormal Involuntary Movement Scale [AIMS], and Barnes Akathisia Rating Scale [BARS]) were to be assessed at in-clinic visits. To account for normal growth in a pediatric population, Z-scores for height, weight, and BMI were derived (Centers for Disease Control and Prevention, 2025). A safety follow-up assessment took place 21 (±2) days after the last dose of study treatment (either after the double-blind or open-label studies).
Statistical analysis
Across both studies, the safety sample included all enrolled participants who received ≥1 dose of study medication and the efficacy sample included all participants in the safety set who had baseline and ≥1 post-baseline assessment of the ABC-I subscale score (during the double-blind treatment phase and the open-label extension, respectively).
The double-blind efficacy analyses were based on the observed case (OC), without imputation of any missing observations or assessments; mean change from double-blind baseline in ABC-I subscale and CGI-S scores were analyzed using a restricted maximum likelihood (REML)-based repeated measures approach including treatment, trial center, baseline body weight stratum, visit, and treatment-by-visit interaction as fixed factors and baseline-by-visit interaction as a covariate. A hierarchical testing procedure was specified in the double-blind study to maintain the type I error at 0.05 for the analysis of the key secondary efficacy variable.
In the open-label extension, changes from open-label baseline to week 26 in ABC-I and CGI-S were assessed as secondary endpoints and summarized descriptively (OC).
Sample size estimation
For the double-blind study, it was estimated that a sample size of 51 participants per arm would provide at least 85% power at a nominal 2-sided alpha level of 0.05 to detect a 6.0-point reduction in change from double-blind baseline in ABC-I subscale score for brexpiprazole versus placebo, assuming a standard deviation (SD) of 10. The sample size for the open-label study depended on completion rates from the prior study, and no formal sample size calculation was performed.
Results
Study disposition and baseline characteristics
Of the 260 participants screened, 119 were randomized to double-blind treatment (brexpiprazole n = 60, placebo n = 59), 115 received ≥1 dose of study medication (safety sample), and 114 were included in the efficacy sample (Fig. 1). Of the 104 participants who completed the double-blind study, 95 participants enrolled in the open-label extension study and received ≥1 dose of study medication, and 70 participants completed the open-label extension study. The mean dose at week 8 for the double-blind trial was 1.15 mg for participants who weighed <50 kg at baseline and 2.21 mg for participants who weighed ≥50 kg at baseline. The mean dose (prior placebo/prior brexpiprazole) at the end of the open label trial was 1.30/1.37 mg for participants weighing <50 kg and 2.33/2.42 mg for participants weighing ≥50 kg. The main reasons for early discontinuation in the double-blind study were guardian withdrew consent (n = 2 with brexpiprazole and n = 1 with placebo) and adverse events (AEs) (n = 2 with brexpiprazole and n = 1 with placebo). AEs were the main cause of withdrawal in the open-label extension study (n = 6).
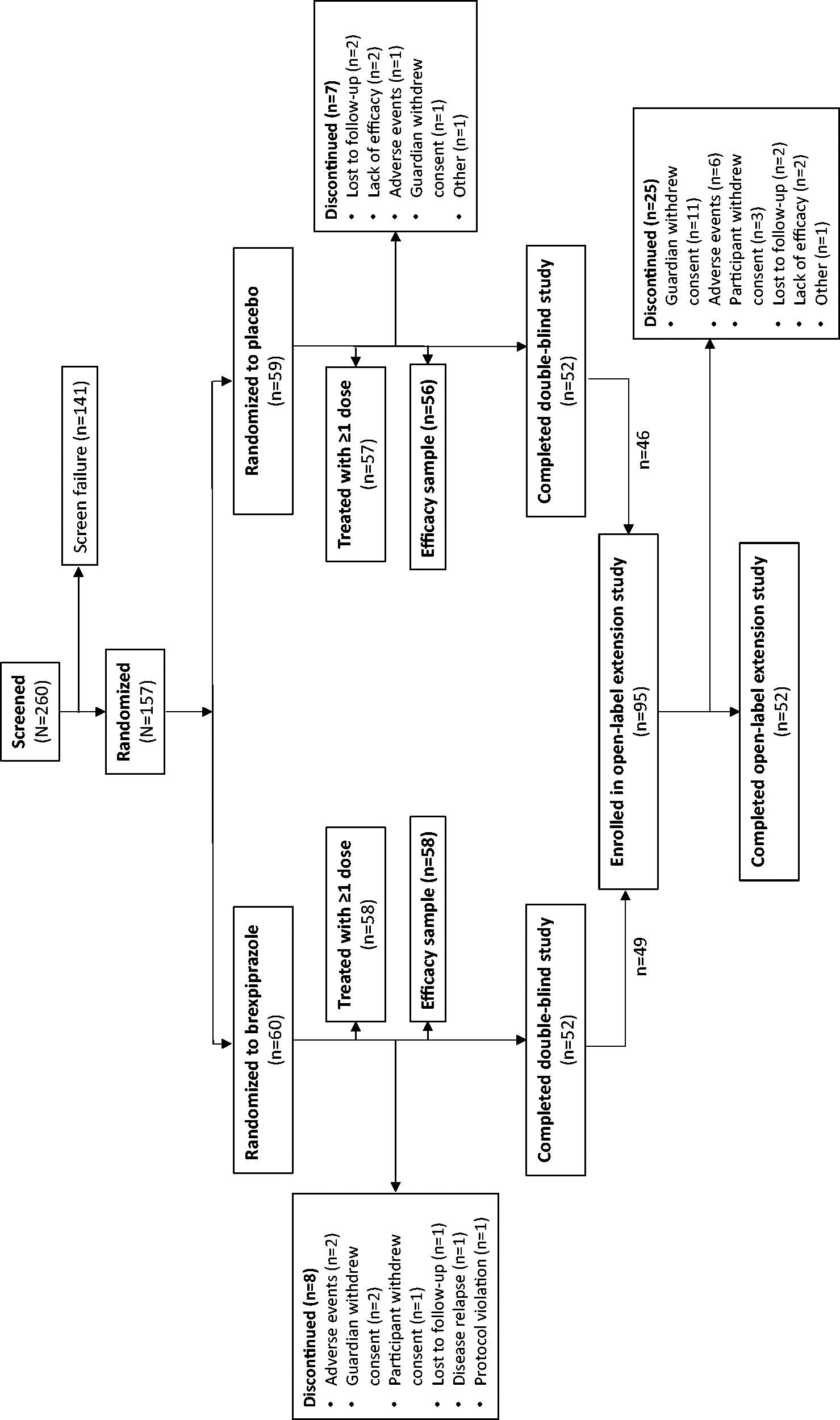
Study disposition.
Baseline characteristics were similar across both studies and treatment arms and are presented for the double-blind study in Table 1. The mean ± SD age at double-blind baseline was 9.9 ± 3.0 years, with 96 (80.7%) children aged between 5 and 12 years old and 23 (19.3%) adolescents aged between 13 and 17 years old. Most participants (n = 104, 87.4%) were male; participants of Black or African American origin accounted for 10.9% of the sample population and participants of Hispanic or Latino ethnicity accounted for 12.6% of the sample population. Participants entering the double-blind study had a mean ABC-I score of 29.1 points (of a maximum 45) and a mean CGI-S score of 4.8 (corresponding to moderately affected).
Baseline Characteristics of the Enrolled Population (Start of Double-Blind Study)
Data for the safety sample.
ABC-I, Aberrant Behavior Checklist–Irritability subscale; CGI-S, Clinical Global Impressions–Severity scale; SD, standard deviation.
Efficacy findings
Brexpiprazole did not show statistically significant improvement compared with placebo on the primary endpoint (change from double-blind baseline to week 8 in ABC-I score), and the hierarchical efficacy analysis ended at this point (Table 2). Numerical reductions from double-blind baseline to week 8 in mean ABC-I subscale score were seen in both the brexpiprazole and placebo-treated groups (Supplementary Fig. S1); at week 8, the least-squares (LS) mean ± standard error change from baseline was −10.1 ± 1.3 in the brexpiprazole-treated group compared with −8.9 ± 1.3 in the placebo-treated group. The LS mean treatment difference (brexpiprazole vs placebo) was –1.22 (95% confidence interval: –4.49, 2.05) (p = 0.46). There was no notable difference between the brexpiprazole and placebo groups in the change from baseline in CGI-S score at any of the double-blind study visits (Supplementary Fig. S2).
Change from Baseline to Week 8 in the Double-Blind Study
ABC-I, Aberrant Behavior Checklist–Irritability subscale; CGI-S, Clinical Global Impressions–Severity scale; CI, confidence interval; LS, least squares; SD, standard deviation; SE, standard error.
In the open-label phase, the baseline mean ± SD ABC-I subscale score was 19.1 ± 9.9, and scores were similar between the prior brexpiprazole and prior placebo groups (18.5 ± 9.2 vs 19.6 ± 10.8, respectively). At the end of the open-label extension study (week 26), 62/94 participants completing the week 26 visit had a mean ± SD reduction from open-label baseline of −6.1 ± 8.2 in ABC-I subscale score (Supplementary Fig. S3). At week 26, participants showed an improvement of −0.7 ± 1.0 from open-label baseline on the CGI-S scale, with a trend to greater improvements in participants previously treated with placebo than those previously treated with brexpiprazole (Supplementary Fig. S4).
Safety and tolerability
Table 3 summarizes TEAE reporting across the double-blind and open-label treatment periods. In the double-blind study, 51.7% participants treated with brexpiprazole had ≥1 mild or moderate TEAE compared with 35.1% treated with placebo; no severe or serious TEAEs were reported. The incidence of TEAEs considered potentially treatment related was 29.3% in the brexpiprazole group and 15.8% in the placebo group. However, the only potential treatment-related TEAE that occurred in ≥5% of participants was somnolence (12.1% for brexpiprazole vs 5.3% for placebo). Two participants discontinued treatment with brexpiprazole (both were on a dose of 2 mg) due to moderate TEAEs (emotional breakdown with self-blaming/guilt and increased blood pressure), and one participant discontinued placebo due to aggression and excessive hunger. Mild extrapyramidal-related events were reported in four participants (akathisia and tremor in two participants each, and restlessness and drooling in one participant each) in the brexpiprazole group and two participants (psychomotor hyperactivity and restlessness in one participant each) in the placebo group, with none leading to study discontinuation. For the two participants with mild akathisia, onset occurred at weeks 2–3. No clinically significant changes from double-blind baseline to week 8 were observed in SAS, AIMS, and BARS scores, and C-SSRS scores showed no suicidal behavior or ideation in either treatment arm. Weight Z-score increases of ≥0.5 from double-blind baseline adjusted for natural growth of pediatric patients were recorded for 11 (19.0%) participants in the brexpiprazole group and 3 (5.3%) participants in the placebo group; weight gain was reported in one participant treated with brexpiprazole as a treatment-related TEAE. There were no meaningful differences between the brexpiprazole and placebo groups for clinical laboratory results (including metabolic parameters; see Supplementary Table S1), vital signs, and electrocardiograms (ECGs).
Treatment-Emergent Adverse Events in the Double-Blind and Extension Studies
TEAE, treatment emergent adverse event.
In the open-label extension study, about half of the participants (n = 47) reported ≥1 TEAE, which were mostly mild or moderate in severity; the incidence of TEAEs considered by the investigator as potentially treatment related was 27.4%. Potential treatment-related TEAEs that occurred at an incidence of ≥5% were increased weight, increased appetite, and somnolence. Serious TEAEs occurred in three (3.2%) participants treated with open-label brexpiprazole (affective disorder, dyskinesia, suicidal ideation), and mild or moderate EPS-related TEAEs were reported in six participants (6.3%) (restlessness in two participants, and akathisia, dyskinesia, psychomotor hyperactivity, and tremor in one participant each). No clinically significant changes from double-blind baseline were observed in SAS, AIMS, and BARS scores. The serious event of suicide ideation was not judged by the investigator as related to brexpiprazole treatment, and no suicidal behavior was reported during the treatment or follow-up period based on the C-SSRS. Z-scores >0.5 for body weight at any post baseline visit were observed in 26.3% participants. Of these, 13 (13.7%) participants reported increased weight as a TEAE (11 were judged by the investigator as potentially treatment related); none were categorized as serious TEAEs, but one participant discontinued brexpiprazole treatment. There were no clinically meaningful changes in clinical laboratory results, vital signs, or ECGs.
Discussion
This phase 3, randomized, controlled clinical trial did not demonstrate a statistically significant improvement for brexpiprazole compared with placebo for the primary endpoint (mean change from double-blind baseline to week 8 in ABC-I subscale score) in children and adolescents with ASD exhibiting moderate irritability. The key secondary endpoint, mean change in CGI-S score, and subgroup analyses also failed to demonstrate signals of efficacy for brexpiprazole relative to placebo. The safety and tolerability profile of brexpiprazole (0.25–3 mg) was consistent with trials in other pediatric and adult indications.
While the magnitude of improvement from double-blind baseline with brexpiprazole was similar to that previously reported with other antipsychotics including aripiprazole, risperidone, and lurasidone (mean ABC-I reductions of –10.1 with brexpiprazole vs –12.4 to –14.4 with aripiprazole 5–15 mg, −13.5 to –14.9 with risperidone [0.5–3.5 mg, ASD subpopulation], and –9.4 with lurasidone 60 mg), participants in the placebo group showed a larger improvement than seen in prior studies (mean ABC-I reductions of –8.9 in the current study vs –3.5 to –8.4 in the aripiprazole, risperidone, and lurasidone studies) (Loebel et al., 2016; Marcus et al., 2009; McCracken et al., 2002; Owen et al., 2009). A systematic review and meta-regression analysis of 86 clinical trials in participants with ASD found that the placebo response in ASD trials was substantial and predicted by higher levels of baseline irritability as assessed using the ABC-I (Siafis et al., 2020). As such, it may be that the considerable changes in daily routine caused by the COVID-19 pandemic inflated baseline irritability with subsequent impact on the placebo effect. Indeed, while the authors observed the placebo response to have generally decreased over time in studies of ASD, this effect was not apparent when ABC-I was included in the multivariable meta-regression. In the current study, both groups showed continued improvements versus baseline in CGI-S with double-blind treatment (highlighting the considerable placebo effect). There was, however, a tendency for patients previously treated with placebo to show greater improvement, when switched to brexpiprazole in the open-label study than those continuing with brexpiprazole.
Safety results in both studies were consistent with the known safety and tolerability profile of brexpiprazole and indicated that brexpiprazole at doses between 0.25 and 3 mg was generally well tolerated in children and adolescents when taken for up to 26 weeks. In line with the pediatric schizophrenia safety findings (Ward et al., 2024), the most common TEAEs were somnolence and weight increase. The U.S. current labeling for brexpiprazole describes the risk for EPS and notes that dystonia is more frequent in younger individuals and males. Across the double-blind and open-label studies, the overall incidence of EPS-related TEAEs after brexpiprazole treatment was similar to that seen in the pediatric schizophrenia study (7% and 6% vs. 6%, respectively) (Ward et al., 2024). All TEAEs related to EPS were considered mild, and none of them led to discontinuation. Weight gain affected 13.7% of participants on open-label treatment and led to discontinuation in one participant. The proportion of participants with >0.5 increase in body weight at any open-label visits (26.3%) was also similar to that reported in pediatric schizophrenia studies (21% over 2 years of follow-up) (Atkinson et al., 2024).
Several potential study limitations should be noted, including the smaller number of females and adolescents in both studies and the small proportion of non-White participants. The inclusion of an active control group (i.e., risperidone or aripiprazole) would have helped understand whether the study was a failed trial rather than a negative trial. As with prior studies, the study design did not include a single-blind, placebo run-in period, which has been recommended for studies of ASD-related irritability to reduce the placebo response rate (Loebel et al., 2016). While the risk of metabolic syndrome is a known class safety issue with atypical antipsychotics, none of the participants in the open-label study had fasting glucose or lipid profile examined post baseline, so metabolic parameters could not be evaluated over the longer term. Finally, while this trial was purposefully designed as a hybrid trial of in-person and virtual visits (to decrease study burden and associated irritability), the COVID-19 pandemic necessitated a protocol amendment that increased the number of virtual visits and that affected some measurements including BMI (e.g., some participants reported a reduction in height, which is likely due to measurements with shoes-on vs shoes-off).
Conclusions
In conclusion, in this randomized, phase 3, placebo-controlled study with open-label extension, treatment with brexpiprazole did not demonstrate significant efficacy versus placebo for the treatment of irritability associated with ASD.
Clinical Significance
While this study does not support the use of brexpiprazole for treatment of irritability associated with ASD, it extends the brexpiprazole safety experience to the younger population of children aged 5–12 years old in whom brexpiprazole TEAEs were consistent with its known safety and tolerability profile.
Footnotes
Acknowledgments
The authors thank all participants, their guardians, and the study investigators in the two studies. They also thank Anita Chadha-Patel (ACP Clinical Communications Ltd, funded by H. Lundbeck A/S) for her support in the preparation, revisions, and editing of this article.
Authors’ Contributions
C.W.: Conceptualization, methodology, resources, writing original draft, and visualization. A.C.: Methodology, investigation, and writing—review and editing. K.M.: Supervision, project administration, and writing—review and editing. D.C.: Data curation, formal analysis, and writing—review and editing. K.G.L.: Data Curation, formal analysis, and writing—review and editing. A.S.: Project administration and writing—review and editing. P.S.: Project administration and writing—review and editing. N.H.: Conceptualization, methodology, resources, and writing—review and editing. J.K.: Methodology, investigation, and writing—review and editing.
Disclosures
C.W., K.M., D.C., and A.S. are employed by Otsuka Pharmaceutical Development & Commercialization, Inc. A.C. received research support from Aardvark, Allergan, Axsome, Takeda/Shire, Emalex, Akili, Corium, Ironshore, Arbor, Neurocentria, Otsuka, Pfizer, Purdue, Rhodes, Sunovion, Tris, KemPharm, and Supernus; was on the advisory board of Takeda/Shire, Akili, Arbor, Cingulate, Ironshore, Otsuka, Pfizer, Purdue, Adlon, Sunovion, Tris, Supernus, and Corium; received consulting fees from Arbor, Ironshore, Alora, Aytu, Purdue, Rhodes, Sunovion, Tris, KemPharm, Supernus, Corium, Tulex Pharma, and Lumos Pharma; received speaker fees from Takeda/Shire, Corium, Ironshore, Tris, and Supernus; and received writing support from Takeda/Shire, Arbor, Ironshore, Neos Therapeutics, Purdue, Rhodes, Sunovion, Supernus, and Tris. N.H. and K.G.L. are employed by H. Lundbeck A/S, and P.S. is employed by Lundbeck US. J.K., is employed by Core Clinical Research, which was contracted to perform this study.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.