
Abstract
Objectives:
To study the safety and efficacy of the long-acting methylphenidate formulation PRC-063 in adolescents with attention-deficit/hyperactivity disorder (ADHD).
Methods:
Adolescents 12 to ≤17 years who met Diagnostic and Statistical Manual of Mental Disorders (DSM)-5 criteria for ADHD and had a baseline ADHD Rating Scale DSM-5 (ADHD-5-RS) score ≥24 participated in a randomized, double-blind, placebo-controlled, fixed-dose, parallel-group study. Participants were randomized 1:1:1:1:1 to receive placebo or one of four doses of PRC-063 once daily for 4 weeks. The primary endpoint was change from baseline in least-squares mean clinician-rated ADHD-5-RS total score for PRC-063 (all doses combined) versus placebo. Other efficacy assessments included Conners third Edition: Self-Report (C3SR) and Clinical Global Impression-Improvement (CGI-I). A subset of double-blind study participants entered a subsequent open-label, dose-optimized study. Safety outcomes in both studies included treatment-emergent adverse events (TEAEs).
Results:
Three hundred fifty-four participants were included in the primary analysis. The least-squares mean change from baseline in ADHD-5-RS total score was −15.17 for PRC-063 versus −10.98 for placebo (least-squares mean difference −4.2, p = 0.0067). For individual PRC-063 doses, improvements in ADHD-5-RS total score versus placebo were significant for 45 mg (p = 0.0155) and 70 mg (p = 0.0401), but not for 25 or 85 mg. A significant improvement for PRC-063 versus placebo was recorded for C3SR Inattention (p = 0.0168), but not for the other C3SR subscales. About 52.7% of participants randomized to PRC-063 were responders based on CGI-I versus 32.4% of those randomized to placebo (p = 0.0004). Further improvements in ADHD symptoms based on ADHD-5-RS were observed from 1 month through 6 months of open-label treatment (p < 0.0001). There were two serious adverse events (both during the open-label study), one of which (aggressive behavior) was assessed as related to study drug. The only TEAEs that occurred in >10% of participants during double-blind treatment were decreased appetite (20.1%) and headache (15.0%). Most TEAEs were of mild or moderate severity.
Conclusion:
PRC-063 significantly improved ADHD symptomatology in adolescents. It was generally well tolerated, with an AE profile consistent with other long-acting stimulants. NCT02139111 and NCT02168127.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder that is often first seen in childhood, with core symptoms of inattention, impulsivity, and hyperactivity. In epidemiological studies, the prevalence of ADHD in adolescence has ranged from 5.8% in Brazil (Rohde et al. 1999) to 8.5% in Finland (Smalley et al. 2007) and 8.6% in the United States (Merikangas et al. 2010). ADHD is associated with significant behavioral and functional impairments, including academic, occupational, social, and emotional difficulties; legal challenges; risk of accidents and injury; and high-risk behaviors such as smoking, substance abuse, and risky sexual behavior (Shaw et al. 2012; Bhat and Hechtman 2016; Erskine et al. 2016; Harpin et al. 2016). These high-risk behaviors emerge during adolescence (Barkley et al. 1990; Howard et al. 2016), at a time when parental supervision declines and medication adherence can decrease (Molina et al. 2009).
ADHD often requires pharmacological interventions and is typically treated with stimulants. Long-acting methylphenidate and amphetamine are generally recommended as first-line pharmacological agents (Pliszka 2007; CADDRA 2018; NICE 2018; Wolraich et al. 2019; Barbaresi et al. 2020). It remains important to conduct randomized clinical trials in adolescents (13–17 years) as a distinct population because adolescent ADHD is common, impairing, and treatment responsive (Wilens et al. 2006). The recently approved long-acting mixed amphetamine salt preparation Mydayis® (SHP465) demonstrated efficacy when administered to children and adolescents, although with an onset of action of 2 hours (Brams et al. 2018; Wigal et al. 2019).
PRC-063 (marketed as Adhansia XR® in the United States and as FOQUEST® in Canada) is a once daily extended-release formulation of methylphenidate hydrochloride designed to provide a rapid onset (1 hour) and extended duration of action (up to 16 hours in adults) (Childress et al. 2020; Wigal et al. 2020). It is an oral multilayer-release beaded formulation, which delivers a bimodal plasma methylphenidate concentration–time profile. After morning administration in adults, the methylphenidate concentration reaches an initial peak at ∼1.5 hours. This is followed by a moderate decline, and then a gradual increase after 4–6 hours, resulting in a second, higher peak at ∼12 hours postadministration. Methylphenidate levels subsequently decline slowly, with residual levels of ∼18% at 24 hours postadministration. Steady state is achieved after 3 days of dosing (Katzman et al. 2020). In adolescents aged 13–17 years, the first peak methylphenidate concentration occurs at ∼2 hours, and the second at ∼11 hours (Adhansia XR USPI).
A double-blind adult simulated workplace environment study demonstrated significant improvements in mean Permanent Product Measure of Performance (PERMP) scores across the day with PRC-063 relative to placebo. These improvements were observed from 1 hour postdose through 16 hours postdose, confirming both the rapid onset and extended duration of action (Wigal et al. 2020). Similarly, a double-blind laboratory classroom study in children 6–12 years of age showed significant improvements in the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) rating scale scores with PRC-063 compared with placebo, with treatment effects observed from 1 hour postdose up to and including 13 hours postdose, the last time measured (Childress et al., 2020). Onset and duration of effect for PRC-063 have not been studied specifically in the adolescent population.
In addition to these pharmacodynamic studies, the efficacy of PRC-063 in a large community-based sample was recently reported (Weiss et al. 2021). Eligible adults diagnosed with ADHD participated in a 4-week randomized, double-blind, parallel-group, fixed-dose study of PRC-063 versus placebo with the option of continuing to a 6-month open-label extension study. Compared with placebo, PRC-063 gave a greater reduction in ADHD Rating Scale Diagnostic and Statistical Manual of Mental Disorders (DSM)-5 (ADHD-5-RS) total score during the double-blind study, with symptom improvement continuing during the open-label extension when patients were dose optimized.
Adolescents need a quick onset of action for symptom relief before and at the start of the school day, as well as an extended duration of action to facilitate homework in the evening and to help prevent high-risk behaviors after school. This study reports on the safety and efficacy of PRC-063 in a randomized, double-blind, parallel-group, fixed-dose 4-week study in adolescents. Eligible subjects were offered the opportunity to participate in a second 6-month open-label extension study to assess the long-term safety and tolerability of PRC-063. Functional impairment, sleep, executive functioning, and quality of life were also assessed as secondary outcomes and will be reported separately.
Methods
Study conduct
The studies were conducted at sites in the United States and Canada between 2014 and 2015. For each study site, the study protocols were approved by an independent ethics committee or institutional review board, as appropriate. The studies were conducted in accordance with the Declaration of Helsinki, International Council for Harmonisation Good Clinical Practice (GCP), and all applicable local, state, and federal regulations. For each of the studies, study participants provided voluntary written informed assent, and their parents/guardians written informed consent, before any study procedures.
Double-blind study (NCT02139111)
This was a randomized, double-blind, placebo-controlled, fixed-dose, parallel-group study conducted at 42 sites in the United State and Canada.
Participants
Adolescents 12 to ≤17 years of age who met DSM-5 criteria (APA 2013) for diagnosis of any of the three presentations of ADHD (hyperactive/impulsive, inattentive, or combined) were screened for study entry. Eligible subjects had a baseline ADHD-5-RS score ≥24, assessed after a 7-day washout of ADHD treatment (if applicable), and were either treatment-naïve or dissatisfied with their current ADHD pharmacotherapy. Participants had to have age-appropriate intellectual functioning (IQ ≥80 based on the Wechsler Abbreviated Scale of Intelligence (Wechsler 2011) or Kaufman Brief Intelligence Test, Second Edition (Bain and Jaspers 2010)), provide a negative pregnancy test (if female), and demonstrate that they could successfully swallow the largest capsule size used in the study.
Subjects were excluded from the study if they had a primary diagnosis of a comorbid psychiatric condition that was unstable and required treatment, were allergic to or had a history of serious adverse reactions to methylphenidate, or were known to be nonresponsive to methylphenidate. Other exclusion criteria included substance or alcohol abuse; history of a cardiac condition for which stimulants would be contraindicated; history of strokes, epilepsy, or migraine headaches; use of guanethidine, pressor agents, monoamine oxidase inhibitors, coumadin anticoagulants, anticonvulsants, phenylbutazone, tricyclic antidepressants, selective serotonin reuptake inhibitors, or herbal remedies; clinically significant laboratory and/or electrocardiogram (ECG) abnormalities; elevated blood pressure; current suicide risk (as judged by the investigator); and use of an investigational drug or experimental medical device within 30 days of the planned treatment start date. Participants were permitted to remain on a stable dose of melatonin, at the investigator's discretion.
Study design
The study had four phases: (1) screening, 1-week washout, and baseline/randomization to either PRC-063 or placebo; (2) forced-dose titration over a 2-week period; (3) a 2-week dose maintenance period; and (4) a 14-day safety follow-up period, except for participants who entered the 6-month open-label study. The forced-dose titration and dose maintenance period were double-blind and placebo controlled.
After the 1-week washout period, eligible subjects were randomized 1:1:1:1:1 to receive placebo or 25, 45, 70, or 85 mg/day PRC-063. The titration schedule was predetermined. Subjects randomized to receive 25, 45, or 70 mg/day started at 25 mg/day, and subjects randomized to 85 mg/day started at 45 mg/day. Participants had their dose increased weekly, as applicable, until the final dose was reached (maximum of two dose increases) (Fig. 1). Randomization schedules were generated by Y-Prime, Inc., using an integrated web response system. PRC-063 and placebo were supplied in bottles, each containing 10 capsules. To maintain blinding, placebo and PRC-063 at each dose were identical in appearance. No emergency unblinding occurred during the study. Compliance was evaluated by counting and recording the number of dispensed capsules and the number of returned capsules. Noncompliance was defined as missing ≥2 doses from a single bottle of study medication.
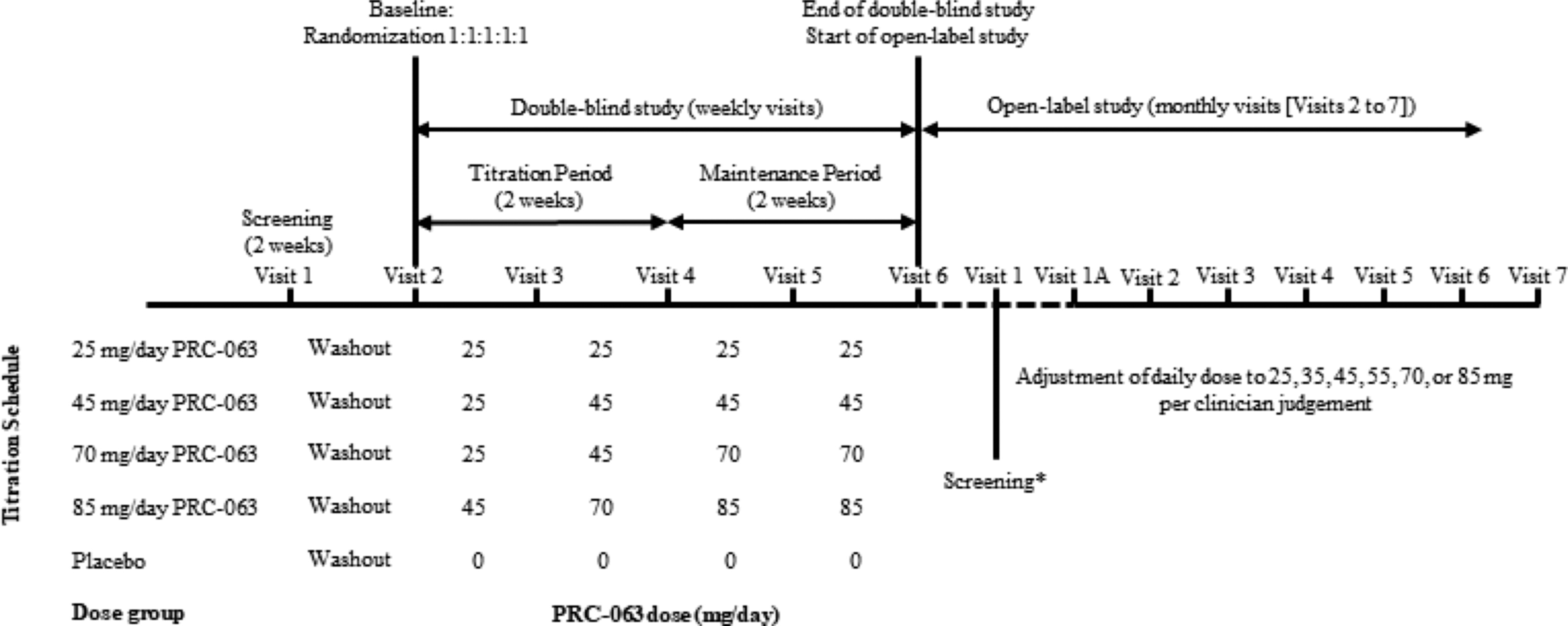
Study design for the double-blind and open-label studies. The starting dose for the open-label study was chosen at Visit 1 and treatment at that dose began the following morning. Visit 1A was scheduled for 5–10 days after Visit 1. If at Visit 1A the PRC-063 dose was judged not to be optimized, additional dose optimization visits could be scheduled. *Visit 1 (screening) in the open-label study could coincide with Visit 6 in the double-blind study.
Outcome measures
The primary endpoint was the change from baseline in least-squares (LS) mean clinician-rated ADHD-5-RS total score for PRC-063 (all doses combined) versus placebo after 4 weeks of double-blind treatment (Visit 6). The ADHD-5-RS is a DSM-5-based 18-item clinician-administered scale to rate the severity of ADHD symptoms (APA 2013). Items are scored on a 4-point scale from 0 = not present to 3 = severe, giving a maximum total score of 54. The ADHD-5-RS was completed by the clinician at each study visit from the baseline (randomization) visit onward. A secondary analysis of investigator-rated ADHD-5-RS looked at change from baseline for each PRC-063 dose versus placebo.
Data for secondary efficacy assessments were obtained from multiple sources. Parents rated adolescents on the ADHD-5-RS, and parent-rated ADHD-5-RS scores were analyzed for change from baseline for PRC-063 (all doses combined) versus placebo. The Conners 3rd Edition: Self-Report (C3SR) Short Form was also used to evaluate change from baseline for PRC-063 versus placebo. The C3SR is a self-assessment instrument that can be reliably used by youth aged 8–18 years (Conners 2008). It comprises subscales to assess inattention and hyperactivity/impulsivity, as well as executive functioning, learning problems, defiance/aggression, and family relations. The C3SR was completed at each study visit from the baseline visit onward. Clinicians rated disease severity at baseline using the Clinical Global Impression (CGI)-Severity (CGI-S) and improvement using the CGI-Improvement (CGI-I) (Guy 1976). For CGI-I, the percentage of patients who were responders (those whose illness was much improved or very much improved) was calculated.
Safety endpoints
Treatment-emergent adverse events (TEAEs) were assessed at every postscreening study visit. Vital signs were assessed at all study visits. Clinical laboratory tests and 12-lead ECG were evaluated at screening and the final study visit. Columbia Suicidality Severity Rating Scale (C-SSRS) assessments were made at all study visits. In addition, participants underwent physical examinations at screening and the final study visit.
Sleep problems are a potential concern when using a long-acting stimulant in adolescents, who as a group tend toward “eveningness” or circadian delay (Becker et al. 2020). Overall sleep quality was assessed by adolescent self-report on the Pittsburgh Sleep Quality Index (PSQI) (Buysse et al. 1989) at screening, baseline, and the final study visit, with scores >5 indicating poor sleep quality.
Statistical analyses
The primary efficacy analysis used the full analysis population, which comprised all randomized subjects who received at least one dose of study drug and who had any clinican-rated ADHD-5-RS assessment (observed data). The analysis was repeated on the per-protocol population: all participants who had a clinican-rated ADHD-5-RS assessment during the maintenance period with no major protocol deviations (observed data).
Sample size calculations were based on detecting the mean difference in change from baseline scores between all doses of PRC-063 combined and placebo for the primary efficacy variable: clinician-rated ADHD-5-RS total score. Assuming an effect size of 0.62, with a common standard deviation (SD) of 12.8, a total of 300 completed participants were needed (60 in each of the 5 treatment groups) to maintain a two-sided family-wise type 1 error rate of 0.05, with 80% power. Based on a potential dropout rate of 17%, the study planned to randomize 360 subjects. While the effect size of stimulants based on ADHD-RS-IV total score was reported to be 0.83 in a meta-analysis (Cerrillo-Urbina et al. 2018), we were conservative in our assumptions, based on the forced-dose, randomized design, which was expected to give a lower effect size than a trial using a dose optimization design.
The primary and secondary efficacy analyses of clinician-rated ADHD-5-RS total score were based on analysis of covariance (ANCOVA) models including terms for study treatment and baseline clinician-rated ADHD-5-RS total score as covariates. Differences in LS mean change from baseline to the end of double-blind treatment, together with corresponding two-sided 95% confidence intervals, were calculated for the separate PRC-063 dose levels and all doses of PRC-063 combined versus placebo. The comparison of different PRC-063 doses versus placebo involved multiple pairwise comparisons, so Dunnett's adjustments with a family-wise type I error rate set at 0.05 (two-sided) were used to correct for this. A post hoc mixed-effects model for repeated measures including terms for study treatment, study visit, study treatment by study visit interaction, and baseline clinician-rated ADHD-5-RS total score as covariates was used to verify the results of the primary analysis.
Due to multiple protocol violations and GCP issues, it was necessary to exclude efficacy data for the 13 participants enrolled at one study site from the primary efficacy analysis. A sensitivity analysis including data from this study site was performed.
As a further sensitivity analysis to assess the impact of missing data, the primary efficacy analysis for the full analysis population was repeated using Markov Chain Monte Carlo (MCMC) multiple imputation. Missing data were imputed 20 times.
In secondary analyses, ADHD-5-RS subscale scores were analyzed in the same way as ADHD-5-RS total score, using the full analysis population (observed data). CGI-I scores were summarized categorically by visit, and compared between placebo and PRC-063 (all doses combined) using the Wilcoxon rank-sum test. Changes from baseline in C3SR subscale scores and global PSQI score were compared between PRC-063 and placebo using ANCOVA models with the baseline score as a covariate.
Safety endpoints are presented using descriptive statistics for the safety population, which comprised all participants who received at least one dose of study drug.
Open-label extension study (NCT02168127)
Participants
This was a 6-month open-label extension study conducted at 34 sites in the United States and Canada.
Participants who completed the double-blind study (irrespective of whether they had been randomized to PRC-063 or placebo) were eligible to enroll in the open-label study if they continued to satisfy the eligibility criteria for the double-blind study. Enrollment of eligible participants continued until half of the sample in the double-blind study had been enrolled in the open-label extension study. Blinding for the double-blind study was not broken until after completion of the open-label study.
Study design
On the day after they completed the double-blind study, participants were started on PRC-063 at a dose determined by the investigator. Seven days later, they attended a dose evaluation visit. Thereafter, participants continued to be titrated to their optimal dose, which the investigator determined on a by-participant basis using their clinical judgment. Six daily PRC-063 doses were available: 25, 35, 45, 55, 70, and 85 mg. Participants subsequently attended monthly study visits until 6 months postenrollment (Fig. 1). Dose adjustments were made as required. A safety follow-up assessment was performed 14 days after the end of the 6-month open-label treatment period.
ADHD-5-RS, PSQI, vital signs, C-SSRS, and TEAEs were assessed at the monthly study visits. Clinical laboratory tests (including serum pregnancy tests) were conducted at the final monthly study visit.
ADHD-5-RS total score and change from (1) baseline in the double-blind study and (2) the end of double-blind treatment are presented by study visit. Mean scores at baseline in the double-blind study and at the end of double-blind treatment were calculated for the subset of participants who entered the open-label study. Baseline and follow-up scores were compared by paired t-test. TEAEs are summarized according to the last PRC-063 dose the participant received before the TEAE start date.
Results
Double-blind study
Participants
Of 450 subjects screened for study entry, 83 were screen failures and 367 were randomized, including 13 participants enrolled at a site excluded from the primary efficacy analysis (Fig. 2). Exclusion of this site gave 354 randomized subjects, of whom 323 completed the study. The target of 60 participants per treatment group was met. For the 31 participants who discontinued the study, “Withdrawal by subject” was the most common reason. The overall percentage of participants who discontinued was marginally higher among participants who received PRC-063 than among participants who received placebo (9.2% vs. 7%). The rate of discontinuation due to AEs was 3.5% for PRC-063 and 0% for placebo. For participants who received PRC-063, the overall number of discontinuations for any reason was higher in the 85 mg dose group (nine participants) than in the other dose groups (five or six participants) (Table 1).
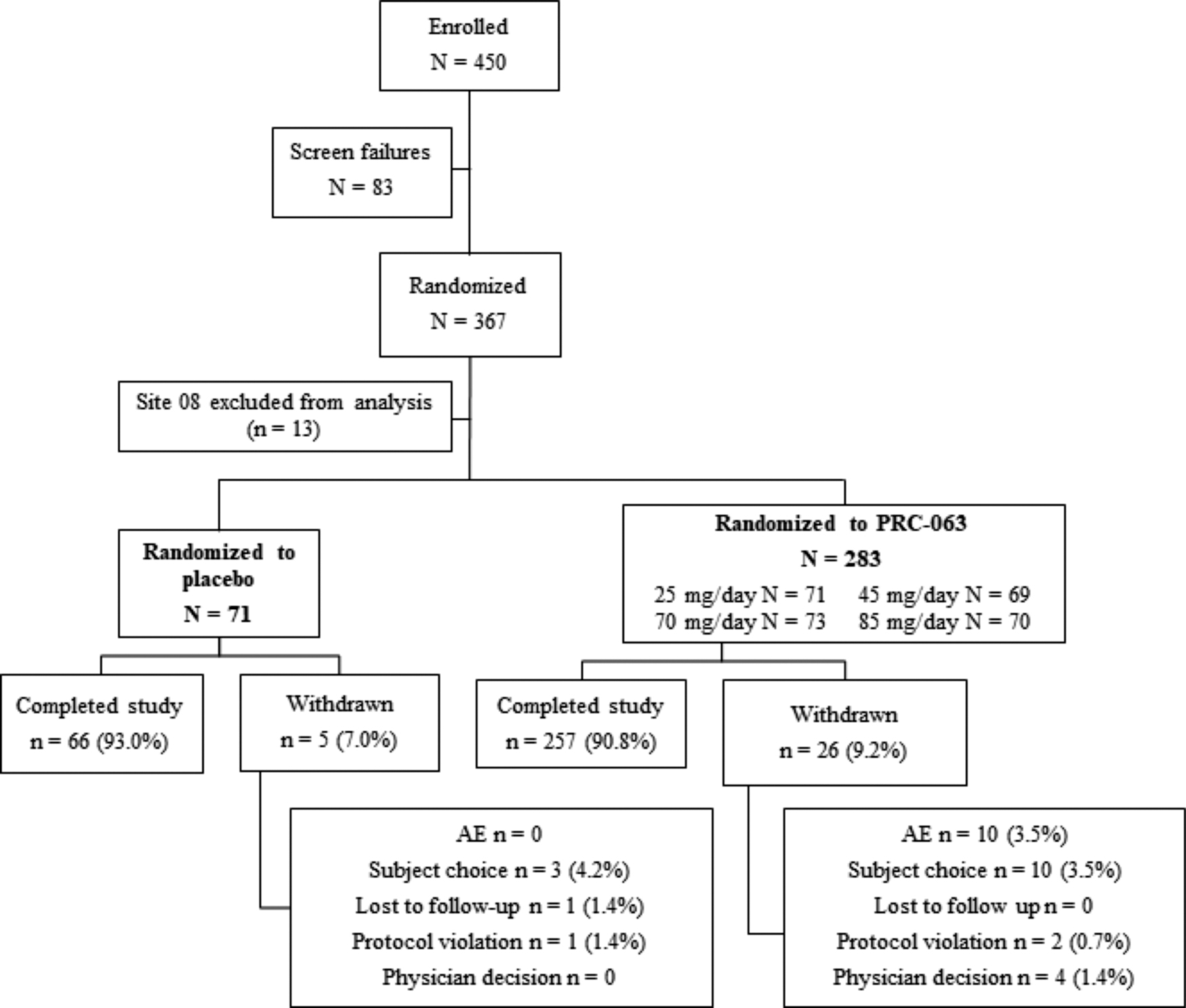
CONSORT diagram for the double-blind study. Site 08 was excluded because of major protocol violations. AE, adverse event.
Baseline Characteristics and Disposition for the Double-Blind Study
Missing for n = 1 subject.
ADHD-5-RS, Attention-Deficit/Hyperactivity Disorder Rating Scale Diagnostic and Statistical Manual of Mental Disorders-5; CGI-S, Clinical Global Impression-Severity; SD, standard deviation.
Mean (SD) participant age at baseline was 14.2 (1.6) years for PRC-063 and 14.1 (1.5) years for placebo, with participants evenly distributed between the 12- to 13- and 14- to 15-year age groups, and with fewer participants in the 16- to 17-year age group (Table 1). Approximately two-thirds of participants were male and 72% were diagnosed with combined presentation ADHD at screening. In both treatment groups, 66% of participants were ADHD treatment naïve at baseline. Mean (SD) CGI-S score at baseline was 4.5 (0.7) (range 3–7), with most participants identified as moderately (46.9%) or markedly (42.4%) ill. The PRC-063 and placebo treatment groups were comparable in terms of body mass index, systolic and diastolic blood pressure, body temperature, heart rate, and respiratory rate.
Exposure
The rate of medication compliance decreased slightly from 91.3% at week 2 to 85.8% at end of study. During the 2-week evaluation period, it was 83.7%.
Efficacy
ADHD-5-RS total score (clinician rated)
Figure 3 shows changes in clinician-rated ADHD-5-RS total score during the double-blind study for the full analysis population. Compared with participants receiving placebo, participants receiving PRC-063 (all doses combined) showed a statistically significant improvement in ADHD symptoms as measured by ADHD-5-RS total score at the end of double-blind treatment (mean decrease from baseline 40.8% vs. 29.8%; LS mean change from baseline −15.17 vs. −10.98; LS mean difference for PRC-063 vs. placebo −4.2, p = 0.0067). For all individual doses of PRC-063, improvements in ADHD-5-RS total score from baseline were significant (p < 0.0001). Compared with placebo, improvements were higher for the 45 mg (p = 0.0155) and 70 mg (p = 0.0401) PRC-063 doses, but not for 25 or 85 mg. A post hoc mixed-effects model for repeated measures confirmed the results of the primary analysis.
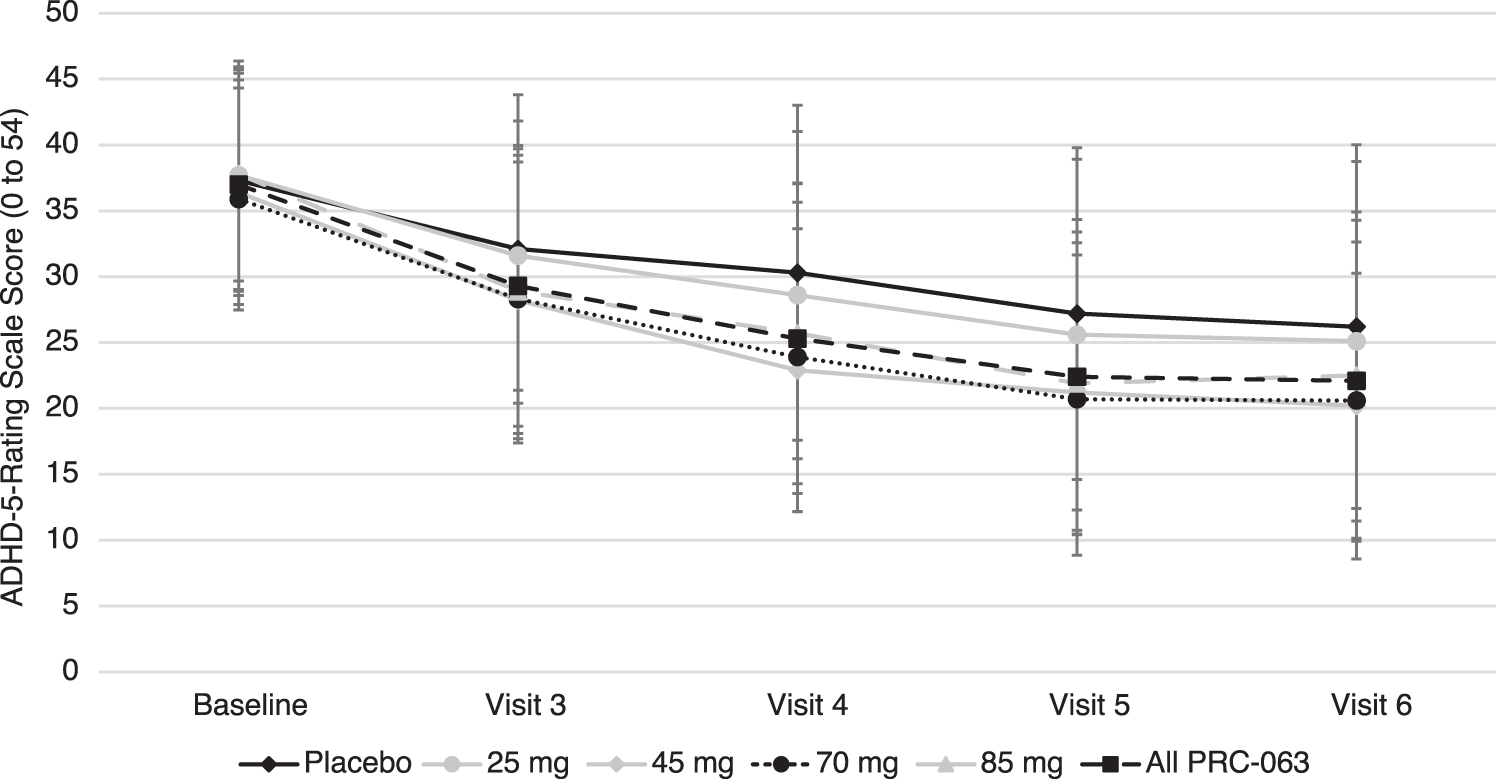
Clinician-rated ADHD-5-RS total score by treatment group from baseline (Visit 2) through Visit 6 during the double-blind study (full analysis population). ADHD-5-RS, Attention-Deficit/Hyperactivity Disorder Rating Scale Diagnostic and Statistical Manual of Mental Disorders-5.
A sensitivity analysis including data from the study site excluded from the primary analysis because of protocol violations and GCP issues gave results consistent with the primary efficacy analysis. In a second sensitivity analysis performed using the MCMC method, there was a significant improvement in LS mean total score at the end of double-blind treatment for PRC-063 compared with placebo (p = 0.0049). Improvements versus placebo were higher for the 45 mg (p = 0.0046), 70 mg (p = 0.0064), and 85 mg (p = 0.0223) PRC-063 doses, but not for 25 mg. Similar results were obtained for the per-protocol population.
ADHD-5-RS total score (parent rated)
Parent-rated ADHD-5-RS total score showed more variability than the clinician-rated assessment. Nonetheless, the improvement in parent-rated ADHD-5-RS total score at the end of double-blind treatment was higher for PRC-063 (all doses combined) than for placebo (LS mean change from baseline −11.29 vs. −7.54, p = 0.0221). As with clinician-rated ADHD-5-RS total score, significant improvements versus placebo were observed for the 45 mg (p = 0.0192) and 70 mg (p = 0.0127) PRC-063 doses, but not for 25 or 85 mg.
Conners 3rd Edition: self-report scale
Of the five subscales measured by the C3SR, the greatest improvements with PRC-063 (all doses combined) relative to placebo were on the Inattention subscale (LS mean change from baseline −11.7 vs. −7.3, p = 0.0168) and the Hyperactivity-Impulsivity subscale (LS mean change from baseline −9.6 vs. −6.6, p = 0.0798). For the Inattention subscale, significant improvements versus placebo were observed for the 45 mg (p = 0.0135), 70 mg (p = 0.0203), and 85 mg (p = 0.0128) PRC-063 doses, but not for 25 mg. For the Hyperactivity-Impulsivity subscale, significant improvements versus placebo were observed for the 45 mg (p = 0.0465) and 70 mg (p = 0.0246) doses, but not for the 25 mg and 85 mg doses. Significant differences for PRC-063 (all doses combined) versus placebo were not observed for the Learning Problems, Defiance/Aggression, and Family Relations subscales (p = 0.3458, 0.6079, and 0.0945, respectively).
Clinical Global Impression-Improvement
Figure 4 shows the distribution of CGI-I scores at end of the double-blind study. About 52.7% of participants receiving PRC-063 were identified as responders, rated as “much” or “very much improved,” as compared with 32.4% of those receiving placebo (p = 0.0004, Wilcoxon rank-sum test comparing ranked scores for PRC-063 vs. placebo). For individual PRC-063 doses, the proportion of responders was 45.1% for 25 mg, 59.4% for 45 mg, 52.1% for 70 mg, and 54.3% for 85 mg.
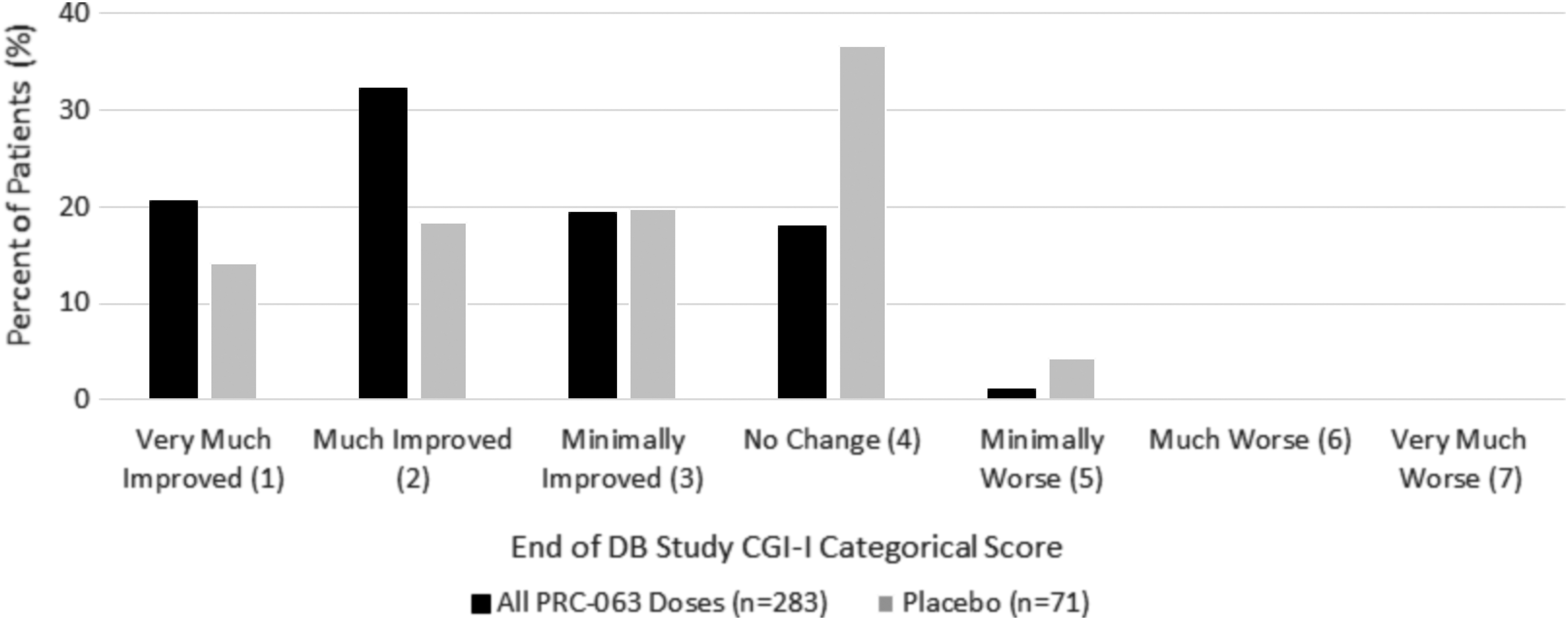
CGI-I at the end of the DB study. CGI-I, Clinical Global Impression-Improvement; DB, double blind.
Safety and tolerability
Adverse events
Eighty-eight TEAEs were reported by 36 participants receiving placebo (48.6%) and 476 TEAEs were reported by 192 participants receiving PRC-063 (65.5%). Considering individual PRC-063 doses, the proportion of participants reporting one or more TEAEs was 60.3% for 25 mg/day, 65.3% for 45 mg/day, 65.8% for 70 mg/day, and 70.8% for 85 mg/day. TEAEs led to withdrawal in 10 participants who received PRC-063, distributed relatively equally across doses (2, 3, 2, 3 participants on 25, 45, 70, and 85 mg/day doses, respectively). The most common TEAEs (those occurring in ≥5% of participants) are listed in Table 2. Most TEAEs were mild or moderate in severity. No deaths or serious adverse events (SAEs) were reported.
Treatment-Emergent Adverse Events in the Double-Blind Study (Safety Population)
SAE, serious adverse event; TEAE, treatment-emergent adverse event.
Treatment-related TEAEs were reported by 24 participants receiving placebo (32.4%) and 154 participants receiving PRC-063 (52.6%). The treatment-related TEAEs occurring in ≥5% of participants receiving PRC-063 were decreased appetite (19.8%), headache (11.6%), irritability (8.2%), insomnia (6.5%), weight loss (6.1%), initial insomnia (5.1%), and nausea (5.1%).
Pittsburgh Sleep Quality Index
Mean (SD) global PSQI score was 5.7 (3.3) at baseline and 5.4 (3.4) at the end of treatment for PRC-063 (all doses combined). For placebo, mean (SD) global PSQI score was 6.0 (3.7) at baseline and 5.4 (3.7) at the end of treatment. There was no significant difference between PRC-063 (all doses combined) and placebo in mean global PSQI score at the end of treatment (p = 0.6110).
Suicidality
One report of suicidal behavior and four reports of suicidal ideation were captured using the C-SSRS. All of these reports were from participants randomized to PRC-063. Intensity of ideation was “1—wish to be dead” for two participants, “2—nonspecific active suicidal thoughts” for one participant, and “3—active suicidal ideation with any methods” (no intent to act) for one participant. The suicidal ideation of intensity 3, which occurred in a participant randomized to 70 mg/day PRC-063, was recorded as a TEAE of moderate severity and was assessed as related to study drug. The participant was withdrawn from the study. The other reports of suicidal behavior and ideation did not result in any action regarding study treatment or study participation.
Other safety assessments
There were no clinically significant changes in vital signs (body weight, body mass index, systolic or diastolic blood pressure, body temperature, heart rate, or respiratory rate) (Table 3). Mean body weight decreased by 0.11 to 1.33 kg in the different PRC-063 dose groups and increased by 1.47 kg in participants who received placebo. Modest increases in mean systolic blood pressure (range 0.83–2.55 mmHg) and diastolic blood pressure (0.82–3.70 mmHg) were recorded in the different PRC-063 dose groups. Clinical laboratory tests, ECG, and physical examinations revealed no clinically significant abnormalities.
Vital Signs: Mean Change from Baseline in the Double-Blind Study
Open-label extension study
Participants and exposure
Excluding site 08 from the efficacy analysis, 178 participants who completed the double-blind study (139 of whom received PRC-063 and 39 placebo) entered the open-label extension study (Fig. 5). Of these participants, 122 completed the study.
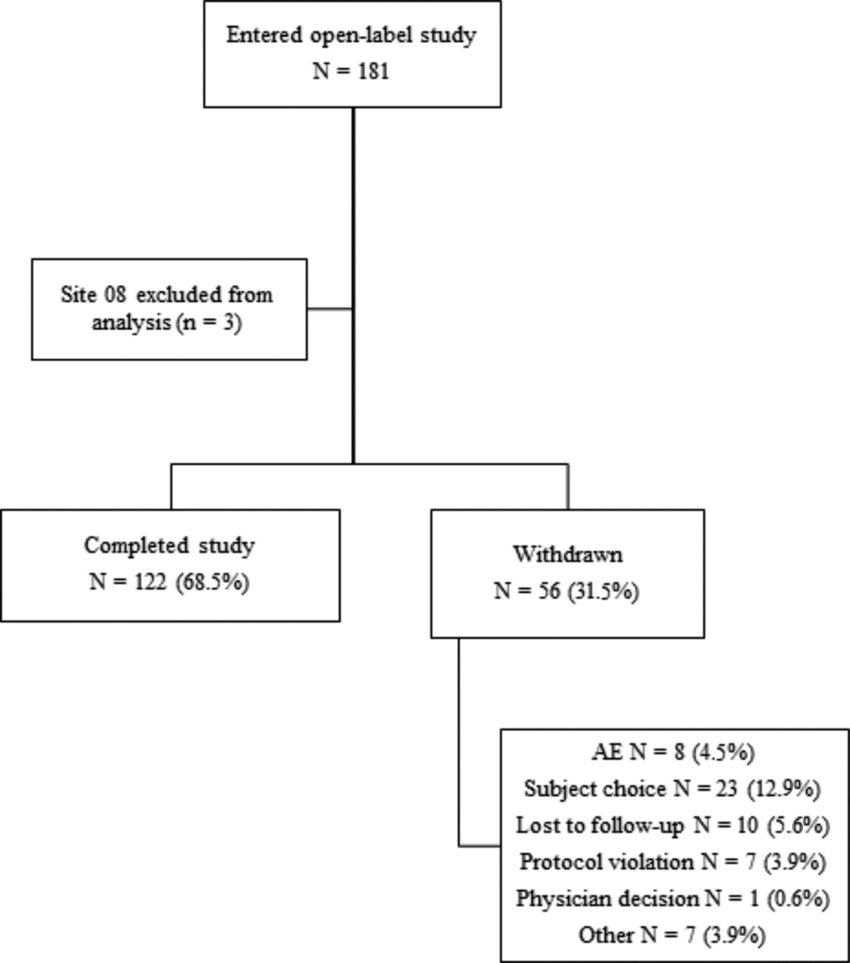
CONSORT diagram for the open-label study. Site 08 was excluded because of major protocol violations. AE, adverse event.
In the open-label study, participants were exposed to PRC-063 for a mean of 152.0 days (range 7-217 days). The mean starting dose of PRC-063 was 35.7 mg/day (range 25–70 mg/day). By the end of the first month of open-label treatment, the mean dose of PRC-063 had increased to 56.6 mg/day (range 25–85 mg/day) (Table 4). The mean dose was 61.4 mg/day after 2 months and 63.5 mg/day after 3 months. From 3 months through 6 months, the mean dose remained relatively constant. Overall, the mean daily optimized dose was 65.1 mg/day (Table 4). Of the 139 participants who had received PRC-063 in the double-blind study, 32 (23.0%) were correctly dosed (Table 5). Sixty-eight participants (48.9%) were optimized to a higher dose during the open-label study, and 39 participants (28.1%) were optimized to a lower dose during the open-label study. All 37 open-label study participants who had been randomized to 25 mg in the double-blind study were underdosed in the double-blind study. Only 6 participants (3.4%) in the open-label study were optimized to 25 mg (Table 4). For 31.5% of patients, 85 mg was the optimized dose.
PRC-063 Dose by Month in the Open-Label Study
Overall represents participants' optimal dose or the last dose taken before study exit.
Number and Percentage of Participants Dosed Correctly During the Double-Blind Study
Total number of participants in the open-label study who received PRC-063 in the double-blind study (including participants from the site excluded from the primary efficacy analysis).
Efficacy
Figure 6 shows changes in clinician-rated ADHD-5-RS total score during open-label treatment. Compared with the mean (SD) baseline score of 36.6 (9.26) in the double-blind study, mean (SD) clinician-rated ADHD-5-RS total score decreased by 33.9% to 24.2 (12.60) at the end of double-blind treatment and by 53.8% to 16.9 (10.01) at the end of the first month of open-label treatment (p < 0.0001). Thereafter, mean clinician-rated ADHD-5-RS total score remained stable throughout the 6-month open-label study, and was significantly lower compared with baseline in the double-blind study and the end of double-blind treatment (p < 0.0001). The mean (SD) change in ADHD-5-RS total score between baseline in the double-blind study and the end of open-label treatment was −22.0 (10.90), a decrease of 60.7%. At the completion of 6 months of open-label treatment, 69.3% of patients still on treatment had an ADHD-5-RS score of ≤18.
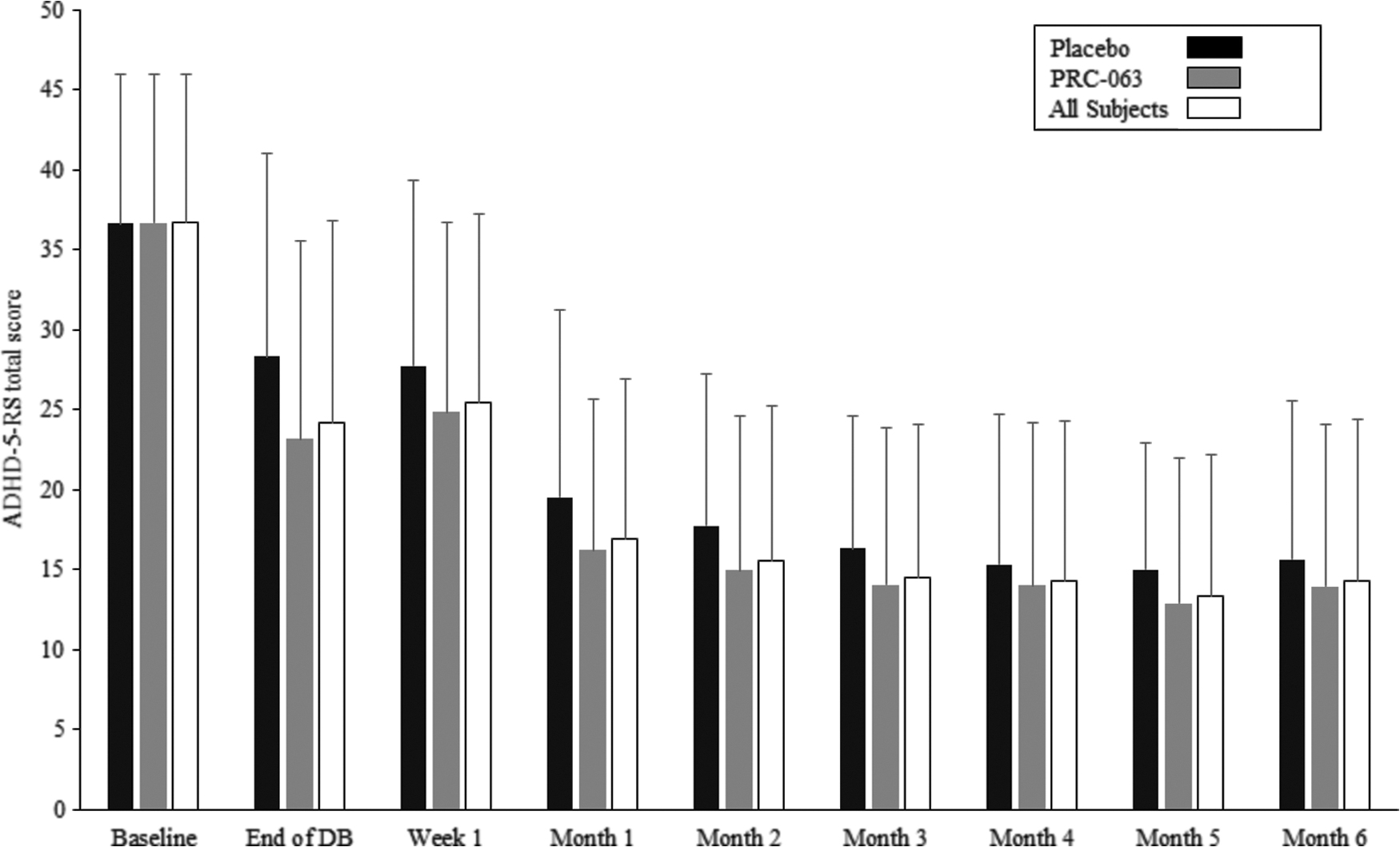
Clinician-rated ADHD-5-RS total score over the course of the open-label study by treatment allocation in the double-blind study (full analysis population). ADHD-5-RS, attention-deficit/hyperactivity disorder Rating Scale Diagnostic and Statistical Manual of Mental Disorders-5.
Safety and tolerability
Four hundred ten TEAEs were reported by 133 participants (74.7%). TEAEs led to withdrawal of eight participants. TEAEs occurring in ≥5% of participants are listed in Table 6.
Treatment-Emergent Adverse Events Occurring in ≥5% of Participants in the Open-Label Study (Safety Population)
TEAE, treatment-emergent adverse event.
Treatment-related TEAEs were reported by 99 participants (55.6%). Treatment-related TEAEs occurring in ≥5% of participants were decreased appetite (14.0%), headache (12.4%), insomnia (9.6%), weight loss (8.4%), and initial insomnia (5.6%).
There were two SAEs: asthma exacerbation in a participant optimized to 70 mg/day PRC-063 (assessed as not related to study drug) and aggressive behavior in a participant optimized to 55 mg/day PRC-063 (assessed as related to study drug). In both cases, the participant was withdrawn from the study.
For participants in the double-blind study who entered the open-label extension study, mean (SD) global PSQI score was 5.5 (3.4) at the end of the double-blind study, 4.4 (2.9) after 1 month of open-label treatment, and 4.4 (3.3) after 6 months of open-label treatment.
There was one report of suicidal ideation of intensity “3—active suicidal ideation with any methods” (no intent to act) at the last study visit in a participant taking 70 mg PRC-063. The report was recorded as a TEAE that was judged to be unrelated to study drug and that was resolved. No action was taken regarding study treatment.
Discussion
PRC-063 significantly improved ADHD symptomatology in adolescents relative to placebo, as measured by clinician-rated ADHD-5-RS total score, CGI-I, parent-rated ADHD-5-RS total score, and adolescent self-report of ADHD symptoms. The 40.8% improvement from baseline in mean clinician-rated ADHD-5-RS total score with PRC-063 in the double-blind study is similar to the improvements reported in children and adolescents treated with SHP465 (Brams et al. 2018). Previous research concluded that a 40%–50% decrease in ADHD-RS total score from baseline is representative of clinical improvement (CGI-I score of 1–2) (Goodman et al. 2010; Weiss et al. 2019). Overall, patients showed a 60.7% improvement from baseline in the double-blind study to the end of open-label treatment. This improvement suggests that when PRC-063 is used in accordance with treatment guidelines and product labeling, it can give robust improvement, and even remission of symptoms, in adolescents with ADHD.
At the end of the double-blind study, clinicians rated 52.7% of patients receiving PRC-063 as “much” or “very much improved” from baseline, based on CGI-I. This percentage may have been affected by the fixed-dose design of the double-blind study, which resulted in only 23% of patients being correctly dosed (and almost half being underdosed) during double-blind treatment, based on the optimized dose they subsequently received in the open-label study. National and international methylphenidate treatment guidelines recommend that patients with ADHD, particularly those who are naïve to methylphenidate treatment, should be treated with low doses initially and have their dose gradually increased to achieve an optimal response while minimizing the risk of side effects (Huss et al. 2017).
This study included clinician-, parent-, and adolescent-rated ADHD symptoms. Parent-reported changes were the smallest, but the parent-rated ADHD-5-RS scores were nonetheless improved with PRC-063 as compared with placebo. Adolescents were able to self-report improvement in attention and showed a trend toward improvement in hyperactivity/impulsivity, which suggests that some of the positive illusory bias of childhood is less significant in youth. Adolescents did not report improvement in learning, defiance/aggression, or family relations. While these domains may not have improved over the course of the study, it is likely that adolescents have limited insight into externalizing symptoms or awareness of how these symptoms impact their families. Nonetheless, there is a relative paucity of clinical trials in adolescents, and this study suggests that adolescents can reliably self-report ADHD symptoms. Adolescents may be more aware of their own ADHD symptoms than their parents, who may have limited opportunities to observe their children's symptoms during the day.
A high placebo response rate was observed in the double-blind study, as has been reported in some previous clinical trials of stimulants for ADHD in children and adolescents (Wilens et al. 2006; Waxmonsky et al. 2011; Brams et al. 2018). Compared with flexible-dose trials, placebo effects can be higher in psychiatric trials with fixed-dose designs like the one we used in the double-blind study (Khan et al. 2003). Also, there is evidence that the placebo response can increase if the number of active treatment arms (and hence the probability of receiving an active treatment) is increased (Doering et al. 2014). It is possible that adolescents are more sensitive to placebo effects than young children, who do not have pretreatment expectations of drugs, or adults, who are more aware of expected side effects and thus less adequately blinded.
PRC-063 was generally well tolerated in the presented studies. There were no deaths and only two SAEs, both during the open-label study. Most TEAEs were of mild or moderate severity, and the only TEAEs that occurred in >10% of participants during double-blind treatment were decreased appetite and headache. The AE profile was consistent with previous reports for other long-acting stimulants in children and adolescents (McGough et al. 2006; Wilens et al. 2006; Coghill et al. 2013, 2014; Newcorn et al. 2017). Across the double-blind and open-label studies, five adolescents reported suicidal ideation and one reported suicidal behavior, all while receiving PRC-063. Although small in number, these reports suggest that adolescents receiving stimulant treatment for ADHD may be at increased risk of suicide. Given the high rates of suicidal ideation and behavior in the adolescent population in general (Lim et al. 2019) and in young people with ADHD (Chen et al. 2019), this further emphasizes the need for clinicians to be vigilant in assessing suicide risk in their patients with ADHD.
It is noteworthy that while rates of insomnia as an AE were higher for PRC-063 than for placebo, they were lower than in adults treated with PRC-063 in fixed-dose and dose-optimized studies (Weiss et al. 2021). There was, however, no difference between PRC-063 and placebo in overall sleep quality at the end of the double-blind study, and global PSQI scores improved during the open-label dose-optimized study. This suggests that while a subset of adolescent patients may experience insomnia as an adverse event, PRC-063 does not worsen sleep quality overall.
The reported findings need to be understood in the context of the study limitations. Notably, the double-blind study was only powered to determine the overall effect of PRC-063 relative to placebo, and individual dose groups were too small to evaluate dose effectiveness. Nonetheless, several inferences about dose effects can be made from the open-label study. Across efficacy outcomes, significant improvements versus placebo were observed with the 45 and 70 mg PRC-063 doses, but not with 25 or 85 mg, except in analyses where dropouts were excluded. All participants in the open-label study who received 25 mg in the double-blind study were optimized to a higher dose in the open-label study, which suggests that the lack of an effect for the 25 mg dose was due to underdosing.
The overall dropout rate was higher for the 85 mg/day group than for other dose groups in the double-blind study. In a sensitivity analysis that accounted for this higher dropout rate, the improvement in ADHD-5-RS total score was significantly higher for the 85 mg PRC-063 dose than for placebo. The fixed-dose design of the double-blind study may have resulted in some participants (most of whom were treatment naïve) being titrated too rapidly to an 85 mg dose. In the open-label study, where titration over time as tolerated was possible, almost half of patients were optimized to a higher dose than they had received in the double-blind study. Notably, almost a third of participants were optimized to the 85 mg dose in the open-label study. These observations support guideline-based recommendations for titrating stimulants slowly from a low dose to account for interindividual differences in dose responses between patients and to limit untoward effects. In the open-label study, the mean dose increased at months 1 and 2 relative to the start of the study, with a minor increase observed at month 3. Thereafter, the mean dose was relatively consistent. This suggests that most participants took ∼2 to 3 months to have their dose optimized. A significant limitation of the double-blind study was our failure to allow adequate time for titration to the highest dose used, which may have impacted our ability to demonstrate a response for this dose group.
The fixed-dose design of the double-blind study has inherent limitations. Overall, 77% of participants were randomized to a dose that was either too low or too high, potentially leading to the treatment response being underestimated and AE rates being overestimated. In the open-label study, where doses were optimized, we observed substantial clinical improvement beyond what was observed in the double-blind study. Finally, exclusion of adolescents with unstable psychiatric conditions and other comorbidities meant that our patient sample was not fully reflective of the spectrum of patients encountered in normal clinical practice.
Conclusions
In conclusion, the presented studies demonstrate that the once daily beaded methylphenidate formulation PRC-063 was efficacious versus placebo in the treatment of ADHD in adolescents. Significant improvements in ADHD symptomatology were observed based on the primary and secondary outcome measures and across three different sets of informants: clinicians, parents, and adolescents themselves. PRC-063 was generally well tolerated, with an AE profile consistent with other long-acting stimulants. While the studies failed to determine dose-optimized response rates, they do illustrate the potential for maximal symptom improvement with individualized dose optimization achieved over an adequate titration period. Future research might include a double-blind, dose-optimized study to look at the effect size for PRC-063 versus placebo under conditions that more closely reflect normal clinical practice. Such a study would provide valuable information on dose response to guide titration by clinicians, and would provide insight into how many adolescents actually require and tolerate higher PRC-063 doses. Adolescence is a period of high risk for patients with ADHD, and further studies of response to stimulants in the adolescent age group and their impact on risk are much needed.
Clinical Significance
With a rapid onset and extended duration of action, PRC-063 represents a new long-acting methylphenidate treatment option for adolescents with ADHD. PRC-063 was well tolerated, and the AE profile was reflective of side effects typically associated with methylphenidate use.
Footnotes
Acknowledgments
The authors thank Stephen Gilliver, PhD, of Evidera for providing medical writing support, which was funded by Purdue Pharma in accordance with Good Publication Practice (GPP3) guidelines (
Data Management and Statistical Analysis
Statistics & Data Corporation, Tempe, AZ.
Disclosures
M.D.W. has received consulting fees/honoraria from Tris, Purdue, Adlon, Takeda, Huron, Mundipharma, and CBPartners; support for travel to meetings, article preparation, or other purposes from World Federation of ADHD, Eunethydis, Canadian Attention Deficit Disorder Resource Alliance (CADDRA), Children and Adults with Attention Deficit Disorder (CHADD), American Professional Society for ADHD and Related Disorders (APSARD), Israeli Federation of ADHD, Purdue CA, Purdue US, Rhodes Pharmaceutical. Akili. Takeda, Global Medical Education, and Boston Children's Hospital; payment for lectures from Global Medical Education, Centre for ADHD Awareness Canada (CADDAC), and CADDRA; and royalties from Multi Health Systems and Johns Hopkins University Press. A.J.C. has received consulting fees from Acadia, Aevi Genomics, Akili Interactive, Alkermes, Allergan, Arbor Pharmaceuticals, AstraZeneca, Forest Laboratories, Ironshore, Janssen, KemPharm, Lundbeck, Neos Therapeutics, NLS Pharma, Novartis, Noven, Otsuka, Purdue, Shionogi, Shire, Sunovion, Supernus, Takeda, Tris Pharma, and Vanda; honoraria from Acadia, Alkermes, Allergan, Arbor Pharmaceuticals, AstraZeneca, Janssen, Lundbeck, Neos Therapeutics, Novartis, Otsuka, Shionogi, Shire, Sunovion, Takeda, Tris Pharma, and Vanda; and research funding from Aevi Genomics, Akili Interactive, Alkermes, Allergan, Arbor Pharmaceuticals, AstraZeneca, Forest Laboratories, Ironshore, Janssen, KemPharm, Lilly, Lundbeck, Otsuka, Neos Therapeutics, Novartis, Noven, Purdue, Rhodes Pharmaceuticals, Shionogi, Shire, Sunovion, Supernus, Takeda, Tris Pharma, and Vanda. S.H.K. has received consulting fees from Akili Interactive, Arbor, Holmusk, Ironshore, Jazz, KemPharm, Otsuka, Rhodes, Shire, Sunovion, and Tris; has received research support from Akili Interactive, BehaVR, Bose, KemPharm, Limbix, Neos, OnDosis, Rhodes, Sana Health, Shire, Sunovion, Tali Health, and Tris; and has equity interests in Akili Interactive. G.A.E.D. is an employee of Purdue Pharma (Canada), the company that sponsored this research.