
Abstract
Objective:
Major depressive disorder (MDD) presents a significant psychosocial burden, and there is an unmet need for additional treatment options in pediatric patients. Here, we report the results of two phase 3 multicenter, randomized, double-blind, placebo- and active-controlled, parallel-group studies evaluating the efficacy and safety of levomilnacipran extended release in children and adolescents with MDD.
Methods:
In the first study, LVM-MD-11, patients aged 12–17 years received daily doses of levomilnacipran 40 mg (n = 134), levomilnacipran 80 mg (n = 138), fluoxetine 20 mg (n = 134), or placebo (n = 141). In the second study, LVM-MD-14, patients aged 7–17 years received levomilnacipran 40 to 80 mg (n = 166), fluoxetine 20 mg (n = 166), or placebo (n = 160) daily. Primary and secondary efficacy endpoints were changes in Children's Depression Rating Scale-Revised (CDRS-R) total score and Clinical Global Impressions-Severity (CGI-S) score, respectively.
Results:
In LVM-MD-11, there were no significant differences in change in CDRS-R total score between patients treated daily with placebo (least squares mean [LSM] change in CDRS-R total score −22.9) versus levomilnacipran 40 mg (−23.3; p = 0.8035) or 80 mg (−22.6; p = 0.8681). Similarly, in LVM-MD-14, there were no significant differences in LSM change in CDRS-R total score with placebo (−21.3) versus levomilnacipran 40 to 80 mg daily (−23.0; p = 0.2215). There were also no significant differences between the fluoxetine and placebo groups in either study for changes in CDRS-R total score. Changes in CGI-S score were not significant between placebo and levomilnacipran 40 to 80 mg daily or between placebo and fluoxetine. Levomilnacipran was generally well tolerated.
Conclusions:
The high placebo response in this study prevented the detection of an effect of levomilnacipran in children and adolescents.
Clinical Trial Registration numbers: NCT02431806 and NCT03569475.
Introduction
Depressive disorders are a leading cause of disease burden worldwide and can cause serious mental illness in children and adolescents (Findling et al., 2020). These conditions affect up to 2% of prepubertal children and 8% of adolescents (Zalsman et al., 2006). Major depressive disorder (MDD) is commonly associated with persistent feelings of worthlessness, low self-esteem, suicidal ideation, or suicide attempts (American Psychiatric Association, 2013; Findling et al., 2020), and MDD in children and adolescents can be chronic and recurrent (Mullen 2018; Zalsman et al., 2006). The psychosocial burden of MDD in the formative years can compromise development, and untreated MDD can affect social functioning and impact academic achievements (Davies et al., 2018). Many parents and caregivers are unaware of the serious consequences of delayed treatment, and pediatric MDD is often underdiagnosed and undertreated (Mullen, 2018).
Treatment for children and adolescents with MDD includes a combination of psychotherapy and pharmacotherapy (Findling et al., 2020). Currently, the selective serotonin reuptake inhibitors (SSRIs) fluoxetine and escitalopram are approved by the U.S. Food and Drug Administration (FDA) for the treatment of MDD in children and adolescents (Selph and McDonagh 2019). Although several studies have supported the use of SSRIs for the treatment of MDD in pediatric patients, some children and adolescents with MDD do not experience clinical benefit with these drugs (Emslie et al., 2002; March et al., 2009). Notably, some clinical outcomes have been similar between patients treated with SSRI monotherapy and those treated with placebo (Selph and McDonagh 2019). Furthermore, antidepressants, including fluoxetine and escitalopram, have been associated with an increased risk of suicidal ideation and behavior in patients under the age of 25 years (AbbVie Inc., 2023a; Eli Lilly 2024; Hammad et al., 2006). Given the substantial impact of MDD in children and adolescents, as well as the limitations and potential risks of SSRIs in treating these patients, there is a major unmet need to identify additional drugs to treat this challenging disease safely and effectively.
Currently, no serotonin and norepinephrine reuptake inhibitors (SNRIs) are approved for the treatment of MDD in children and adolescents. Previous trials of SNRIs including venlafaxine extended release (ER) and duloxetine in pediatric patients did not demonstrate activity versus the comparator, and venlafaxine was associated with hostility and suicidal ideation (Atkinson et al., 2014; Emslie et al., 2007; Emslie et al., 2014). Levomilnacipran is an SNRI that exhibits more potent inhibition of norepinephrine versus 5-hydroxytryptamine reuptake, with higher selectivity for inhibition of norepinephrine reuptake than venlafaxine and duloxetine (Auclair et al., 2013). This pharmacological profile suggests that treatment of patients with MDD with an SNRI such as levomilnacipran may confer additional clinical benefits associated with noradrenergic transmission, including improved alertness, energy, attention, and anhedonia (Montgomery and Briley 2011). The levomilnacipran ER formulation allows once-daily dosing (Bakish et al., 2014). Although levomilnacipran is approved for the treatment of adults with MDD in the United States (AbbVie Inc., 2023b), the efficacy and safety profile of levomilnacipran in children and adolescents is currently under investigation.
Here, we report the results of two studies, LVM-MD-11 and LVM-MD-14, in which the efficacy, safety, and tolerability of levomilnacipran were evaluated in children and adolescents with MDD.
Methods
Study designs
Both LVM-MD-11 and LVM-MD-14 are phase 3 multicenter, randomized, double-blind, placebo- and active-controlled, parallel-group studies. Patients were enrolled at 59 U.S. sites in study LVM-MD-11 and 47 U.S. sites in LVM-MD-14. Both trials had a 10-week duration, with a 1-week screening/washout period followed by 8 weeks of double-blind treatment and 1 week of double-blind down-tapering of treatment (Fig. 1).
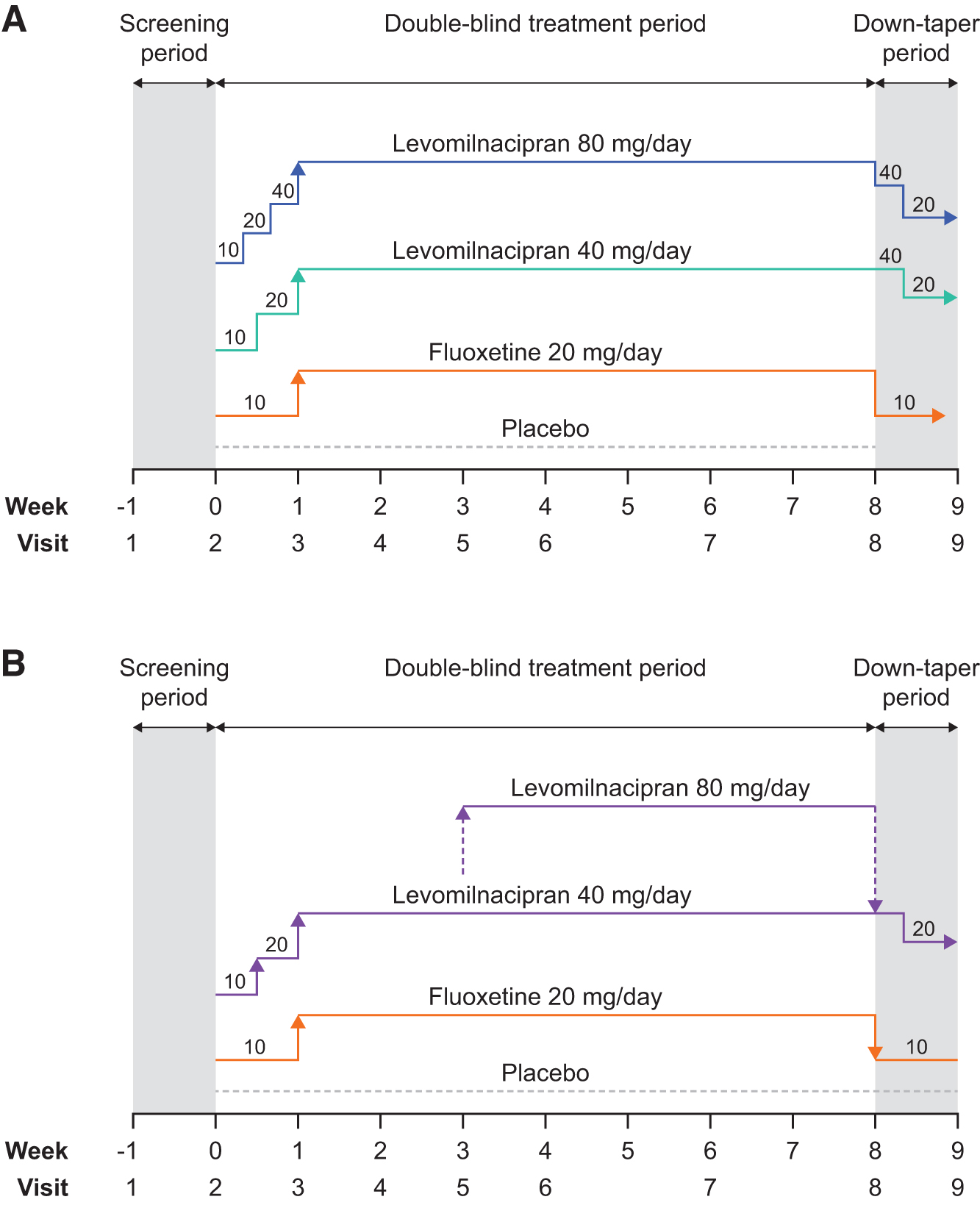
Dosing schedule in the
Patient populations
Eligible patients for LVM-MD-11 were children and adolescents aged 12–17 years who met the relevant Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-4-TR) criteria for MDD (American Psychiatric Association, 2000). Patients enrolled in LVM-MD-14 were aged 7–17 years and diagnosed with MDD according to criteria in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) (American Psychiatric Association, 2013).
Patients enrolled in both studies had diagnoses confirmed according to the Kiddie Schedule for Affective Disorders—Present and Lifetime (K-SADS-PL) (Kaufman et al., 1997), a score of 40 or more on the Children's Depression Rating Scale—Revised (CDRS-R) total score (Poznanski et al., 1984) at Visits 1 and 2, a Clinical Global Impressions-Severity (CGI-S) score ≥4 at Visits 1 and 2, plus normal physical examinations, electrocardiography, and clinical laboratory findings.
In both studies, efforts were made to ensure reasonable representation with respect to gender, race, and ethnicity to reflect the proportions in the disease population. Exclusion criteria included diagnosis of an axis I disorder that was the primary focus of treatment other than MDD according to DSM-4-TR (for LVM-MD-11) or DSM-5 (for LVM-MD-14), prior diagnosis of intellectual disability, and significant risk of suicide (any suicide attempt within the past year or significant risk at screening/baseline visit as judged by the investigator based on the psychiatric interview or information collected in the Columbia-Suicide Severity Rating Scale [C-SSRS]). Patients who had received prohibited concomitant medications or any psychoactive drug within 5 half-lives before baseline, or psychotherapy or behavior therapy within 1 month before Visit 1, were also excluded.
Dosing schedule
In both LVM-MD-11 and LVM-MD-14, patients were randomized to receive placebo, levomilnacipran, or fluoxetine (Fig. 1). Once-daily doses of levomilnacipran were based on the adult pharmacokinetic (PK) modeling data (AbbVie, Inc., 2023b): In LVM-MD-11, patients were randomized to receive a fixed dose of levomilnacipran 40 mg, levomilnacipran 80 mg, fluoxetine 20 mg, or placebo. The levomilnacipran 40-mg group received a 10 mg dose of levomilnacipran on Days 1 to 2, 20 mg on Days 3 to 7, and 40 mg daily during Weeks 2 to 8. The levomilnacipran 80 mg group received 10 mg of levomilnacipran on Days 1 to 2, 20 mg on Days 3 to 4, 40 mg on Days 5 to 7, and 80 mg daily during Weeks 2 to 8. A flexible dosing schedule was used in LVM-MD-14: Patients randomized to levomilnacipran received a once-daily dose of 10 mg on Days 1 to 3, 20 mg on Days 4 to 7, and 40 mg during Weeks 2 to 8. The decision to increase levomilnacipran dosing to 80 mg daily at Week 3 was based on response and tolerability. In both studies, patients randomized to fluoxetine received once-daily fluoxetine 10 mg on Days 1 to 7 and 20 mg during Weeks 2 to 8.
All randomized patients completing the 8-week double-blind period then entered the down-tapering schedule (shown in Fig. 1). All patients were required to complete the termination visit (Visit 8) and safety follow-up visit (Visit 9). Patients who prematurely discontinued the study also entered the down-tapering period and were required to complete an early termination visit and safety follow-up visit.
Outcome measures
The primary efficacy endpoint of each study was change in CDRS-R total score from baseline to end of Week 8. The secondary efficacy endpoint was changed in CGI-S from baseline to Week 8. Rate of CDRS-R remission (defined as CDRS-R total score ≤28) and CGI-I were also assessed. Safety evaluations included adverse events and serious adverse events (SAEs), defined as any untoward medical occurrence that resulted in death or life-threatening circumstances, hospitalization, persistent/significant disability, or birth defects. An adverse event was considered a treatment-emergent adverse event (TEAE) if it began or worsened during the double-blind treatment period or between the date of the first dose and 30 days after last dose of the double-blind study drug (inclusive) if the patient did not enter the double-blind down-taper period.
Each patient underwent physical examination, and vital signs (including height, weight, and electrocardiography) were recorded. Clinical laboratory tests were performed, and liver function was assessed for drug-induced liver injury according to Hy's law criteria (alanine aminotransferase or aspartate aminotransferase elevation ≥3 times the upper limit of normal, total bilirubin elevation ≥2 times the upper limit of normal, and alkaline phosphatase <2 times the upper limit of normal). Patients were also assessed according to the C-SSRS (Posner et al., 2011). Sparse blood samples were collected at Visits 5, 6, and 7 for PK analyses, which allowed for assessment of population PK and compliance. Efficacy assessments were conducted by experienced clinicians meeting and maintaining training requirements and qualifications standards established by the sponsor and rater training vendor.
Statistical analyses
Descriptive safety analyses were performed on the safety population from each study, which comprised randomized patients who received ≥1 dose of the double-blind investigational product. Efficacy analyses were based on the intention-to-treat (ITT) populations, which comprised patients in the safety population who had a baseline and ≥1 postbaseline assessment of the CDRS-R total score. Both primary and secondary efficacy analyses used a mixed effects model for repeated measures, with treatment group, study center, visit, and treatment group-by-center interaction as the fixed effects, and baseline value and baseline value-by-visit interaction as the covariates. In LVM-MD-11, the matched parallel gatekeeping procedure was applied to control for the overall type I error rate for multiple comparisons across primary and secondary hypotheses for the two dose groups of levomilnacipran compared with placebo. In LVM-MD-14, a fixed sequence procedure was applied to control for the overall type I error rate for multiple comparisons across primary and secondary hypotheses for the levomilnacipran flexible-dose group compared with placebo.
The CGI-I score was analyzed using the MMRM approach, similar to the one used for the primary efficacy parameter, except that the baseline CGI-S score was used as the baseline variable. A post hoc CGI-I responder analysis based on dichotomized CGI-I values was performed; results are presented as summaries without modeling or statistical comparison given the post hoc nature of these analyses. CDRS-R remission rate was analyzed with a generalized linear mixed model based on logit link function, with random intercept and fixed terms for treatment group, visit, treatment-by-visit interaction, and baseline score. If the model did not converge, a logistic regression model with treatment group and the baseline score as explanatory variables was used. For the logistic regression analysis, the postbaseline missing data were imputed using the last observation carried forward approach.
Ethics
The study was conducted according to the ethical principles that have their origin in the Declaration of Helsinki. Study protocols were approved by the institutional review board/independent ethics committees before the studies commenced. All patients provided informed assent to participate, and the patients' parent/caregiver gave written informed consent. The only compensation provided to patients for study participation was for travel costs.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
Patient disposition
In study LVM-MD-11, the first patient visit occurred in June 2015 and the last patient visit occurred in August 2019. In total, 807 patients were screened for enrollment, and 552 of those were randomized for treatment (Supplementary Fig. S1A). The number of patients screened and randomized by each study site is indicated in Supplementary Table S1. Of 547 patients who received ≥1 dose of the double-blind investigational product (safety population), 134 patients received levomilnacipran 40 mg daily, 138 received levomilnacipran 80 mg daily, 134 received fluoxetine 20 mg daily, and 141 received placebo; 546 patients were included in the ITT population. In total, 448 patients (81.9% of the safety population) completed the double-blind treatment period, and 431 patients (78.8%) completed the down-tapering period.
In study LVM-MD-14, the first patient visit occurred in July 2018 and the last patient visit occurred in March 2021. In total, 748 patients were screened for enrollment and 501 were randomized for treatment (Supplementary Fig. S1B). The number of patients screened and randomized by each study site is indicated in Supplementary Table S2. Of 492 patients who received ≥1 dose of the double-blind investigational product (safety population), 166 patients received levomilnacipran 40 to 80 mg daily, 166 received fluoxetine 20 mg daily, and 160 received placebo; 487 patients were included in the ITT population. In total, 436 patients (88.6% of the safety population) completed the double-blind treatment period, and 417 patients (84.8%) completed the down-tapering period.
Reasons for premature treatment discontinuation in both studies are shown in Supplementary Table S3.
Patient demographics and clinical characteristics
Patient demographics and characteristics at baseline in the safety populations for each study are presented in Table 1. The mean age was 14.7 years in LVM-MD-11 and 13.5 years in LVM-MD-14. Most study participants were female (65%–66%) and White (61%).
Baseline Demographic and Clinical Characteristics in Studies LVM-MD-11 and LVM-MD-14 (Safety Population)
Other includes reports of Asian, American Indian, Alaska Native, Hawaiian, Pacific Islander, and multiple races.
Calculated from the ITT population: LVM 40 to 80 mg daily, n = 162; fluoxetine 20 mg daily, n = 166; placebo, n = 159; total, n = 487.
ITT, intention-to-treat; LVM, levomilnacipran; MDD, major depressive disorder; SD, standard deviation.
Around two-thirds of patients in both studies had experienced MDD for less than 3 years. Duration of MDD was >5 years in 13.9% and 11.6% of patients enrolled in LVM-MD-11 and LVM-MD-14, respectively. At the time of screening, the mean duration of the current MDD episode was 9.4 months in LVD-MD-11 and 11.5 months in LVD-MD-14.
Primary efficacy endpoints: Change in CDRS-R total score
In LVM-MD-11, mean baseline CDRS-R total score ranged from 59.4 to 61.8 across all treatment groups. Similarly, the mean CDRS-R total score ranged from 60.8 to 60.9 across all treatment groups in LVM-MD-14 (Table 2). A steady reduction in CDRS-R total score was observed across all treatment arms from baseline to Week 8 of the double-blind treatment period. Figure 2A shows the least squares mean (LSM) change in CDRS-R total score by visit for patients in LVM-MD-11. No significant differences were observed in the LSM change in CDRS-R total scores between placebo (LSM change −22.9) and levomilnacipran 40 mg daily (LSM change −23.3; p = 0.80) or levomilnacipran 80 mg daily (LSM change −22.6; p = 0.87; Table 2). Similar reductions in LSM change in CDRS-R total score were observed from baseline to Week 8 across the treatment arms in LVM-MD-14 (Fig. 2B), and no significant differences were observed between patients receiving placebo (LSM change −21.3) and levomilnacipran 40 to 80 mg daily (LSM change −23.0; p = 0.22; Table 2).
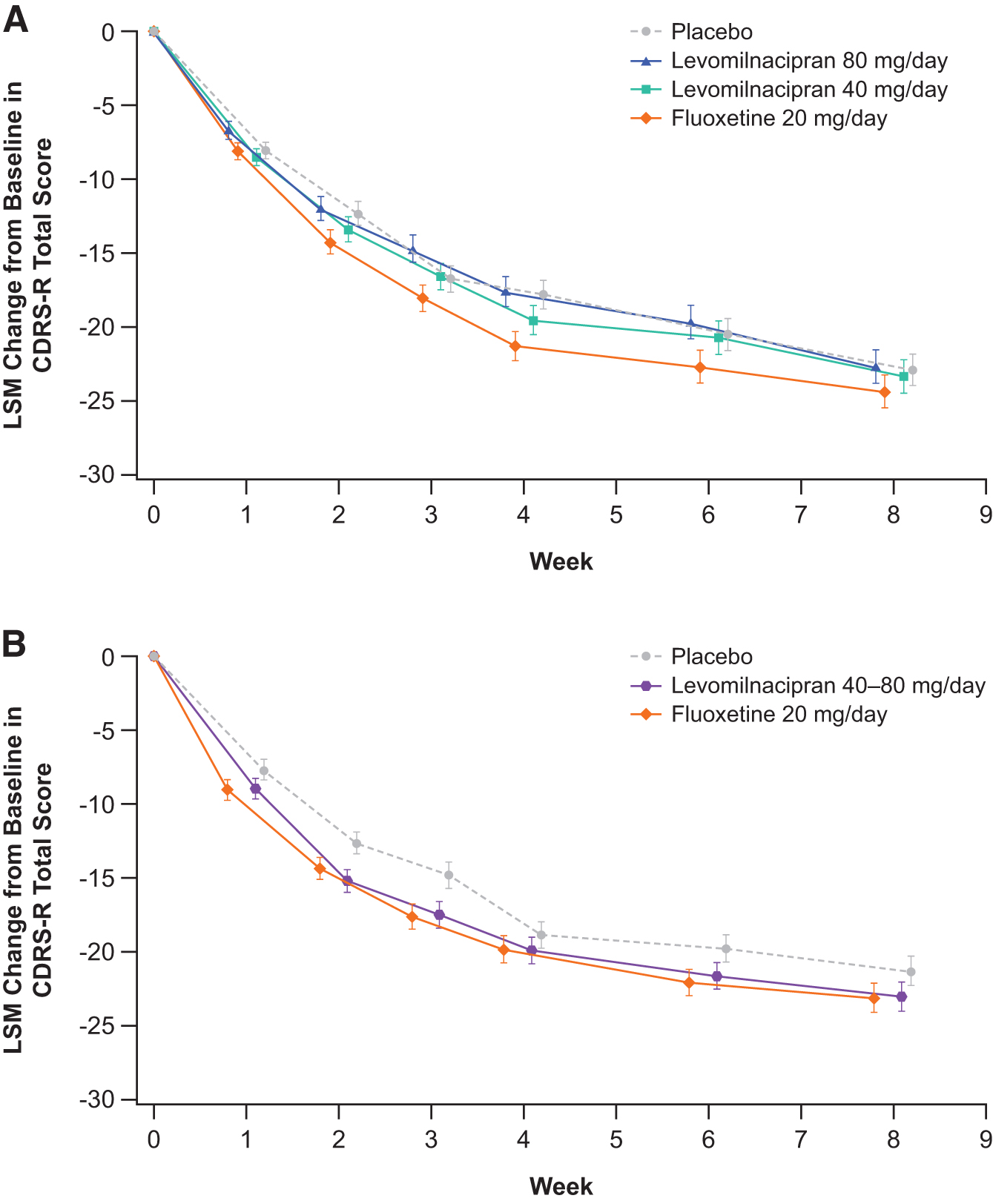
LSM change from baseline in CDRS-R total score by visit (ITT population) in
Primary Efficacy Endpoints: Change in Children's Depression Rating Scale-Revised Total Score from Baseline to Week 8 (Intention-to-Treat Population)
The estimates and the p-values for treatment comparisons are based on a mixed effects model for repeated measures, with treatment group, study center, visit, and treatment group-by-visit interaction as fixed effects, and baseline and baseline-by-visit as covariates using an unstructured covariance matrix.
Adjusted p-values obtained using the matched parallel gatekeeping procedure.
CI, confidence interval; LSM, least-squares mean; LVM, levomilnacipran; SD, standard deviation; SE, standard error.
In both studies, there were no significant differences in change in CDRS-R total score with fluoxetine 20 mg compared with placebo (LSM change −24.4, p = 0.34 in LVM-MD-11; LSM change −23.1, p = 0.20 in LVM-MD-14; Fig. 2 and Table 2). The proportion of patients in remission in each group is shown in Supplementary Table S4.
Secondary efficacy endpoints: Change in CGI-S score
In both studies, the median CGI-S score was reduced from 5 at baseline to 3 by Week 8 of the double-bind treatment period in all treatment groups (Table 3).
Secondary Efficacy Endpoints: Change in Clinical Global Impressions-Severity Score from Baseline to Week 8 (Intention-to-Treat Population)
The estimates and the p-values for treatment comparisons are based on a mixed effects model for repeated measures, with treatment group, study center, visit, and treatment group-by-visit interaction as fixed effects, and baseline and baseline-by-visit as covariates using an unstructured covariance matrix.
CI, confidence interval; LSM, least squares mean; LVM, levomilnacipran; SD, standard deviation; SE, standard error.
Among patients who received placebo in LVM-MD-11, the LSM change in CGI-S score was −1.5, and no significant differences were observed with levomilnacipran 40 mg (LSM change in CGI-S score −1.5; p = 0.88) or levomilnacipran 80 mg (LSM change in CGI-S score −1.5; p = 0.92) compared with placebo (Table 3). In LVM-MD-14, no significant difference in LSM change in CGI-S score was observed between levomilnacipran 40 to 80 mg (LSM change in CGI-S score −1.6) and placebo (LSM change in CGI-S score −1.5; p = 0.68; Table 3). An LSM change in CGI-S score of −1.7 was observed in patients receiving fluoxetine 20 mg daily in both studies, and no significant differences were observed between the fluoxetine and placebo groups (Table 3). CGI-I scores by visit are shown in Supplementary Table S5 and CGI-I responder status by visit is shown in Supplementary Table S6.
Safety and tolerability
In general, the safety profile of levomilnacipran in LVM-MD-11 and in LVM-MD-14 was comparable with that of fluoxetine. In LVM-MD-11, TEAEs were observed in 60.4% of the levomilnacipran 40 mg group, 58.7% of the levomilnacipran 80 mg group, 52.2% of the fluoxetine group, and 45.4% of the placebo group (Table 4). In LVM-MD-14, TEAEs were observed in 47.0% of the levomilnacipran 40 to 80 mg group, 48.8% of the fluoxetine group, and 44.4% of the placebo group (Table 4). Higher frequencies of nausea, vomiting, and tachycardia were reported in patients receiving levomilnacipran than in patients who received placebo or fluoxetine (Table 4).
Adverse Events Occurring During the Double-Blind Treatment Period (Safety Population)
AE, adverse event; LVM, levomilnacipran; SAE, serious adverse event; TEAE, treatment-emergent adverse event.
Adverse events led to discontinuation of the study treatment in 30 patients in LVM-MD-11 (5.5%) and 8 patients in LVM-MD-14 (1.6%). Reasons for discontinuation were mostly isolated events and included tachycardia, nausea, vomiting, depression, and headache (Supplementary Table S7). The incidence of SAEs, including suicidal ideation and/or suicide attempt, was low and similar across treatment groups (Table 4). In LVM-MD-11, SAEs were reported in two patients (1.5%) who received levomilnacipran 40 mg (suicide attempt and overdose) and four patients (3.0%) who received fluoxetine 20 mg (suicidal ideation, suicide attempt, intentional overdose, and fecaloma; Table 4). In LVM-MD-14, SAEs were reported in two patients (1.2%) in the placebo group (suicide attempt and major depression), one patient (0.6%) who received levomilnacipran 40 to 80 mg (major depression), and four patients (2.4%) who received fluoxetine 20 mg (two cases of suicidal ideation, one of bipolar I disorder, and one of intentional self-injury). There were no deaths during either study.
In both LVM-MD-11 and LVM-MD-14, changes in hematology, chemistry, and urinalysis parameters from baseline to the end of the study were small and similar across treatment groups. In LVM-MD-11, mild increases in blood pressure occurred in one patient (0.7%) who received levomilnacipran 40 mg and one patient (0.7%) who received levomilnacipran 80 mg, while no cases of increased blood pressure occurred with fluoxetine 20 mg. In LVM-MD-14, three cases of increased blood pressure (two mild [1.2%]; one moderate [0.6%]) occurred with levomilnacipran 40 to 80 mg, and one (0.6%) mild case occurred with fluoxetine 20 mg. Increases in blood pressure did not occur in the placebo groups of LVM-MD-11 or LVM-MD-14. No patients met potential Hy's law criteria for drug-induced liver injury by the end of either study.
Discussion
The psychosocial burden of MDD in pediatric patients, along with the potential lack of efficacy and safety implications of existing SSRIs in these populations, means that there is a significant unmet need for additional treatment options for MDD in children and adolescents. Currently, no SNRIs are approved for use in children with MDD, and the studies LVM-MD-11 and LVM-MD-14 were designed to determine the efficacy and safety of the SNRI levomilnacipran in children and adolescents with MDD.
The primary efficacy endpoint (change in CDRS-R total score) was not met in either LVM-MD-11 or LVM-MD-14. In patients aged 12–17 years enrolled in LVM-MD-11, there was no significant difference in the change in CDRS-R total score between patients treated with levomilnacipran 40 or 80 mg daily, and those who received placebo. Similarly, among patients with MDD aged 7–17 years enrolled in LVM-MD-14, levomilnacipran 40 to 80 mg daily showed no significant change in CDRS-R total score compared with placebo. In addition, no significant differences between levomilnacipran 40 or 80 mg daily versus placebo were observed in the change in CGI-S score or clinical remission rates. There were also no clinically significant differences in CGI-I score or, based on a post hoc analysis, CGI-I responder rate versus placebo.
While these studies were not powered to measure the differences in efficacy between levomilnacipran and fluoxetine for the treatment of children and adolescents with MDD, fluoxetine 20 mg daily, the standard of care in pediatric MDD, was included as an active control for validation of both studies. Interestingly, the magnitude of change in CDRS-R total score with fluoxetine was similar to that reported in the pivotal trial for children and adolescents with MDD (Emslie et al., 2002). However, in the current studies, the primary and secondary efficacy endpoints for fluoxetine were not significantly different versus placebo, implying lack of assay sensitivity in these studies and making it difficult to draw conclusions on efficacy.
Placebo response is an important factor to recognize in psychiatric clinical trials, and it has previously been reported that children and adolescents with MDD may be more responsive to placebo than adult patients (Meister et al., 2020; Mossman et al., 2021). Placebo-related improvements in CDRS-R score in LVM-MD-11 and LVM-MD-14 are consistent with previous reports from clinical trials of vilazodone and fluoxetine for pediatric and adolescent patients with depressive disorders (Emslie et al., 2002; Findling et al., 2020). However, the effect of placebo on CDRS-R total score in the current studies was larger than that observed in the pivotal study with fluoxetine (Emslie et al., 2002). It is likely, therefore, that the large effect of placebo on improvements in efficacy endpoints in LVM-MD-11 and LVM-MD-14 reduced the detection of differences between levomilnacipran and placebo groups. The absence of a placebo “run-in” period may have contributed to the placebo response in LVM-MD-11 and LVM-MD-14. It is also possible that the magnitude of the placebo response in children and adolescents treated for MDD has increased over time, as previously reported (Bridge et al., 2009).
Other factors that may contribute (at least in part) to the placebo response in children and adolescents include baseline severity of illness, patient age, the likelihood of receiving placebo in a clinical trial, and, interestingly, the number of participating study sites (Bridge et al., 2009; Meister et al., 2020; Papakostas and Fava 2009). While baseline clinical parameters were comparable between LVM-MD-11, LVM-MD-14, and previous studies (Emslie et al., 2002; Findling et al., 2020), the number of study sites was higher than that reported for the pivotal fluoxetine trial (Emslie et al., 2002). Identification of individual patient-level demographic and clinical characteristics that may be predictive of a placebo response may be one way to increase the sensitivity of clinical trials for MDD in children and adolescents (Bridge et al., 2009). It is also worth noting that the COVID-19 pandemic, which was at its peak during the LVM-MD-14 study, may have impacted trial enrollment and patient responses.
The double-blind periods of LVM-MD-11 and LVM-MD-14 were only 8 weeks, and it cannot be discounted that the observed effect of levomilnacipran treatment may have continued to increase over time with longer double-blind treatment periods. Another limitation of this study is the requirement that patients have a single primary diagnosis of MDD; while comorbidities including anxiety and attention deficit hyperactivity disorder were allowed, they could not be the primary focus of any treatment. Further, long-term studies and meta-analyses are required to identify the response to treatment of MDD more sensitively in children and adolescents (Meister et al., 2020).
Levomilnacipran was generally well tolerated. The TEAEs reported more frequently in the levomilnacipran versus placebo and fluoxetine groups were nausea, vomiting, and tachycardia, consistent with TEAEs observed in adults with MDD (AbbVie Inc., 2023b). Preexisting hypertension should be controlled before initiation of treatment with levomilnacipran in adult patients, and caution should be exercised in treating patients with preexisting hypertension, cardiovascular, or cerebrovascular conditions that may be compromised by increases in blood pressure. Discontinuation or other appropriate medical intervention should be considered for patients who experience a sustained increase in blood pressure while receiving levomilnacipran (AbbVie Inc., 2023b).
Conclusions
In these phase 3, double-blind, placebo-controlled trials, treatment with levomilnacipran 40 to 80 mg once daily was generally well tolerated. However, the efficacy of levomilnacipran was not statistically superior to placebo for the treatment of MDD in children and adolescents due to the high rates of response in the placebo arms of both studies compared with the placebo response seen in previous studies of antidepressants.
Footnotes
Acknowledgments
The authors would like to thank the patients, their families, and all investigators involved in these studies. The authors wish to acknowledge statistical support provided by Bin Xue, MS, as well as clinical pharmacology support provided by Christine Neumar, PhD, and Nathan Teuscher, PhD (both at Certara, iDD Consulting during their active involvement in these studies). Medical writing and editorial support, including assisting authors with the development of the outline and initial draft and incorporation of comments, was provided by Keri Wellington, PhD, Valerie Moss, PhD, CMPP, and Rosie Henderson, MSc, of Onyx (a division of Prime, London, UK), supported by AbbVie Inc. The sponsor was involved in the study design, collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors.
Authors' Contributions
M.A. was involved in study recruitment and data acquisition, writing—original draft, writing—review and editing; W.Z.R. was involved in data curation, formal analysis, methodology, software, supervision, validation, visualization, writing—original draft, writing—review and editing; M.G. was involved in data interpretation, writing—review and editing; E.G. was involved in investigation, supervision, validation, writing—original draft, writing—review and editing; D.T.R. was involved in conceptualization, data curation, formal analysis, methodology, project administration, resources, supervision, writing—original draft, writing—review and editing.
Disclosures
W.R., M.G., E.G. are employees of AbbVie; D.T.R. is a former employee of AbbVie; M.A. declares no conflicts of interest.
Funding Information
This study was sponsored by AbbVie.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.