
Abstract
Objectives:
To evaluate the efficacy and safety of once-daily serdexmethylphenidate/dexmethylphenidate (SDX/d-MPH) capsules (Azstarys™) compared with placebo in children with attention-deficit/hyperactivity disorder (ADHD) in a randomized, double-blind, dose-optimized laboratory classroom study.
Methods:
Children ages 6–12 with ADHD were enrolled. During a 3-week, open-label, Dose Optimization Phase, subjects initiated treatment with 39.2 mg/7.8 mg/day of SDX/d-MPH and were titrated weekly to an optimal dose (maximum dose of 52.3/10.4 mg). During the double-blind Treatment Phase, subjects were randomized to receive their optimal dose of SDX/d-MPH or placebo for 7 days. On day 7, efficacy was assessed in the laboratory classroom using the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Rating Scale and Permanent Product Measure of Performance (PERMP). To evaluate safety, adverse events (AEs), vital signs, and electrocardiograms were assessed, and suicide risk was assessed.
Results:
A total of 149 subjects completed the study. In the primary efficacy analysis, the mean postdose change from baseline in SKAMP-Combined scores averaged over the laboratory classroom day was significantly improved with SDX/d-MPH versus placebo (least-squares mean treatment difference [95% confidence interval]: −5.41 [−7.10 to −3.71]; p < 0.001). A significant treatment effect for SDX/d-MPH compared with placebo was observed from 1 to 10 hours postdose. A post hoc analysis more comparable with that conducted in similar studies indicated a 0.5- to 13-hour onset and duration of efficacy. Both average postdose PERMP-Attempted and PERMP-Correct score changes from baseline were significantly improved among those treated with SDX/d-MPH versus placebo (p < 0.001 for both). No serious AEs were reported. During the Dose Optimization Phase, two-thirds of subjects reported AEs; the most common being insomnia and decreased appetite.
Conclusions:
SDX/d-MPH showed significant improvement in ADHD symptoms compared with placebo in children 6–12 years of age, with a rapid onset and extended duration of treatment effect. SDX/d-MPH was safe, with AEs comparable with those observed with other stimulant treatments.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a chronic neurobehavioral disorder that affects children, adolescents, and adults. The worldwide prevalence of ADHD in children and adolescents is estimated to be between 3.4% and 7.2% based on meta-analyses (Polanczyk et al. 2007, 2015; Willcutt 2012; Thomas et al. 2015). Studies performed in the United States report higher rates of diagnosis, likely due in part to variability in diagnostic processes (Wolraich et al. 2014; Danielson et al. 2018). ADHD is characterized by symptoms of impulsivity, hyperactivity, and/or inattention, which can lead to impairments in academic, social, emotional, behavioral, occupational, and executive functioning (American Psychiatric Association 2013). The symptoms of ADHD and associated impairments can lead to poor long-term outcomes, including diminished self-esteem, educational and employment difficulties, relationship problems, increased risk of substance abuse, and reduced quality of life, if not appropriately managed (Barkley et al. 1996; Murphy and Barkley 1996; Mannuzza et al. 1997; Biederman et al. 1998; Harpin 2005; Matza et al. 2005; Lee et al. 2011; Wilens 2011; Agarwal et al. 2012; Harpin et al. 2013; Mattos et al. 2013; Erskine et al. 2016). Moreover, health outcomes, including mortality, are adversely impacted (Dalsgaard et al. 2015; Instanes et al. 2018). ADHD symptoms are associated with several of the top causes of overall mortality, including cigarette smoking, obesity, premature accidental death, and suicide (Stickley et al. 2016; Balazs and Kereszteny 2017; Cortese and Tessari 2017; CADDRA 2018; van Amsterdam et al. 2018).
Among pharmacological treatments for ADHD, psychostimulants have demonstrated the largest effect size in randomized, controlled trials and are the first-line treatments for ADHD; however, there remain therapeutic challenges with these agents (Swanson et al. 2001; Pliszka and AACAP Work Group on Quality Issues 2007; CADDRA 2018; Cortese et al. 2018; Wolraich et al. 2019). Extended-release (ER) stimulants are often preferred because of their duration of effect, reduced need for multiple doses, tendency for better tolerability, and decreased potential for abuse and diversion compared with short-acting stimulants (CADDRA 2018). Despite the availability of various once-daily methylphenidate products, children/adolescents and their caregivers have reported inconsistent or incomplete symptom control while taking ADHD medications with associated negative impacts, including poor school performance, behavioral problems, and difficulty with peer relations (Sikirica et al. 2015). It is common for clinicians to add a short-acting stimulant or prescribe other adjunct nonstimulant treatments when ADHD symptoms are not adequately controlled throughout the day with monotherapy (Childress and Tran 2016; CADDRA 2018; Wolraich et al. 2019).
Although emphasis is often placed on treating ADHD symptoms while in the classroom, the duration of effect usually needs to extend outside of school hours to improve overall quality of life. Inadequate symptom control at the beginning of the day can contribute to a stressful morning routine for children and their caregivers, whereas an insufficient duration of efficacy leading to poor symptom control toward the end of the day can interfere with evening activities, including homework, extracurricular activities, and family life (Sallee 2015; Sikirica et al. 2015). Parents of children and adolescents with ADHD have reported that the early morning routine and evening homework periods were the times of the day when uncontrolled ADHD symptoms were most severe, even when receiving stimulant treatment at a stable dose. Parents also reported that uncontrolled ADHD symptoms in the early morning led to stress, feelings of overwhelm, and adverse effects on the parent–child relationship, with parents often waking the child for earlier medication administration in an attempt to avoid early morning dysfunction (Sallee 2015). This suggests that a single pharmacological treatment that provides both a rapid onset of symptom control in the morning and sustained duration of symptom control throughout the waking day is an unmet need in the treatment of pediatric ADHD.
Serdexmethylphenidate/dexmethylphenidate (SDX/d-MPH) is a recently approved ADHD product (Azstarys™) containing a molar ratio of 70% serdexmethylphenidate (SDX), a novel prodrug of d-methylphenidate (d-MPH), and 30% d-MPH HCl. SDX, a Schedule IV controlled substance, is pharmacologically inactive until gradually converted to active d-MPH in the lower intestinal tract. The pharmacokinetic profile of SDX/d-MPH exhibits a single d-MPH concentration peak, followed by a gradual elimination curve characterized by a long half-life (T1/2: 11.7 hours) (Corium 2021). After oral administration, early exposure to d-MPH is governed primarily by the d-MPH component, and mid- to late-day exposure is determined primarily by the gradual conversion of inactive SDX to active d-MPH. Whereas most once-daily stimulant products utilize a formulation-based approach to impart delayed- and/or ER properties, SDX/d-MPH utilizes a prodrug approach to produce a unique, extended-duration pharmacokinetic profile. A similar approach to prolonging drug exposure has been successfully achieved with the d-amphetamine prodrug, lisdexamfetamine (Vyvanse®).
The objective of this study was to evaluate the efficacy (including onset and duration of effect), safety, and tolerability of SDX/d-MPH capsules compared with placebo in children (6–12 years of age) with ADHD using a laboratory classroom protocol. The setting used in this study mimics a school classroom and allows for repeated assessments of efficacy throughout the day (Wigal and Wigal 2006). This study design has been used extensively to evaluate the overall efficacy and time-dependent pharmacodynamic effects of stimulants throughout the day (Wigal and Wigal 2006; Muniz et al. 2008; Wigal et al. 2014, 2017; Childress et al. 2017, 2020).
Methods
Study design
This was a multicenter, dose-optimized, randomized, double-blind, placebo-controlled, parallel, laboratory classroom study in children 6–12 years of age with ADHD (Clinical Trial NCT03292952). Recruitment began on November 20, 2017. The study was conducted at five sites in the United States starting in December 2017, with the last follow-up visit occurring on May 16, 2018. All study sites were experienced with the laboratory classroom protocol used in this study, and rater training was conducted to standardize the rating process across sites. As shown in Figure 1, the study consisted of a Screening Period, an open-label Dose Optimization Phase, a double-blind Treatment Phase, and a follow-up visit. Participants who met criteria for screening underwent a 5-day washout period of their current stimulant ADHD treatments before dose optimization of SDX/d-MPH.
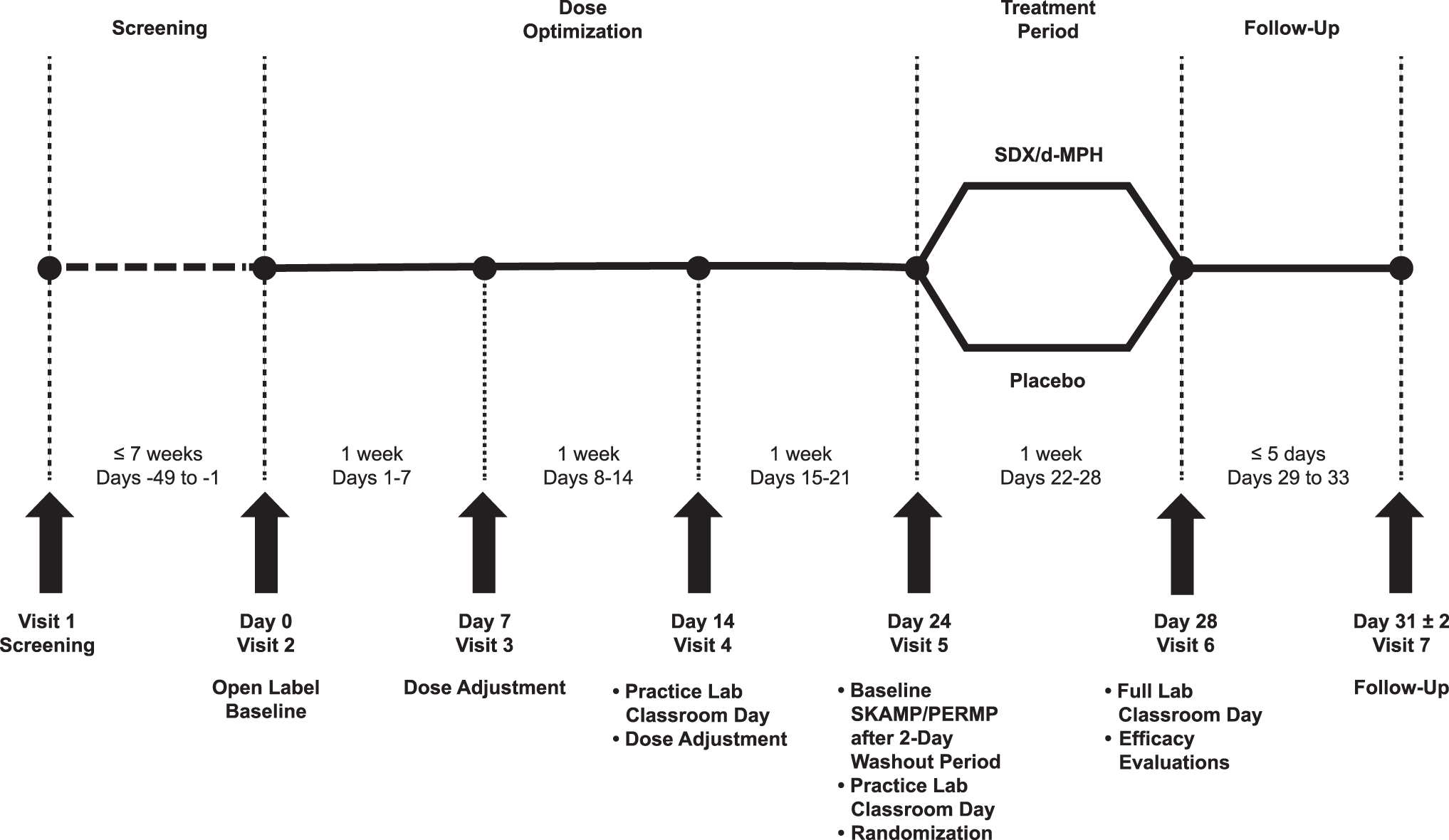
Study design. The study days listed for Visits 3, 4, and 5 may have deviated ±3 days for individual subjects such that the total duration of the Dose Optimization Phase was 21 ± 3 days (duration ranging from 18 to 24 days). Any deviation of the targeted 21-day Dose Optimization Phase was carried over into the actual days for the subsequent visits: Visit 6 at days 28 ± 3 and Visit 7 ranging from days 29 ± 3 to days 33 ± 3.
During the 3-week, open-label Dose Optimization Phase, subjects were titrated with SDX/d-MPH capsules, at doses of 26.1 mg/5.2 mg, 39.2 mg/7.8 mg, and 52.3 mg/10.4 mg (molar equivalent to 20, 30, and 40 mg of total d-MPH HCl, respectively), based on tolerability and best individual dose response in the opinion of the investigator. The starting dose for all subjects was 39.2 mg/7.8 mg. At the end of this phase, participants must have demonstrated a reduction in ADHD-Rating Scale-5 (ADHD-RS-5) score of ≥30% from baseline, a clinician-administered Clinical Global Impressions-Improvement (CGI-I) score of 1 or 2 (“Very Much Improved” or “Much Improved”), and acceptable tolerability of the optimized SDX/d-MPH dose to be eligible to enter the double-blind Treatment Phase. During the last day of the Dose Optimization Phase (Visit 5), after a 2-day washout period from SDX/d-MPH, subjects took part in an abbreviated, practice laboratory classroom day during which their final dose of open-label SDX/d-MPH was administered. Baseline Swanson, Kotkin, Agler, M-Flynn, and Pelham-Combined (SKAMP-C) scores were obtained before dosing.
During the Treatment Phase, eligible subjects were randomized on a 1:1 basis to receive their optimized dose of SDX/d-MPH or placebo for 7 days. Randomization was stratified by study site. Neither the subject, the investigator, nor the sponsor knew a given subject's treatment assignment. The appropriate blinded treatment was taken at home, once daily in the morning, on days 22–27. The final dose of the study drug was administered by study staff on the seventh day of the double-blind Treatment Phase (day 28; Visit 6) at the laboratory school. Efficacy and safety assessments were performed on this day according to a laboratory classroom protocol (Wigal and Wigal 2006). Subjects completed a follow-up visit (day 31; Visit 7) to evaluate safety parameters 3 days after the last dose of study drug. Upon completion of the study and after the database was locked according to the sponsor (or designee) operating procedures, the randomization codes were provided to the statistician to unblind the study.
The study was approved by an Institutional Review Board. Before conducting any study-related activities, at least one parent or legal guardian provided voluntary written permission for study subjects to participate in the study. In addition, the subject provided written or verbal assent to participate. This study was conducted per the ethical principles outlined in the 2008 version of the Declaration of Helsinki, and was designed, conducted, recorded, and reported in compliance with the principles of Good Clinical Practice guidelines per the International Council for Harmonization (ICH E6[R2]). The full trial protocol can be accessed at clinicaltrials.gov.
Subjects
Eligible subjects were children (male or female) aged 6–12 years with a primary diagnosis of ADHD (combined, inattentive, or hyperactive/impulsive presentation). ADHD diagnosis was based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (American Psychiatric Association 2013) criteria, and confirmed by the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID). Subjects must have had a score of at least 3 (mildly ill) on the clinician-administered Clinical Global Impressions-Severity (CGI-S) scale and an ADHD-RS-5 total score of at least 28.
Exclusion criteria included known nonresponse to MPH treatment, history of allergic reaction or sensitivity to MPH, history of a substance use disorder, clinically significant medical abnormalities such as cardiovascular abnormalities, and any chronic condition of the central nervous system. Participants were excluded if they had any of the following: bipolar I or II disorder, major depressive disorder, conduct disorder, obsessive-compulsive disorder, any history of psychosis, autism spectrum disorder, disruptive mood dysregulation disorder, intellectual disability, Tourette syndrome, or confirmed genetic disorder with cognitive and/or behavioral disturbances. They were also excluded if found to have clinically significant suicidal ideation or a history of suicide attempt, as assessed by the Columbia-Suicide Severity Rating Scale (C-SSRS).
Study assessments
The primary efficacy assessment was the evaluation of classroom behaviors using the SKAMP Rating Scale. The SKAMP scale is a validated measure of subjective impairment of classroom behaviors in children with ADHD. Trained study personnel rated subjects according to a 7-point Likert scale on 13 items, grouped into subcategories for attention, deportment, quality of work, and compliance (Swanson et al. 1998; Wigal et al. 1998). The SKAMP-C score is the sum of the rating values for each of the 13 items (total range from 0 to 78), with higher scores indicating greater impairment (Wigal and Wigal 2006; Wigal et al. 2013). The primary endpoint was mean change from baseline (measured at predose Visit 5) in all SKAMP-C scores collected postdose and averaged across the laboratory classroom day at Visit 6, with assessments conducted at 0.5, 1, 2, 4, 8, 10, 12, and 13 hours postdose.
The key secondary endpoint was mean change from baseline (predose Visit 5) in SKAMP-C scores measured at each time point, with statistically significant differences used to determine onset and duration of efficacy. The onset of effect was defined as the first postdose assessment time showing a statistically significant (p < 0.05) difference in SKAMP-C score between SDX/d-MPH and placebo. The duration of treatment effect was defined as the length of the time interval between the first time point demonstrating a statistically significant difference between SDX/d-MPH and placebo until the first time point when the treatment difference was no longer statistically significant, or until the last time point of the classroom day.
Other secondary endpoints included change from baseline in scores measured at each time point and the average of the scores collected across the laboratory classroom day at Visit 6 for SKAMP-Deportment (SKAMP-D) and SKAMP-Attention (SKAMP-A) scores, as well as Permanent Product Measure of Performance (PERMP) scores. In addition, Weekly Rating of Evening and Morning Behavior—Revised (WREMB-R) scores (total score, and morning and evening subscores) were assessed at Baseline (Visit 2), Visit 5, and Visit 6.
The SKAMP-A subscale score is calculated by taking the average of items 1–4 of the SKAMP scale, while the SKAMP-D subscale score is obtained by averaging items 5–8. The PERMP test is a skill-adjusted, 10-minute math test used to assess attention in children with ADHD (Wigal and Wigal 2006). Subjects were instructed to work at their seats and complete as many problems as possible in 10 minutes. Performance was evaluated using two scores: the number of problems attempted (PERMP-A) and the number of problems correct (PERMP-C). The 11-item WREMB-R is a parent-rated questionnaire developed to assess behaviors for their severity during the morning hours (3 items) and evening hours (8 items), with possible scores for each item ranging from 0 (no difficulty) to 3 (a lot of difficulty) (Carlson et al. 2007).
ADHD-RS-5 scores were collected as an exploratory endpoint designed to assess changes in ADHD symptom severity from week to week during the Dose Optimization Phase. The ADHD-RS-5 is an 18-item scale based on DSM-5 criteria of ADHD that rates symptoms on a 4-point scale (American Psychiatric Association 2013). Each item is scored using a combination of severity and frequency ratings with a range of 0 (reflecting no symptoms or a frequency of never or rarely) to 3 (reflecting severe symptoms or a frequency of very often).
On the morning of the laboratory classroom day (Visit 6), marked differences in mean predose SKAMP-C scores were observed between the SDX/d-MPH and placebo treatment groups. To control for these differences, a post hoc analysis of the primary (mean score across all time points) and key secondary (onset and duration of efficacy) endpoints was performed using predose Visit 6 scores as the SKAMP-C baseline. The predose assessment on the morning of the classroom day has been consistently assigned as baseline in other classroom studies of methylphenidate products (Muniz et al. 2008; Wigal et al. 2013, 2017; Childress et al. 2017, 2020). Post hoc analyses were also conducted for the PERMP endpoint.
Safety assessments
The occurrence of treatment-emergent adverse events (TEAEs) was assessed at each visit, beginning with the first dose of the Dose Optimization Phase and ending with the follow-up or early termination visit. Additional safety evaluations included physical examinations, vital signs, height and weight, electrocardiogram (ECG) parameters, clinical laboratory tests, and the C-SSRS, which were administered at each study visit.
Statistical analysis
Efficacy analyses were conducted in the intent-to-treat (ITT) population. For the primary efficacy assessment, change from baseline in SKAMP-C scores, an analysis using a Mixed-effects Model Repeated Measure (MMRM) model was done to estimate the difference between the SDX/d-MPH group and the placebo group. The MMRM model included subject as random effect and postdose time, treatment, and the interaction of time, treatment, and site as fixed effects. The baseline SKAMP-C score was included as a covariate. The output data included the model-adjusted average change from baseline across all postdose SKAMP-C scores for each treatment group and the corresponding treatment group difference (SDX/d-MPH–placebo) with standard error and 95% confidence intervals (CIs). Secondary endpoints including scores and subscores of the SKAMP and PERMP measures used the same model analysis methods described for the SKAMP-C endpoint. To evaluate treatment group differences on the WREMB-R, changes in mean scores from Visit 2 (baseline) to Visit 6 were analyzed using a two-sample t-test. For all analyses, a p-value of <0.05 was defined to indicate statistical significance.
Determination of sample size was based on the following criteria: assuming a mean difference between test and control for change in SKAMP-C of 0.5 units (Norman et al. 2003; Biederman et al. 2007) and a standard deviation (SD) of 1.0, 126 subjects would need to complete the study to achieve 80% power when testing a significance level of α = 0.05 (two-sided, two-sample z-test). Assuming an ∼20% dropout during the open-label Dose Optimization Phase, 176 subjects were planned for enrollment in that phase. Assuming an ∼10% dropout rate during the double-blind Treatment Phase, ∼140 subjects were planned for randomization into that phase, with the intention of completing the study with ∼126 subjects.
Results
Subject disposition
A total of 155 subjects were enrolled in the open-label Dose Optimization Phase, 4 of whom discontinued due to a TEAE and 1 who discontinued before entering the Treatment Phase because of failure to meet randomization criteria. A total of 150 participants were randomized into the Treatment Phase, with 74 in the SDX/d-MPH group and 76 in the placebo group (Fig. 2). All subjects in the SDX/d-MPH group completed the study through the follow-up visit, while one subject in the placebo group was lost to follow-up.
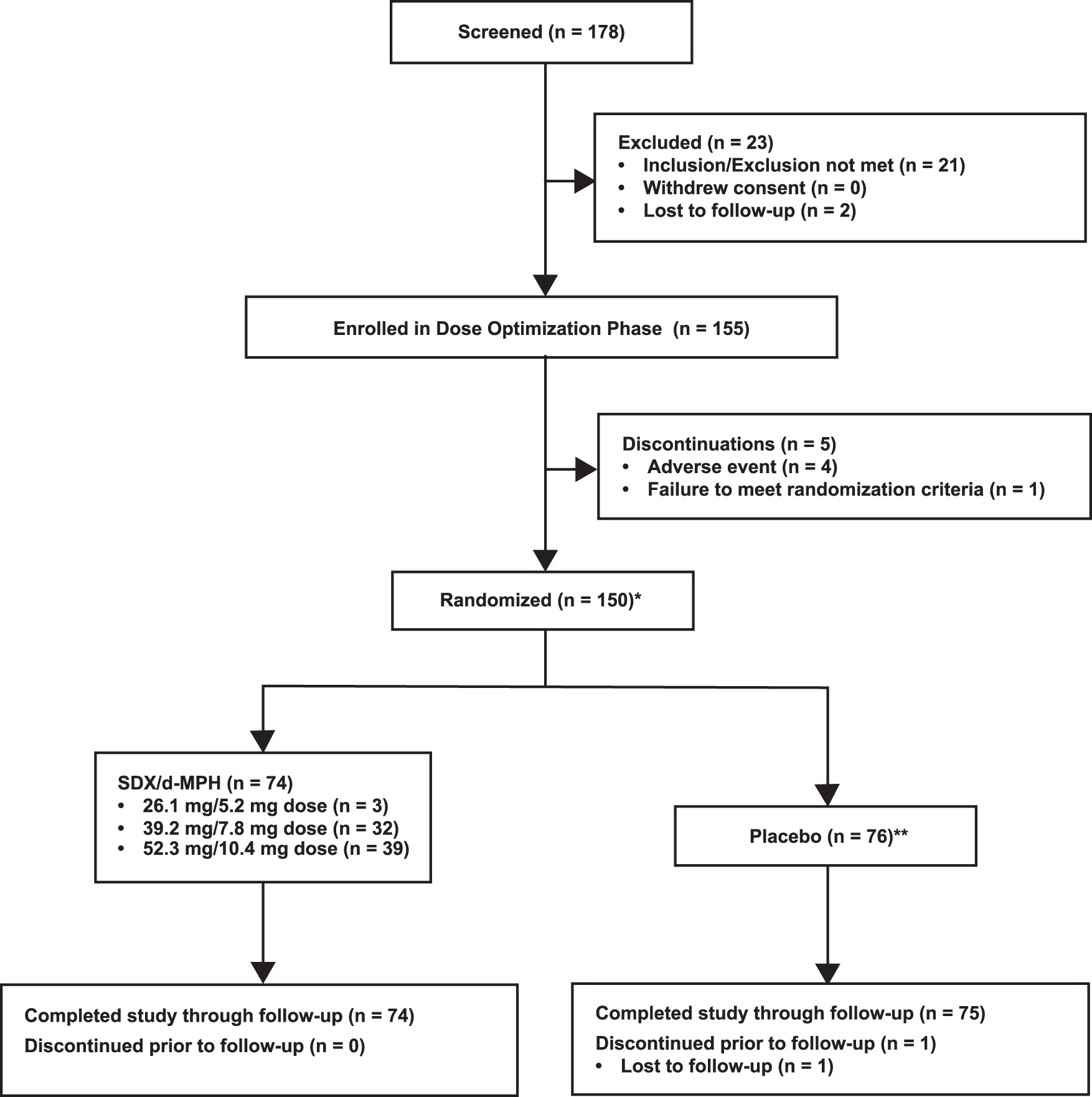
Subject disposition. *All randomized subjects received at least one dose of double-blind study drug. **The site found one subject ineligible after completing the study.
Baseline and demographic characteristics
In the ITT population, the mean age was 9.6 years, most subjects were male (61.3%), and most were diagnosed with a combined ADHD subtype (88.0%). Overall, demographic and baseline characteristics were well balanced, with no significant differences observed between treatment groups (Table 1). Of the 150 subjects, 61 (40.7%) had prior treatment with ≥1 ADHD medications, with MPH products being the most common (24%), followed by amphetamine products (14.7%). Among the SDX/d-MPH group, 29 (39.2%) had prior treatment with an ADHD medication compared with 32 (42.1%) in the placebo group.
Subject Demographics and Baseline Characteristics (Intent-to-Treat Population)
ADHD, attention-deficit/hyperactivity disorder; CGI-S, Clinical Global Impressions-Severity; SD, standard deviation; SDX/d-MPH, serdexmethylphenidate/dexmethylphenidate.
Efficacy results
Primary endpoint
The mean postdose change from baseline (predose Visit 5) in SKAMP-C scores averaged over the laboratory classroom day (Visit 6) was significantly lower (i.e., improved) for the SDX/d-MPH group compared with the placebo group (least-squares [LS] mean treatment difference [95% CI]: −5.41 [−7.10 to −3.71]; p < 0.001; Table 2). A post hoc analysis in which predose Visit 6 was used as the SKAMP-C baseline resulted in similar treatment differences for the primary endpoint (LS mean treatment difference [95% CI]: −7.27 [−9.00 to −5.53]; p < 0.001; Table 2). Notably, on the morning of the laboratory classroom day (Visit 6), mean predose SKAMP-C score change from baseline was higher (i.e., more severe symptoms) in the SDX/d-MPH group compared with the placebo group, a difference that reached statistical significance (LS mean difference [95% CI]: 2.37 [0.07 to 4.68], p = 0.044).
Analysis of Swanson, Kotkin, Agler, M-Flynn, and Pelham-Combined Primary Endpoint (Intent-to-Treat Population)
Prespecified analysis of primary endpoint.
Post hoc analysis of primary endpoint.
LS, least-squares; SD, standard deviation; SDX/d-MPH, serdexmethylphenidate/dexmethylphenidate; SE, standard error; SKAMP-C, Swanson, Kotkin, Agler, M-Flynn, and Pelham-Combined.
The effects of SDX/d-MPH on SKAMP-C scores were a consequence of an improvement in both SKAMP-D and SKAMP-A subscales. The mean postdose change from baseline (predose Visit 5) in SKAMP-D and SKAMP-A scores across all postdose time points was significantly lower (i.e., improved) in the SDX/d-MPH group compared with the placebo group (LS means difference [95% CI] SKAMP-D: −1.38 [−2.00 to −0.76] and SKAMP-A: −1.37 [−1.91 to −0.84]; p < 0.001 for both scores).
Secondary endpoints
Figure 3 shows mean change from baseline (either predose Visit 5 or Visit 6) in SKAMP-C scores during assessments taken at 0.5, 1, 2, 4, 8, 10, 12, and 13 hours postdose on the classroom day (Visit 6). Based on the prespecified analysis (predose Visit 5 as baseline) for determining onset and duration of efficacy, a statistically significant treatment effect for SDX/d-MPH compared with placebo started at 1 hour postdose and continued until 10 hours postdose. As noted above, on the laboratory classroom day (Visit 6), predose SKAMP-C scores were significantly higher (i.e., more severe symptoms) in the SDX/d-MPH group relative to the placebo group, an effect commonly observed in similar trials of stimulant products (Wigal et al. 2013; Childress et al. 2017, 2020). To control for these predose differences in SKAMP-C scores in a manner consistent with other trials, a post hoc analysis was conducted using predose Visit 6 scores as the SKAMP-C baseline. With this more conventional analysis (see Discussion section), statistically significant differences were observed at every time point, resulting in an onset and duration of efficacy of 0.5–13 hours postdose.
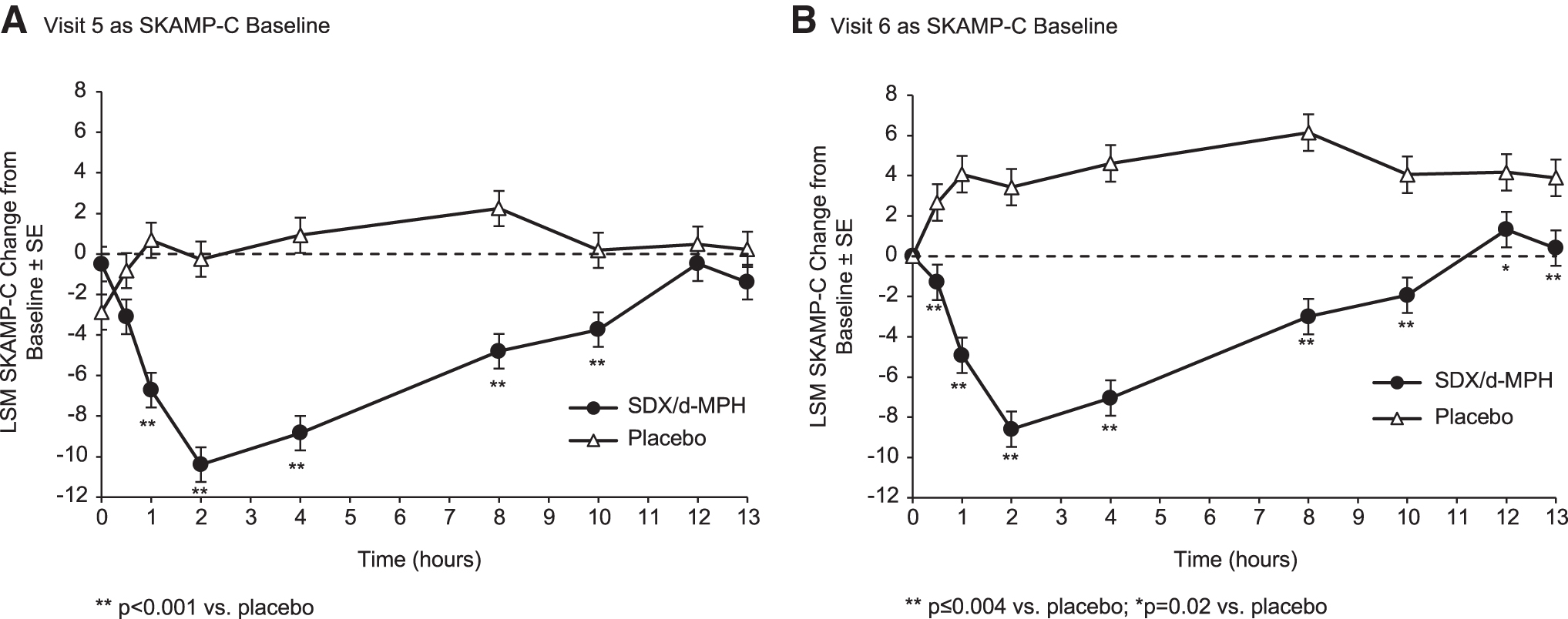
LSM change from baseline in SKAMP-C scores using predose Visit 5
Both average postdose PERMP-A and PERMP-C score changes from baseline (predose Visit 5) on the laboratory classroom day were significantly improved in the SDX/d-MPH group compared with the placebo group (LS means difference [95% CI]; PERMP-A: 23.58 [15.12 to 32.03] and PERMP-C: 24.51 [16.12 to 32.90]; p < 0.001 for both scores). Figure 4 shows mean change from baseline in PERMP-A and PERMP-C scores during assessments taken at 0.5, 1, 2, 4, 8, 10, 12, and 13 hours postdose. Regardless of whether Visit 5 (prespecified) or Visit 6 (post hoc) was used as baseline, statistically significant differences between treatment groups were observed for PERMP-A or PERMP-C at every time point. Therefore, PERMP measures indicated onset and duration of efficacy of SDX/d-MPH from 0.5 to 13 hours postdose.
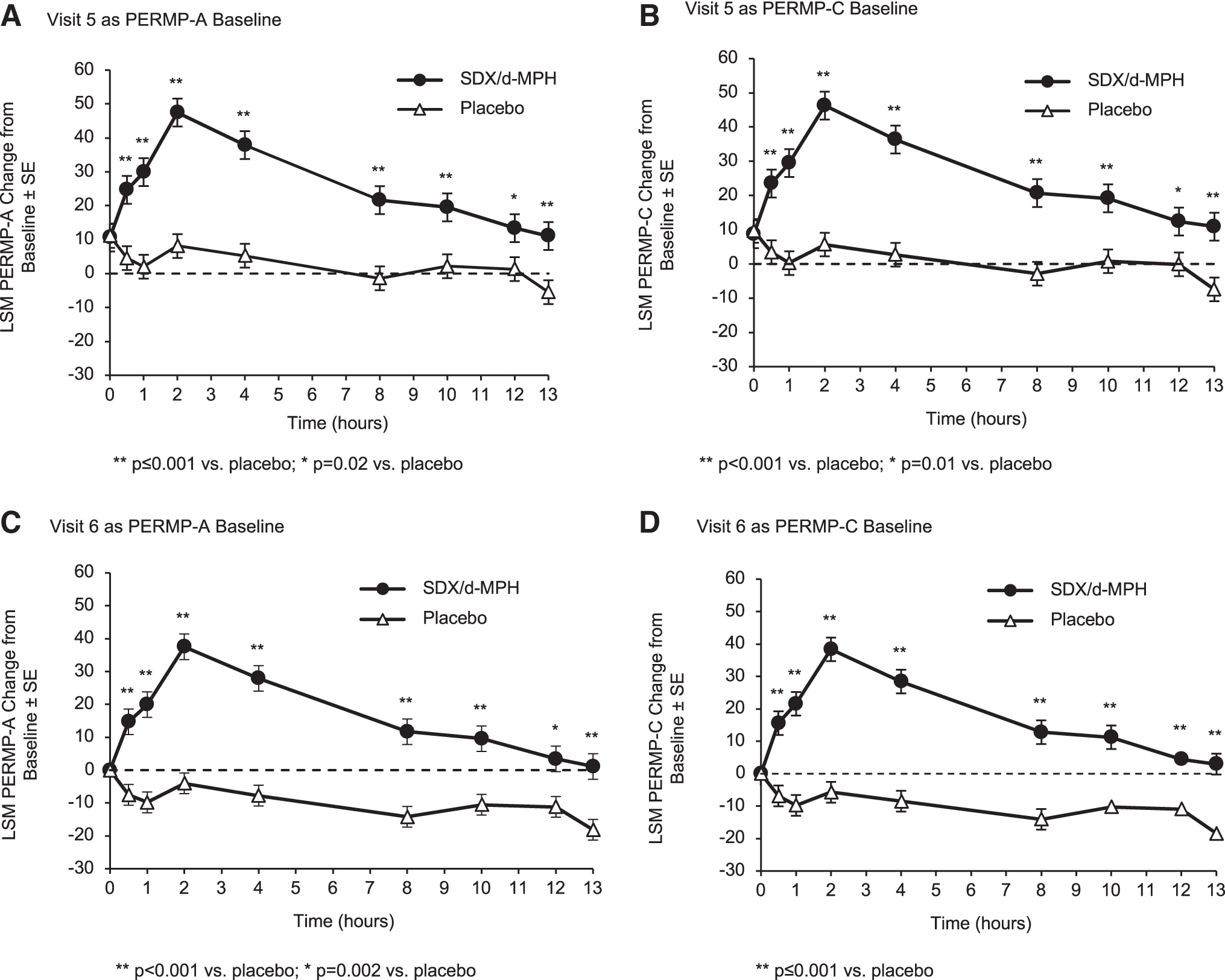
LSM change from baseline in PERMP-A scores and PERMP-C scores using predose Visit 5 as the baseline
During the Dose Optimization and Treatment Phases, morning and evening behavioral symptoms were assessed on the parent-rated WREMB-R scale. At Visit 5 of the Dose Optimization Phase (prerandomization), there were marked improvements in WREMB-R scores, as evidenced by statistically significant reductions from baseline (Visit 2) in overall WREMB-R score (mean difference [SD]: −12.2 [8.6]; p < 0.001) and morning (mean difference [SD]: −2.9 [3.2]; p < 0.001) and evening (mean difference [SD]: −9.3 [6.6]; p < 0.001) subscale scores. Table 3 shows WREMB-R scores during the double-blind Treatment Phase. At Visit 6, mean (SD) changes from baseline in overall WREMB-R scores were significantly greater in the SDX/d-MPH group versus placebo group (−11.7 [9.2] vs. −7.2 [9.1]; p = 0.003). This overall effect was primarily accounted for by improvements on the evening subscale, where improvements from baseline were significantly greater in the SDX/d-MPH versus placebo group (−9.2 [6.7] vs. −5.0 [6.6]; p < 0.001) (Table 3). Because improvements during the Dose Optimization Phase were largely maintained in both treatment groups during the Treatment Phase, no significant differences between SDX/d-MPH and placebo were observed on the morning subscale.
Mean Change from Baseline in Weekly Rating of Evening and Morning Behavior—Revised Scores During Treatment Phase
p Values based on paired t-tests of mean differences from baseline for SDX/d-MPH versus placebo.
SD, standard deviation; SDX/d-MPH, serdexmethylphenidate/dexmethylphenidate; WREMB-R, Weekly Rating of Evening and Morning Behavior—Revised.
Exploratory endpoint
During the Dose Optimization Phase, ADHD severity from week to week was also assessed by the ADHD-RS-5. As shown in Table 4, mean overall ADHD-RS-5 scores, as well as the Inattentiveness and Hyperactivity/Impulsivity subscale scores, showed improvement compared with baseline (Visit 2) when measured 1 week after initiating open-label SDX/d-MPH treatment (Visit 3). Additional incremental improvements were observed at Visits 4 and 5 as titration and dose optimization continued for 2 more weeks.
Mean Change in Attention-Deficit/Hyperactivity Disorder Rating Scale-5 Scores During Open-Label, Dose Optimization Phase
SD, standard deviation.
Safety and tolerability results
No serious adverse events (AEs) were reported in this study. In the open-label Dose Optimization Phase, approximately two-thirds of subjects (67.1%) experienced at least one TEAE, with a majority of TEAEs rated as mild (56.8%) or moderate (29.7%) in severity. The most common TEAEs in this phase were decreased appetite (24.5%), insomnia (15.5%), affect lability (11.6%), upper abdominal pain (9.7%), headache (7.7%), and irritability (7.7%). Four subjects experienced AEs leading to drug discontinuation in this phase. One subject experienced insomnia and upper abdominal pain, which were assessed as “probably related” to the study drug. Another subject experienced multiple adverse effects that were deemed “probably related” to the study drug, including bronchospasm, severe insomnia, abdominal pain, dizziness, tearfulness, constricted affect, and dry mouth. One participant experienced headache, while another was withdrawn due to headbanging and affect lability. These AEs were assessed as “possibly related” to the study drug. Resolution of AEs among these four participants occurred after drug discontinuation.
Table 5 shows TEAEs (>2% incidence) during the Treatment Phase that occurred more frequently in the SDX/d-MPH versus placebo group, including headache (5.4%), upper abdominal pain (4.1%), insomnia (2.7%), and pharyngitis (2.7%).
Treatment-Emergent Adverse Events (>2% Incidence) Occurring More Frequently in Subjects Receiving Serdexmethylphenidate/Dexmethylphenidate Versus Placebo During the Treatment Phase (Overall Safety Population)
SDX/d-MPH, serdexmethylphenidate/dexmethylphenidate; TEAE, treatment-emergent adverse event.
Changes in physical examination, vital signs, laboratory tests, and ECG parameters were minimal and comparable between treatment groups. In the Dose Optimization Phase, 3 (1.9%) subjects scored positive on item 1 (wish to be dead) of the “suicidal ideation” section of the C-SSRS, one of whom also scored positive on item 2 (nonspecific active suicidal thoughts). Based on the opinion of the investigator, these subjects were allowed to continue in the study. No subjects reported suicidal thoughts during the Treatment Phase.
Discussion
In this multicenter, randomized, double-blind, placebo-controlled laboratory classroom study, SDX/d-MPH was shown to be efficacious and well tolerated in children aged 6–12 years with ADHD. Efficacy was established for the primary endpoint, the SKAMP-C, and secondary endpoints, including the SKAMP-D, SKAMP-A, PERMP-A, PERMP-C, and WREMB-R. AEs observed with SDX/d-MPH were typical of stimulant therapy, and the majority were rated as mild to moderate in severity.
A key objective of this study was to characterize the onset and duration of efficacy across the laboratory classroom day. When onset and duration of efficacy were evaluated using the prerandomization SKAMP-C baseline (Visit 5), statistically significant differences versus placebo were observed from 1 to 10 hours postdose. When onset and duration of efficacy were evaluated using the methodology consistent with other methylphenidate products—that is, by using the postrandomization SKAMP-C baseline captured on the morning of the laboratory classroom day (Visit 6)—statistically significant differences were observed from 0.5 to 13 hours postdose. The latter approach, a post hoc analysis in this study, is typical for similarly designed studies, and allows for the pharmacodynamic effects of SDX/d-MPH to be compared more readily with other once-daily methylphenidate products (Muniz et al. 2008; Wigal et al. 2013, 2017; Childress et al. 2017, 2020). PERMP-A and PERMP-C scores, two additional secondary endpoints that examined the time-dependent effects of SDX/d-MPH across the day, corroborated the early onset and extended duration of efficacy. These endpoints demonstrated significant differences versus placebo from 0.5 to 13 hours postdose regardless of whether Visit 5 or Visit 6 was used as the PERMP baseline.
On the morning of the laboratory classroom day, baseline SKAMP-C scores were significantly higher (i.e., more severe symptoms) in subjects treated with SDX/d-MPH compared with placebo. This “rebound” effect, manifested as worsening ADHD symptoms near the end of each daily dosing cycle, has been repeatedly observed in similarly designed laboratory classroom studies of stimulant products (Wigal et al. 2013, 2017; Childress et al. 2017, 2020). In seminal studies of stimulant effects on behavior in “hyperkinetic” children, a similar profile of worsening symptoms before the next scheduled dose was noted in both behavioral assessments and objective measures such as activity counts (Katz et al. 1975; Rapoport et al. 1978; Chatoor et al. 1983). Analogously, in behavioral pharmacology studies of rats treated chronically with stimulants, dopamine-mediated behaviors that were initially enhanced after stimulant administration were subsequently depressed below basal levels for 24–72 hours after cessation of chronic treatment (Haenlein et al. 1985; Barrett et al. 1992). These collective findings suggest that after a period of chronic administration, opponent processes that outlast the acute effects of the stimulant may last for several days or more. While the neurobiological mechanisms underlying this apparent “rebound” effect are not entirely clear, one possible explanation may be a homeostatic disruption of basal dopaminergic signaling in response to repeated stimulant administration (Grace 1991; Siciliano et al. 2015).
In classroom studies of stimulant products, subjects are typically randomized into active drug and placebo groups after an open-label titration phase. All subjects participate in identical study procedures before entering the double-blind Treatment Phase, resulting in randomized treatment groups with similar mean dopaminergic signaling and therefore similar mean ADHD symptoms at the beginning of the Treatment Phase. As double-blind treatment continues, however, mean predose dopamine signaling and thus ADHD symptoms typically diverge between study groups as one continues to receive daily stimulant treatment and the other placebo, resulting in disparate SKAMP-C baseline scores on the morning of the laboratory classroom day.
This apparent “rebound” phenomenon has implications for understanding the onset and duration, and likely the overall magnitude of observed efficacy in laboratory classroom studies. Specifically, statistical techniques that control for predose baseline differences between the active drug and placebo groups on the test day are more likely to demonstrate a faster onset and longer duration of efficacy than statistical techniques that fail to account for such differences. This is exemplified in this study by comparing onset and duration of efficacy calculated by two analyses, one of which utilized predose Visit 5 and the other the more conventional predose Visit 6 as the SKAMP-C baseline. The SKAMP-C analysis using change from predose Visit 5 as the endpoint was equivalent to not implementing any statistical adjustment since all subjects had been treated similarly before randomization (at Visit 5), and active and placebo groups consequently had nearly identical baseline scores. The resulting data of this analysis, therefore, describe the improvement of ADHD symptoms with SDX/d-MPH throughout the day relative to symptoms in the morning before treatment initiation with a stimulant. The SKAMP-C analysis using change from predose Visit 6 (which was different between the active and placebo groups) as the endpoint shows the effects of SDX/d-MPH on ADHD symptoms throughout the day relative to the symptoms in the morning during continued treatment with SDX/d-MPH. While arguments can be made regarding which baseline is more clinically meaningful for interpreting the pharmacodynamic effects of stimulants, additional research is warranted to characterize apparent “rebound” effects in subjects treated with stimulant products.
Currently approved formulation-based ER methylphenidate products have differing onsets and durations of efficacy that are due to product-specific factors, such as the drug release technology and the ratio of immediate-release (IR) to ER components that result in various exposure levels and shapes of the pharmacokinetic curve. Despite the availability of these products, patients with ADHD and caregivers commonly report that stimulant medications either take too long to alleviate symptoms, or wear off in the afternoon or early evening (Sallee 2015; Sikirica et al. 2015). Consequently, it is common for clinicians to prescribe multiple daily doses of an ER stimulant, or augment an ER stimulant with an IR stimulant to achieve the desired onset and duration of efficacy (Childress and Tran 2016; CADDRA 2018; Wolraich et al. 2019). SDX/d-MPH, a novel product comprised of the d-MPH prodrug, SDX, and d-MPH in a 70:30 molar ratio, can potentially afford a true once-daily MPH product without the need for augmentation therapy. In this study, SDX/d-MPH produced a rapid onset (0.5 hours) and extended duration of efficacy (13 hours), as evidenced by endpoints, including the SKAMP-C, PERMP-A, and PERMP-C. If this profile is confirmed in real-world settings, SDX/d-MPH will provide the ability to address some of the aforementioned unmet needs in the treatment of ADHD.
Treatment with SDX/d-MPH was safe and generally well tolerated. The AE profile was consistent with that expected of a stimulant product, with the most common AEs during open-label treatment being decreased appetite and insomnia. These and other AEs were infrequently reported during the subsequent double-blind Treatment Phase. This finding is concordant with similarly designed studies of stimulants in which AEs were more common during initial dose titration and less common during double-blind treatment with an optimized dose (Wigal et al. 2009, 2014, 2017; Childress et al. 2015, 2017). Importantly, the introduction of the prodrug, SDX, did not result in any new or unusual AEs not typically reported with methylphenidate products.
This study has limitations. The duration of the double-blind Treatment Phase, while consistent with similar studies, was relatively short. A long-term study of SDX/d-MPH demonstrated the safety and efficacy of SDX/d-MPH for up to 1 year in children with ADHD, albeit in an open-label design (Childress et al. 2021). In addition, the eligibility criteria resulted in a fairly homogeneous sample of children without comorbidities, potentially limiting the generalizability of the findings (Biederman et al. 1991; Gillberg et al. 2004).
Conclusion
In this dose-optimized, randomized, controlled laboratory classroom study, SDX/d-MPH showed significant improvements in ADHD symptoms compared with placebo in children 6–12 years of age, with a rapid onset and extended duration of effect. SDX/d-MPH was safe and generally well tolerated, with adverse effects comparable with those observed with other stimulant medications.
Clinical Significance
SDX/d-MPH is a recently approved ADHD product (Azstarys) containing a molar ratio of 70% SDX, a novel prodrug of d-MPH, and 30% d-MPH. In this study of children (6–12 years of age) with ADHD, SDX/d-MPH was efficacious and generally well tolerated, with a rapid onset and extended duration of effect.
Footnotes
Authors' Contributions
All the authors were actively involved in data interpretation, article writing and editing. In addition, Drs Kollins, Braekman, Guenther, Barrett, Mickle, Marraffino, Cutler and Brams were involved in study concept development and study conduct.
Acknowledgments
Editorial services were performed by Lauren Derhodge, DO, and Simpson Healthcare Executives.
Disclosures
S.H.K. is an employee of Holmusk and a consultant for Akili Interactive, where he is also a shareholder. He has also consulted for Arbor Pharmaceuticals, LLC, Ironshore Pharmaceuticals, Inc., KemPharm, Inc., Otsuka America Pharmaceutical, Inc., Shire, Tris Pharma, Inc. He has received research support from Akili Interactive, BehaVR, Bose Corporation, KemPharm, Inc., Limbix Health, Inc., Neos Therapeutics, OnDosis AB, Sana Health, Inc., Shire, Tali Health, Tris Pharma, Inc. R.B. is a full-time employee and shareholder of KemPharm, Inc. S.G. is a full-time employee and shareholder of KemPharm, Inc. A.C.B. is a full-time employee and shareholder of KemPharm, Inc. T.C.M. is a full-time employee and shareholder of KemPharm, Inc. C.O. is a full-time employee and shareholder of Corium, Inc. A.M. has received research support from Acadia Pharmaceuticals, Akili Interactive, Allergan, Arbor Pharmaceuticals, LLC, Avanir, Boehringer Ingelheim Pharmaceuticals, Inc., Eisai, Inc., Ironshore Pharmaceuticals & Development, Inc., KemPharm, Inc., Neos Therapeutics, Novartis Pharmaceuticals Corporation, Otsuka America Pharmaceutical, Inc., Purdue Pharma, Roche, Sage Therapeutics, Shire, Sunovion Pharmaceuticals, Inc., Supernus Pharmaceuticals, Inc., Takeda Pharmaceutical Company Ltd., Tonix Pharmaceuticals, Tris Pharma. She has consulted for Ironshore Pharmaceuticals & Development, Inc., KemPharm, Inc., and Supernus Pharmaceuticals, Inc. A.J.C. has received grants for clinical research from Aevi Genomic Medicine, Inc., Akili Interactive, Arbor Pharmaceuticals, LLC, Ironshore Pharmaceuticals, Inc., KemPharm, Inc., Lundbeck, Neos Therapeutics, Inc., Noven Pharmaceuticals, Inc., Otsuka America Pharmaceuticals, Inc., Purdue Pharma, LP, Shire, Sunovion Pharmaceuticals, Inc., Supernus Pharmaceuticals, Inc., Takeda Pharmaceutical Company, Ltd., Tris Pharma, Inc., and served as an advisor, consultant, or speaker for Adlon Therapeutics, LP, Aevi Genomic Medicine, Inc., AiCure, Akili Interactive, Arbor Pharmaceuticals, LLC, Attentive Therapeutics, Inc., Ironshore Pharmaceuticals, Inc., KemPharm, Inc., H Lundbeck, AS, MedAvante-ProPhase, Inc., Neos Therapeutics, NLS, Noven, Otsuka America Pharmaceuticals, Inc., Purdue Pharma, LP, Shire, Sunovion Pharmaceuticals, Inc., Supernus Pharmaceuticals, Inc., Takeda Pharmaceutical Company, Ltd, Tris Pharma, Inc. M.N.B. has served as an advisor, consultant, or speaker for Allergan, Arbor Pharmaceuticals, LLC, Janssen Global Serves, LLC, H Lundbeck, AS, OWP Pharmaceuticals, Inc., Neos Therapeutics, Otsuka America Pharmaceuticals, Inc., Shire, Sunovion Pharmaceuticals, Inc., Teva Pharmaceutical Industries, Ltd., Tris Pharma, Inc., Vertical Pharmaceuticals, LLC.