
Abstract
Objective:
To determine the safety and efficacy of PRC-063, a once-daily, multilayer, extended-release (ER) formulation of methylphenidate (MPH) hydrochloride, in the treatment of attention-deficit/hyperactivity disorder (ADHD) in children in a randomized, double-blind, parallel group, dose-optimized, placebo-controlled phase 3 study.
Methods:
Boys and girls aged 6–12 years diagnosed with ADHD were enrolled. During a 6-week, open-label, dose-optimization phase, subjects began treatment at 25 mg/day of PRC-063 and were titrated until an optimal dose (maximum 85 mg/day) was reached. During the double-blind period, subjects were randomized to receive treatment with their optimal dose of PRC-063 or placebo for 1 week. Efficacy was assessed in a laboratory classroom setting on the final day of the double-blind treatment using the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Rating Scale and Permanent Product Measure of Performance (PERMP). Safety was assessed measuring adverse events (AEs), vital signs, and electrocardiograms.
Results:
The study was completed by 147 subjects. In the primary efficacy analysis, significant improvements were demonstrated with PRC-063 versus placebo (p < 0.0001) when SKAMP-Combined scores were averaged over the 13-hour full-day laboratory classroom (least squares mean difference = −8.6, 95% confidence interval = −10.6 to −6.6). Mean average PERMP-Total scores were also significantly improved with PRC-063 versus placebo at all time points postdose (p < 0.01). The onset of treatment effect was present by 1-hour postdose (the first time point measured) and duration of efficacy was up to and including 13 hours postdose. AEs reported in ≥5% of subjects during the dosing optimization period were decreased appetite, abdominal pain upper, affect lability, weight decreased, headache, irritability, and insomnia.
Conclusions:
PRC-063 was effective in improving attention and reducing symptoms of ADHD versus placebo and had a rapid onset and extended duration of effect. AEs were consistent to those reported with other ER MPH treatments. Clinical Trial Registry: NCT03172481.
Introduction
Attention-deficit/hyperactivity disorder (ADHD), characterized by developmentally inappropriate levels of inattention, hyperactivity, and impulsivity, is the most common neurobehavioral disorder of childhood (Subcommittee on Attention-Deficit/Hyperactivity et al. 2011). The general prevalence of ADHD is estimated at between 5% and 7% in children and adolescents, based on systematic reviews and parent-reported diagnosis (Polanczyk et al. 2007; Thomas et al. 2015; Danielson et al. 2018). Children with ADHD experience a greater risk of academic underperformance and difficulties in school; struggle with family and peer relationships; and are at increased risk of comorbidities such as oppositional defiant disorder (ODD), conduct disorder, and anxiety (Peasgood et al. 2016).
Treatment guidelines recommend a comprehensive and multimodal approach to treating ADHD (Pliszka and AACAP Work Group on Quality Issues 2007; CADDRA 2018; National Institute for Health and Care Excellence 2018; Wolraich et al. 2019). Long-acting psychostimulants (methylphenidate [MPH] and amphetamine) are recommended as first-line pharmacological agents. Numerous long-acting, extended-release (ER) medications are currently available for the treatment of ADHD (Childress 2016; CADDRA 2018). The pharmacokinetic profiles of these ER formulations are distinct, and factors such as onset of action, duration of action during the day, timing of active medication release, and the predicted plasma concentration are dependent on the immediate-release (IR) to controlled-release (CR) ratio of the formulation (Childress 2016; CADDRA 2018).
Currently approved once-daily long-acting MPH treatments for ADHD have demonstrated a 9- to 12-hour duration of action (McCracken et al. 2003; McGough et al. 2006; Childress et al. 2015, 2017). Despite the long duration of action of these agents in clinical trials, caregivers and children and adolescents with ADHD commonly report that medication is generally only effective during school hours (Sikirica et al. 2015). Guidelines recommend second-line or adjunctive treatment options for augmenting long-acting formulations early or late in the day, or early in the evening (CADDRA 2018). These recommendations suggest the use of a fast-acting, long-lasting MPH treatment option for ADHD that provides symptom control during and beyond the school day.
PRC-063, currently marketed as FOQUEST® in Canada and as Adhansia XR® in the United States, is a new formulation of MPH. PRC-063 capsules are formulated using a multilayer-release (MLR®) bead technology. Each bead is identical in composition and appearance and consists of multiple layers of drug and drug release-controlling excipients where the components of the formulation are arranged in concentric layers to achieve the desired release profile.
Each bead has an outer, IR layer of MPH consisting of ∼20% of the MPH dose, absorption of which begins immediately upon ingestion and leads to an initial peak plasma concentration of MPH occurring at ∼1.5 hours postdose (Katzman et al. 2020). After absorption of the IR layer, the CR component of the bead containing ∼80% of the MPH dose is exposed. The CR layer includes a pH-sensitive polymer coating that does not permit significant release of MPH until the beads have passed through the stomach. In pH >7, the pH sensitive polymer dissolves, allowing water to penetrate and erode the CR polymer coating for subsequent diffusion of MPH into the gastrointestinal tract. This leads to a second maximum plasma concentration of MPH occurring at ∼12.5 hours postdose (Katzman et al. 2020), ∼4–6 hours later than most long-acting MPH formulations.
The formulation was designed to optimize the balance between the magnitude of the rapidly attained, initial peak MPH concentration that occurs upon administration and the subsequent, more prolonged MPH peak that occurs later in the day. The safety and efficacy of PRC-063 has been established using a wide range of dose strengths, which facilitates individualization of dosing.
This randomized, double-blind, parallel-group, multicenter laboratory classroom study was designed to compare the efficacy and safety of PRC-063 versus placebo in children (≥6 and ≤12 years) with ADHD in a laboratory classroom setting. The laboratory classroom design has been extensively used to evaluate the efficacy of ADHD medications. It simulates a school classroom and facilitates repeated assessments over the course of the day to evaluate the onset and duration of treatment effects (Wigal and Wigal 2006; Wigal et al. 2013; Childress et al. 2015; Robb et al. 2017).
Methods
Study design
This was a randomized, double-blind, parallel group, placebo-controlled, dose-optimized, phase 3 study conducted at six experienced sites in the United States from May to August 2017 (CT identifier NCT03172481). The study consisted of four periods: screening (up to 28 days), 3-day washout, open-label dose-optimization (up to 6 weeks), and a 1-week double-blind, randomized, placebo-controlled period (Fig. 1).
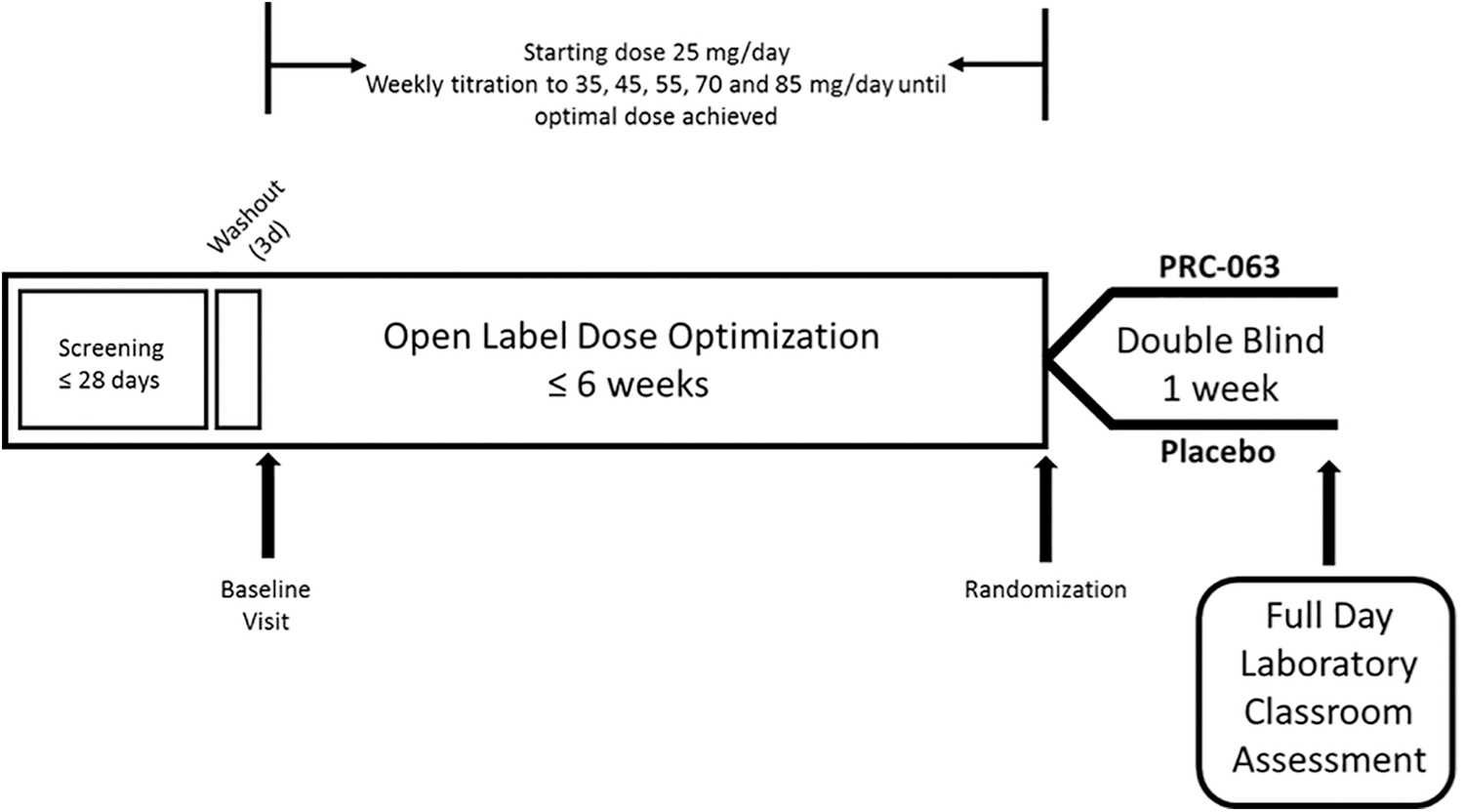
Clinical trial design.
Subjects who met screening criteria entered a 3-day washout period during which subjects taking ADHD treatment discontinued their treatment. After the washout period, subjects returned to the clinic for a baseline visit to have their eligibility confirmed and suicidal ideation and behavior was assessed using the clinician-administered Columbia-Suicide Severity Rating Scale (C-SSRS, Children's Since Last Visit version). The attention-deficit/hyperactivity disorder-Rating Scale-5 (ADHD-RS-5) (DuPaul et al. 2016) and the Clinical Global Impressions: Severity (CGI-S) were administered by the clinician to confirm ADHD symptomatology and to record baseline scores.
Eligible subjects meeting all study criteria completed a level-finding Permanent Product Measure of Performance (PERMP) test and then entered the dose-optimization period. Each subject was initiated at a starting dose of 25 mg/day PRC-063 (Purdue Pharma, Canada), administered once-daily in the morning at home by the parent/guardian. The dose was increased to 35, 45, 55, 70, and 85 mg/day at weekly visits until an optimal dose (i.e., the dose that produced a reduction in ADHD-RS-5 score of ≥30% from baseline accompanied by a score of 1 or 2 on the clinician-administered Clinical Global Impressions: Improvement [CGI-I] and acceptable tolerability) was achieved. Once reached, the optimal dose was maintained for the remainder of the dose-optimization period. Subjects who could not tolerate the lowest dose or who were unable to achieve an optimal dose were discontinued from the study.
Optimized subjects attended a half-day practice laboratory classroom to become familiar with classroom schedules and procedures. Study drug was administered by the clinic staff after all predose assessments were completed. After completing the practice classroom, subjects were randomized in a 1:1 ratio to receive their optimized dose of PRC-063 or matching placebo. Starting from the day after randomization, double-blind study medication was administered once daily in the morning at home by the parent/guardian. Efficacy was assessed in the full-day laboratory classroom setting on the final day of the 7-day double-blind period. Study drug was administered by the clinic staff after all predose assessments were completed. A safety follow-up telephone call was conducted with the subjects ∼7 days after last dose of study medication.
All study subjects provided assent and their parents/guardians provided written informed consent before enrolment. The study was approved by an Institutional Review Board (Schulman IRB, Cincinnati, OH). All documentation and procedures related to the study were executed in accordance with Good Clinical Practice (GCP) guidelines as required by the Declaration of Helsinki 1964 and all its amendments to this date, and the International Conference on Harmonization (ICH) Guideline for GCP (CPMP/ICH/135/95) of the European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products, ICH of Pharmaceuticals for Human Use.
Subjects
Male and female children 6–12 years of age (inclusive) meeting the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5; American Psychiatric Association 2013) (predominantly inattentive, predominately hyperactive-impulsive, or combined) and Kiddie-Schedule for Affective Disorders and Schizophrenia for School Age Children-Present and Lifetime DSM-5 version (K-SADS-PL) criteria for ADHD were enrolled. To be eligible for the study, subjects were required to have an age-appropriate intellectual level (IQ ≥80) and a baseline ADHD-RS-5 score ≥90th percentile normative value for gender and age in at least one category (i.e., total score, inattentive subscale, or hyperactive/impulse subscale).
Key exclusion criteria included blood pressure and pulse greater than the 95th percentile for age and gender, known nonresponse or allergy to MPH treatment, intolerance or hypersensitivity to MPH, history of substance use disorder in the subject or immediate family, a history of seizures during the last 2 years (except simple febrile seizures), Tourette syndrome primary, comorbid psychiatric diagnosis other than ADHD, concurrent medical condition, ODD, positive findings on the C-SSRS at screening, and any clinically significant abnormality in laboratory tests or echocardiogram (ECG) result screening that, in the investigator's opinion, could cause participation in this study to be detrimental to the subject.
Study assessments
The primary endpoint was the mean of all the Swanson, Kotkin, Agler, M-Flynn, and Pelham-Combined (SKAMP-C) scores collected during the full-day laboratory classroom. SKAMP ratings were collected to coincide with PERMP tests performed at baseline (30 minutes predose) and at 1, 2, 4, 6, 8, 10, 12, and 13 hours post-dose during the full-day laboratory classroom. The key secondary efficacy outcome was the time to onset and the duration of efficacy of PRC-063 as measured by SKAMP-C. Secondary efficacy endpoints included: SKAMP-Attention (SKAMP-A), SKAMP-Deportment (SKAMP-D), PERMP, ADHD-RS-5, and CGI.
The SKAMP is a validated, 13-item, trained-rater assessment of ADHD symptoms (i.e., attention and deportment) in a laboratory classroom setting (McCracken et al. 2003). The SKAMP uses a 7-point impairment scale; each item is rated from 0 (no impairment) to 6 (maximum impairment). The SKAMP-C is the sum of scores for all 13 items, with combined scores ranging from 0 to 78, the SKAMP-A subscale contains 7 items that measure attention and work quality (score range: 0–42), whereas the SKAMP-D subscale contains 6 items that measures deportment and compliance (score range: 0–36) (Wigal et al. 1998; Swanson 2000).
The PERMP is a skill-adjusted, 10-minute math test administered as seatwork in a classroom setting (Swanson 2000). Performance was measured by three variables: the number of problems attempted (PERMP-A), the number of problems correct (PERMP-C), and the combined score (PERMP-T).
The ADHD-RS-5 is a clinician-rated global assessment that measures the severity of symptoms of ADHD using 18 items that are grouped into three subscales (hyperactivity/impulsivity, impairment, and attention) (DuPaul et al. 2016). The CGI is a global evaluation of severity (CGI-S) and improvement (CGI-I) over time (Guy 1976).
Safety assessments
Safety assessments included reported and observed adverse events (AEs), vital signs, physical examinations, ECGs, and the C-SSRS. Vital signs, ECGs, and C-SSRS were measured at each clinic visit.
Statistical analysis
The primary and the key secondary efficacy analyses were conducted on the full analysis (FA) population, which included all randomized subjects who received at least one dose of double-blind study medication and who completed mandatory attendance of the full-day laboratory classroom evaluation. The per protocol (PP) population, defined as all subjects in the FA population who completed the full 13-hour full-day laboratory classroom evaluations with no important protocol deviations, was used to provide supportive information.
While conducting the full-day laboratory classroom, it was discovered that at three testing sites Moderate Level tests were given instead of the intended Easy Level tests for the 8-hour time point. While it was decided that all data would be analyzed in the FA population for the primary efficacy variable SKAMP-C, subjects who received the incorrect PERMP test were excluded from the PP population.
The primary efficacy analysis used was a mixed-model repeated-measures analysis that included the full-day laboratory classroom SKAMP-C scores from each time point as the dependent variable. The independent variables in the model included fixed effects for treatment, time, treatment-by-time interaction, investigative site, and covariate terms for the predose SKAMP-C score and predose SKAMP-C score-by-time interaction.
The onset and duration of efficacy of PRC-063 was calculated from the treatment differences in SKAMP-C scores at each time point during the full-day laboratory classroom visit. The onset of efficacy was determined as the first postdose time where the difference between the two treatment groups was statistically significant at the 5% level. If no significant difference was found at any time point, the onset of efficacy was defined as “none.” The offset of efficacy was determined as the last sequential postdose time after onset of efficacy, where the difference between the two treatments was not statistically significant at the 5% level. If all time points were statistically significant at the 5% level between the two treatment groups, the offset of efficacy was defined as >13 hours postdose.
PERMP scores were analyzed in the same manner as the primary endpoint. The ADHD-RS-5 was assessed by analysis of covariance model including fixed effects for treatment, investigative site, and a covariate term for the baseline score. The CGI-S and CGI-I were descriptively summarized by treatment and visit. Summaries were also calculated for CGI-S dichotomized as nonsevere and severe and the category shift from baseline, and for CGI-I dichotomized as responders and nonresponders. No inferential statistics were performed on the CGI-S and CGI-I summaries.
AEs were categorized by system organ class and preferred term-coded according to the MedDRA dictionary (version 20.0). All AEs that changed in severity or relationship to study drug were assigned a new start date and captured as a new event. AEs were assigned to either the dose-optimization period or the double-blind treatment period. AEs occurring in the safety follow-up period following study medication discontinuation were reported under the double-blind treatment period.
Results
Subject disposition
Subject disposition is given in Figure 2. A total of 156 subjects met the entry criteria, were enrolled into the study, and attended the baseline visit of the open-label period. Of these subjects, 148 subjects were randomized into the double-blind period, 147 subjects completed to the full-day classroom visit (FA population), and 140 subjects completed the safety follow-up visit. A total of 112 subjects were included in the PP population, which was defined as all subjects in the FA population who completed the full 13-hour full-day classroom evaluations and had no important protocol deviations.
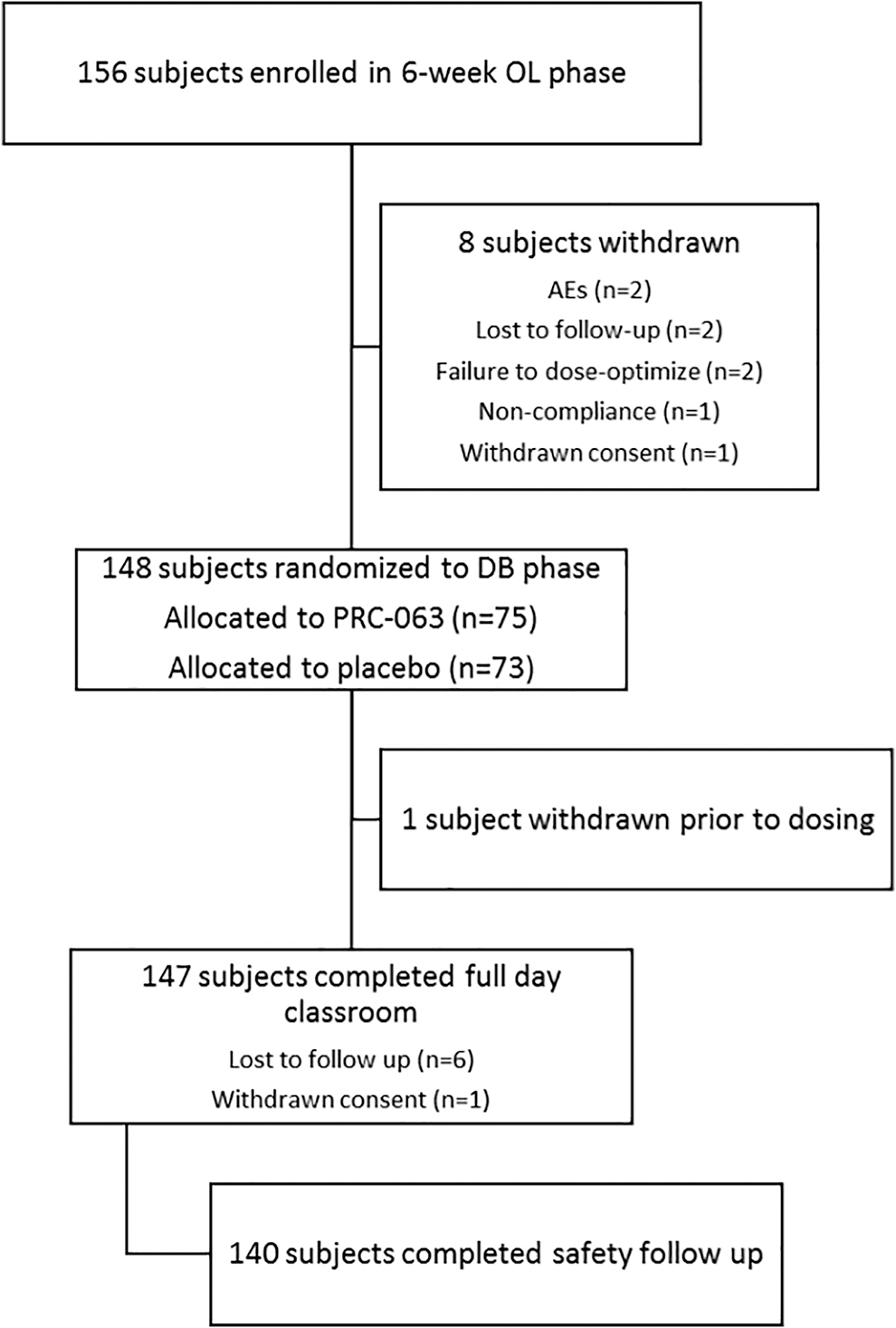
CONSORT diagram of subject disposition. AE, adverse event; DB, double-blind; OL, open-label.
Of the enrolled subjects, a majority were diagnosed with ADHD, combined presentation (n = 131, 84.0%). No subjects were diagnosed as predominantly hyperactive-impulsive. Patient demographics and baseline characteristics were similar between subjects randomized to PRC-063 or placebo (Table 1). The majority of enrolled subjects were boys (65.4%). The mean (± standard deviation [SD]) age of subjects was 9.4 ± 1.88, with a mean body mass index (BMI) (±SD) of 18.5 ± 4.26 kg/m2. The majority of enrolled subjects (n = 99, 63.5%) had prior treatment with psychostimulants for ADHD.
Demographics and Baseline Characteristics
ADHD, attention-deficit/hyperactivity disorder; ADHD-RS-5, attention-deficit/hyperactivity disorder rating scale-5; BMI, body mass index; CGI-S, Clinical Global Impression-Severity; SD, standard deviation.
Dose-optimization period
Figure 3 provides the mean daily dose of PRC-063 by week for the dose-optimization period plotted against the mean (SD) ADHD-RS-5 total score. Of 148 subjects who were dose optimized, the mean (SD) optimized dose was 47.84 (15.12) mg/day, with subjects optimized at each dose level (25 mg: n = 17; 35 mg: n = 30; 45 mg: n = 40; 55 mg: n = 38; 70 mg: n = 16; and 85 mg: n = 7). The mean (SD) ADHD-RS-5 total scores decreased from baseline (42.8 [7.19]) to 15.3 (7.64) at the final dose-optimization visit. Mean (SD) change from baseline values were similar at each optimized dose, from −29.2 (9.95) for those optimized to 25 mg PRC-063 to −26.0 (12.71) for those optimized to 85 mg PRC-063.
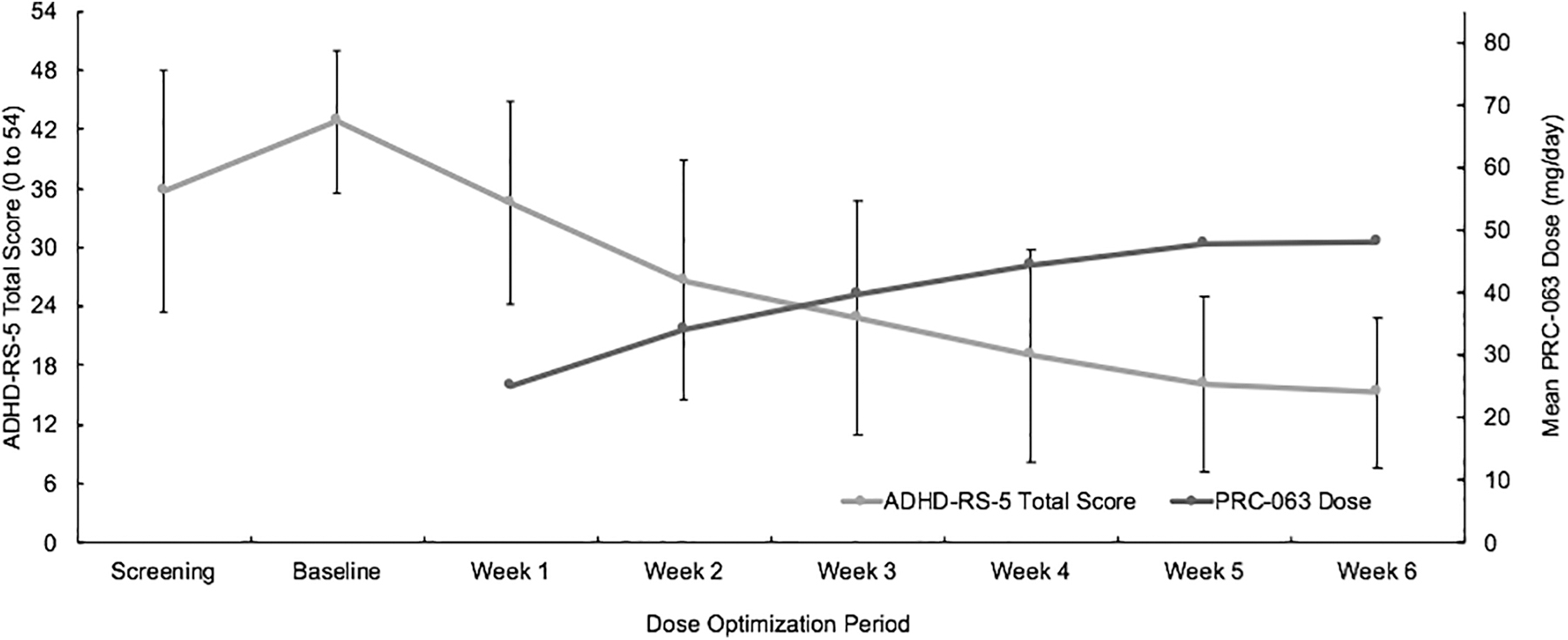
Mean daily dose of PRC-063 by week for the dose-optimization period overlaid by mean (standard deviation) ADHD-RS-5 total score. ADHD-RS-5, attention-deficit/hyperactivity disorder-rating scale 5.
Likewise, clinician ratings of ADHD severity, as measured by CGI-S scores, also showed improvement from baseline. Mean (SD) CGI-S scores decreased from 5.0 (0.76) at baseline to 2.4 (0.86) at the end of the dose-optimization period, with 91.8% of patients rated as 1 (normal, not at all ill), 2 (borderline ill), or 3 (mildly ill). This corresponded with clinician evaluation of improvement on the mean (SD) CGI-I at the last dose-optimization visit, which showed 98.6% of patients were “much improved” (CGI-I = 2) or “very much improved” (CGI-I = 1), and 1.4% of patients were “minimally improved” (CGI-I = 3).
Laboratory classroom
Primary efficacy measure
In the FA population, statistically significant reductions were demonstrated in the PRC-063 group (10.3 ± 0.74) over the placebo group (18.9 ± 0.73, p < 0.0001) when the SKAMP-C scores were averaged over the 13-hour full-day laboratory classroom. The least squares (LS) mean treatment difference (standard error [SE]) was −8.6 (1.02) (95% confidence interval [CI] = −10.6 to −6.6), in favor of PRC-063. A statistically significant response in SKAMP-C score was also observed in the PP population (p < 0.0001). The SKAMP-C predose scores evaluated on the morning of the full-day laboratory classroom indicated that the placebo group displayed less severe behaviors before dosing than the PRC-063 group (p = 0.0367), before dosing.
Key secondary efficacy measures
Figure 4 provides the SKAMP-C scores (LS mean + SE) during the full-day laboratory classroom by time in the FA population. The SKAMP-C scores were significantly lower in the PRC-063 group versus the placebo group (p < 0.0001) at each time point (1, 2, 4, 6, 8, 10, 12, and 13 hours postdose). Therefore, the onset of efficacy was 1 hour and the duration of efficacy was ≥13 hours. Results of the PP analysis support this finding, with significantly reduced SKAMP-C scores in the PRC-063 group versus the placebo group at each time point from 1 to 13 hours postdose (p < 0.0001). The results of the SKAMP-A and SKAMP-D subscales were consistent with SKAMP-C results, with significant reductions in the PRC-063 group over the placebo group at all time points postdose (all, p < 0.0001).
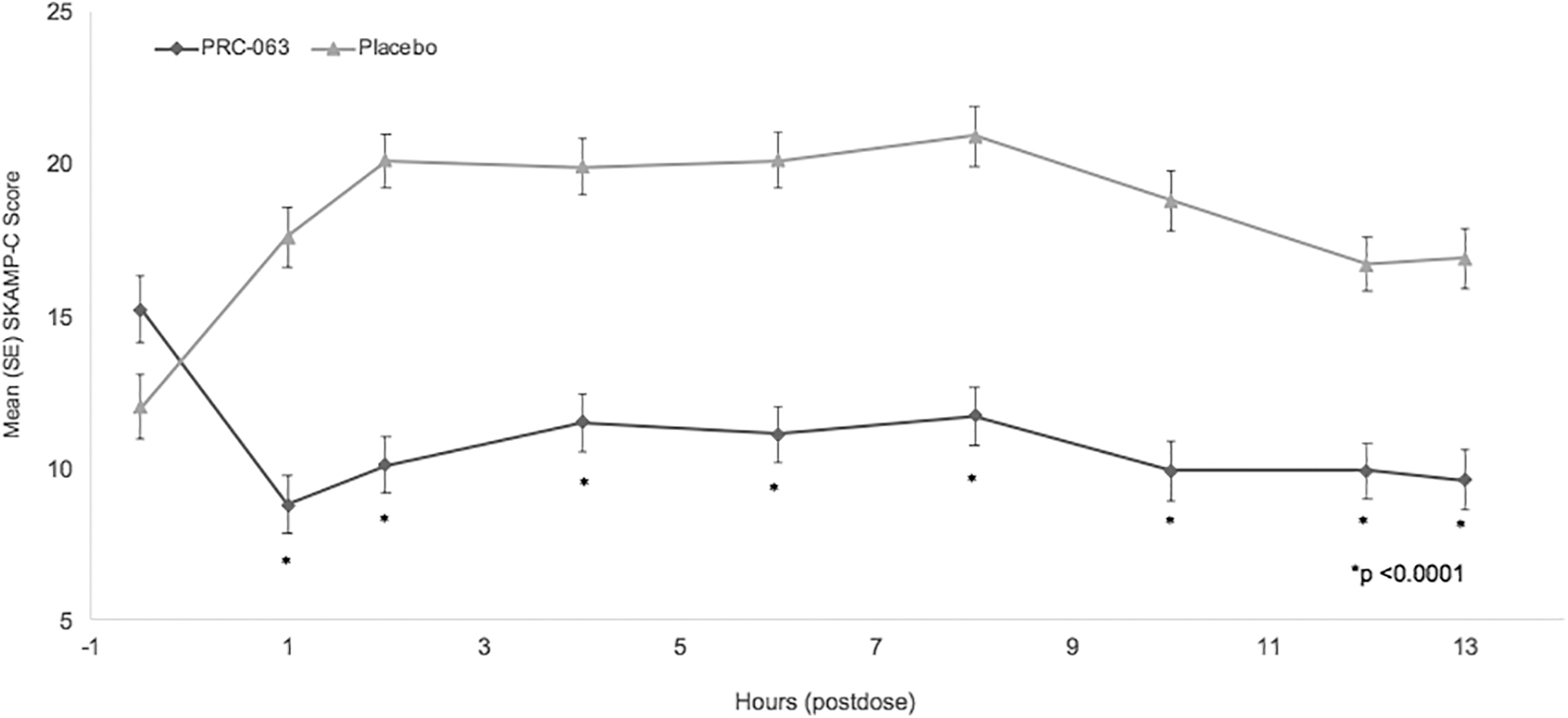
Mean SKAMP-C scores over the full classroom day. LS mean (standard error) SKAMP-C scores were measured predose and at eight postdose time points for subjects in the full analysis set (n = 147). SKAMP-C, Swanson, Kotkin, Agler, M-Flynn, and Pelham-Combined; LS, least squares.
Secondary outcome measures
The mean average postdose PERMP-T scores during the full-day laboratory classroom were significantly better in the PRC-063 group (172.6 ± 5.48) compared with the placebo group (131.1 ± 10.20) for both overall (LS mean treatment difference [SE] = 41.4 [7.58], 95% CI = 26.5–56.4, p < 0.0001) and at each time point (all, p < 0.01) (Fig. 5). The results of the PERMP-A and PERMP-C subscales were consistent with PERMP-T results.
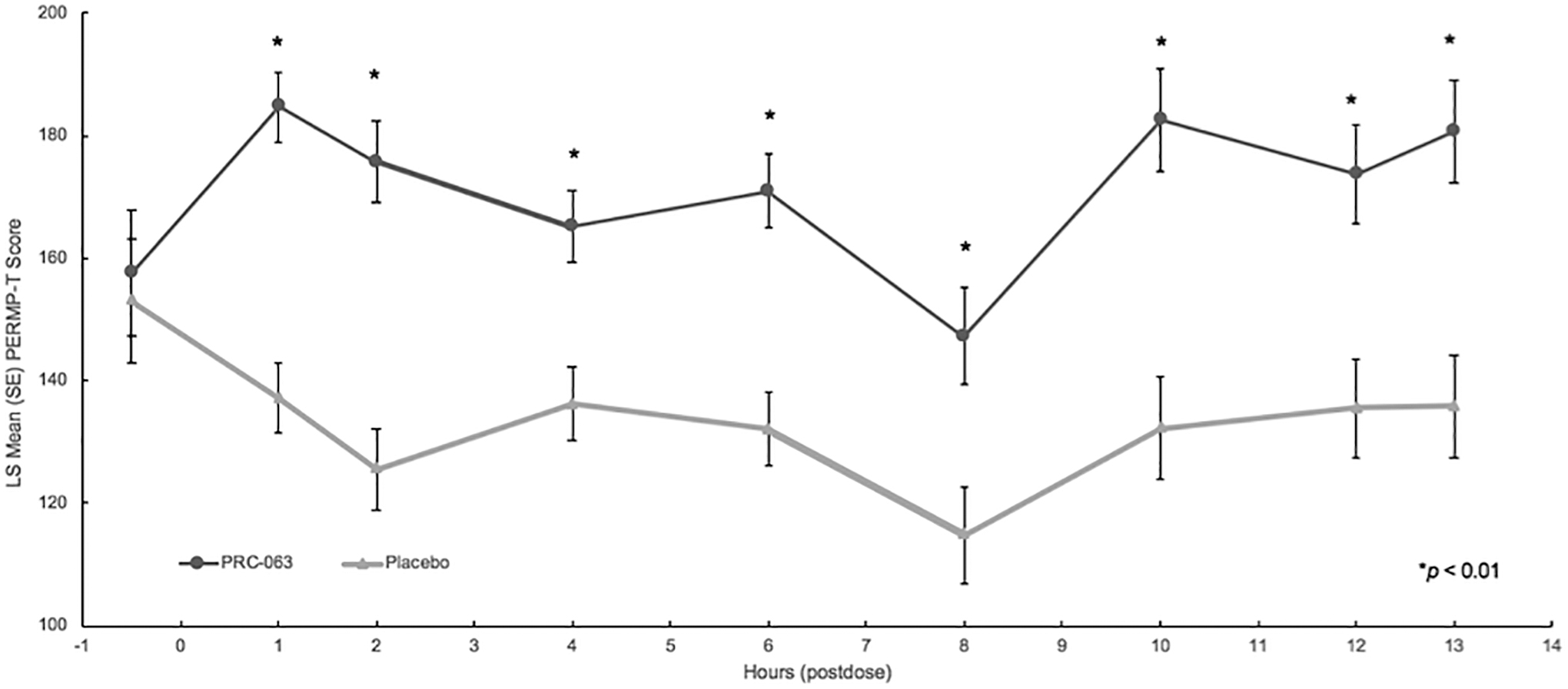
PRC-063 improved PERMP-T scores at each time point during the 13-hour classroom day. LS, least squares; PERMP-T, Permanent Product Measure of Performance-Total.
The clinician-rated ADHD-RS-5 symptoms showed significantly greater improvement for subjects receiving PRC-063 than for those receiving placebo (LS mean treatment difference [SE] = 12.2 [1.88], 95% CI = −15.92 to −8.51, p < 0.0001). During the open-label run-in phase, 73.5% (n = 108) of patients achieved symptomatic remission, defined as an ADHD-RS-5 score of ≤18 (post hoc analysis).
There were 60 subjects (81.1%) in the PRC-063 group versus 28 subjects (38.4%) in the placebo group who shifted from the category of moderate-to-severe illness (CGI-S of 4–7) at baseline to mild illness or less (CGI-S of 1–3) on the CGI-S. At the full-day laboratory classroom, there were 63 subjects (85.1%) in the PRC-063 group who were responders (rated as “much improved” or “very much improved” on the CGI-I) versus 31 subjects (42.5%) in the placebo group.
Safety
A total of 104 of 156 subjects (66.7%) experienced at least one AE during the open-label dose-optimization period. Table 2 provides AEs occurring in ≥5% of subjects during the dose-optimization period. The most common AEs by preferred term were as follows: decreased appetite (n = 55, 35.3%), abdominal pain upper (n = 26, 16.7%), affect lability (n = 22, 14.1%), weight decreased (n = 18, 11.5%), headache (n = 17, 10.9%), irritability (n = 16, 10.3%), and insomnia (n = 16, 10.3%). There was no evidence of an increased incidence of AEs with increased dose level. All AEs were of mild or moderate intensity and no serious adverse events (SAEs) were reported.
Treatment-Emergent Adverse Events Reported in ≥5% of Subjects During the Open-Label, Dose-Optimization Phase
TEAE, treatment-emergent adverse event.
Two subjects each experienced an AE that led to study termination during week 2 of the dose-optimization period while receiving 35 mg: one subject with affect lability and dermatillomania of moderate intensity and one subject with ECG PR interval prolongation of mild intensity, who at screening and baseline visits had asymptomatic ECG abnormality (sinus rhythm with first-degree atrioventricular block).
Table 3 provides AEs occurring in ≥2% (n > 1) of subjects during the 1-week, double-blind, placebo-controlled period. During this week, there was a higher incidence of AEs in the PRC-063 group (n = 18, 24.0%) than in the placebo group (n = 7, 9.6%). The most common AEs in the double-blind phase were increased heart rate (PRC-063: n = 3, 4.0% vs. placebo: n = 1, 1.4%), vomiting (PRC-063: n = 2, 2.7% vs. placebo: n = 0), headache (PRC-063: n = 2, 2.7% vs. placebo: n = 0), and upper respiratory tract infection (PRC-063: n = 2, 2.7% vs. placebo: n = 0).
Treatment-Emergent Adverse Events Reported in ≥2% of Subjects During the Double-Blind Phase
TEAE, treatment-emergent adverse event.
All the AEs in the double-blind week occurred more frequently in the PRC-063 group than in the placebo group with the exception of sinus tachycardia that occurred with a similar incidence in both groups (PRC-063: n = 1, 1.3% vs. placebo: n = 2, 2.7%). All AEs were of mild or moderate intensity. No SAEs were reported and no subjects discontinued owing to an AE during the double-blind period.
There were no clinically meaningful mean changes from screening to end of study in the PRC-063 or placebo groups in hematology, chemistry, or urinalysis parameters.
There were slight, nonclinically meaningful increases from baseline in mean systolic blood pressure (mean change from baseline [SD]: PRC-063 = 2.5 [8.54]), mean diastolic blood pressure (mean change from baseline [SD]: PRC-063 = 5.3 [9.61]), and mean pulse rate (mean change from baseline [SD] = PRC-063 = 7.8 [12.15]) that were observed from baseline to the half day laboratory classroom visit. There was no obvious dose relationship between dose of PRC-063 received and mean change from baseline in diastolic blood pressure or pulse rate. However, the change from baseline in mean systolic blood pressure values showed greater increases in the 70 mg group (4.7 mmHg) and 85 mg group (4.6 mmHg) than in the lower dose groups.
There were no clinically meaningful differences between mean ECG changes from baseline to the full-day laboratory classroom in the PRC-063 group versus the placebo group.
Eighteen subjects (11.5%) experienced an AE of weight decrease (defined as a decrease of >5% of total body weight) during the open-label, dose-optimization period. At the completion of the dose-optimization period, 11 of these subjects continued to experience ongoing weight decrease. Sixteen subjects (10.3%) had a mild intensity weight decrease and two subjects (1.3%) had a moderate intensity weight decrease. By the end of the open-label treatment period, the change from baseline weight in these subjects ranged from −4.0 to +0.5 kg (one subject had weight gain). In most subjects (n = 15), total weight loss was <3.0 kg. Of these, 14 subjects (93%) had a BMI classified as “healthy” at the start of the study (1 subject was classified as “overweight”), and 14 subjects (93%) had a BMI classified as “healthy” at the half day laboratory classroom (1 subject was classified as “underweight”). In the two subjects who lost >3.0 kg, one subject had a screening BMI categorized as “overweight” and the other as “obese.” However, with the recorded weight decrease at the half day laboratory classroom, one subject's BMI classification changed to “healthy” and the other to “overweight.”
There were no mean changes in weight from baseline to the half day laboratory classroom that were classified as clinically meaningful, although the mean change in the 25 mg group was a weight increase (2.284 kg), rather than a weight decrease, as seen in the other dose groups.
Analysis of the C-SSRS showed that there were no subjects who expressed suicidal ideation or suicidal behavior at screening, during the open-label, dose-optimization period, during the double-blind treatment period, or during the follow-up period.
Discussion
Over a 13-hour period in a simulated classroom setting, children with ADHD receiving PRC-063 demonstrated significant improvements in attention and ADHD symptoms compared with those receiving placebo. Some currently approved MPH products have an onset of action within 1 hour, but they have not shown efficacy beyond 12 hours. PRC-063 is the first ER MPH formulation to have demonstrated a 13-hour duration of action in children. This rapid onset and long-acting profile is achieved as a result of the novel formulation, which was developed to provide an initial rapid release of MPH, followed by an extended release throughout the day. PRC-063 was well tolerated with rates of AEs similar to those observed with other ER stimulant treatments.
In similarly designed pediatric classroom studies, the onset of action of other ER stimulant treatments has been reported as 1 hour postdose or less (Wigal et al. 2013, 2014; Childress et al. 2015, 2017) to 1.5–2 hours postdose (e.g., Pelham et al. 2001; McCracken et al. 2003; Swanson et al. 2004; Biederman et al. 2007; Wigal et al. 2013, 2014; Childress et al. 2017). Studies have reported a duration of action of 8 hours (Wigal et al. 2017), 10 hours (Wigal et al. 2013; Childress et al. 2015), 12 hours (Pelham et al. 2001; McCracken et al. 2003; McGough et al. 2006; Biederman et al. 2007; Wigal et al. 2013; Childress et al. 2017), and up to 13 hours for some amphetamine preparations (Wigal et al. 2009; Childress et al. 2018). Of note, some of these studies demonstrated the offset of efficacy, whereas others did not.
In this study, the overall treatment difference, and the onset and duration of PRC-063 were similar or superior to that of currently approved ER ADHD stimulant treatments. Improved attention from baseline in children with ADHD treated with PRC-063 persisted up to and including 13 hours. Despite the duration of action of currently approved long-acting agents, caregivers and children and adolescents with ADHD often report that medication wears off during or after school into the late afternoon or early evening (Sikirica et al. 2015). Moreover, the burden of ADHD may change over time, or according to environmental supports or demands (CADDRA 2018). In this study, efficacy was maintained up to and including 13 hours without significant deterioration. In previous studies examining other long-acting agents, a return to baseline level of attention and functioning has been observed within 10–12 hours (Biederman et al. 2007; CADDRA 2018). Alonger duration of action can be beneficial for caregivers and children and adolescents with ADHD, easing the burden that is experienced later in the school day, during after school activities, and at home. Adult workplace environment studies have demonstrated a duration of action longer than 13 hours among adults treated with PRC-063 (Wigal et al. 2020) and other ADHD stimulants (Wigal et al. 2010).
At the completion of the open-label, dose-optimization period, in the subjects receiving up to 6 weeks of active treatment, the mean ADHD-RS-5 total scores improved from baseline to the half day laboratory classroom in all dose groups, and the CGI-S mean scores shifted from “moderate-to-severe” at baseline in 100% of subjects to “mild illness or less” at the half day laboratory classroom in 91.8% of subjects. In addition, 73.5% of patients achieved symptomatic remission, defined as an ADHD-RS-5 score of ≤18. Taken together, these data demonstrate that when a strictly defined dose-optimization protocol is used, ADHD symptoms can be well-controlled in the majority of subjects (148 met randomization criteria of 156 enrolled subjects [94.9%]). This provides a basis for comparison of subjects who, during the double-blind period, were no longer receiving treatment after they were randomized to placebo.
The results of the primary and key secondary outcome measures were supported by additional secondary outcome measures of the study during both the open-label titration and double-blind periods, which showed significant improvements in clinical impressions and measures of ADHD symptoms by the subject. The secondary endpoints support the efficacy of PRC-063 over placebo at the full-day laboratory classroom.
In this study, before receiving PRC-063 or placebo during the laboratory classroom day, a significant treatment difference (SE) of 3.1 (1.48) (95% CI = 0.2–6.1; p = 0.0367) was observed in the SKAMP-C predose scores, favoring placebo. This finding is consistent with observations in other classroom studies examining other long-acting stimulant preparations (Lopez et al. 2003; Swanson et al. 2004; Wigal et al. 2009, 2014; Childress et al. 2017). By the second treatment time point (between 0.75 and 1.5 hours postdose) in all these classroom studies, the performance of subjects receiving placebo deteriorated, whereas the performance of subjects receiving active medication improved to levels greater than or equivalent to the predose of the placebo group. Although these data suggest a possible morning “rebound” effect for subjects medicated with long-acting stimulants, no study has been designed to examine such an effect and any conclusions should be made with caution.
During this study, treatment with PRC-063 was well-tolerated. There were no SAEs and no deaths. Of the most commonly reported AEs during the open-label, dose-optimization period, the frequencies of decreased appetite, abdominal pain upper, affect lability, and irritability in subjects receiving PRC-063 were similar to those observed in studies of other oral, long-acting MPH and amphetamine products. Weight decrease is often associated with the use of stimulants. In this study, although there were weight decreases of 5% or greater recorded in 18 subjects, all but 1 of those subjects remained at a “healthy” BMI. The subjects with numerically larger weight decreases started the study in the “overweight” and “obese” BMI categories. Of note, the frequencies of insomnia and/or headache in patients receiving PRC-063 were lower than in similar studies of other oral MPH ER products (Wigal et al. 2014, 2017; Childress et al. 2017).
Similar classroom studies that have examined long-acting stimulants for the treatment of ADHD also indicate that the frequency of AEs during the open-label, dose titration period are typically much greater than those reported during the double-blind treatment period of the same study (Wigal et al. 2009, 2013, 2014, 2017; Childress et al. 2015, 2017). The findings from this study were consistent with these previous studies.
There are many factors that affect treatment response to MPH, including genetic variability, interactions with other medications, and comorbidities. Clinically, it is important to individually titrate patients to an optimal dose. The starting dose for all subjects in this study was 25 mg PRC-063 once-daily each morning; subjects subsequently adjusted if required until their next optimal dose was reached. Although no subjects withdrew because of AEs during the first week of treatment, one subject was withdrawn owing to medication noncompliance. This indicates that 25 mg PRC-063 once-daily each morning is an appropriate starting dose in children 6–12 years old.
In addition, 17 subjects (11%) were dose optimized at 25 mg, thus supporting 25 mg/day PRC-063 as the lowest effective dose in children 6–12 years old. In this study, subjects were dose optimized across the range of doses: 35 mg/day (n = 30, 20%), 45 mg/day (n = 40, 27%), 55 mg/day (n = 38, 26%), 70 mg/day (n = 16, 11%), and 85 mg/day (n = 7, 5%). This demonstrates that the wide range of available doses of PRC-063 can facilitate titration to achieve optimal clinical effect in clinical practice.
There are several limitations to this study. Consistent with other phase 3 studies, the study duration was relatively short and the study eligibility criteria, which included children aged 6–12 without comorbidities, could potentially confound the study results. In addition, multiple subjects were provided with the incorrect 8-hour test PERMP tests of the incorrect difficulty level leading to a reduction in the number of correct and attempted problems. These subjects were excluded from the PP population for the confirmatory analysis.
Conclusion
This study successfully demonstrates significant improvements in attention and ADHD symptoms over a 13-hour laboratory classroom in children 6–12 years of age who received optimized oral doses of PRC-063 (ranging from 25 to 85 mg daily) compared with placebo. This study demonstrates that PRC-063 was a well tolerated, safe, and effective treatment of ADHD with rates of AEs similar to those observed with other ER stimulant treatments.
Clinical Significance
PRC-063 represents a new methylphenidate treatment option for children with ADHD, where both a rapid onset and extended duration of effect are required to achieve symptom control. PRC-063 was well tolerated, and AEs observed mainly reflect adverse events commonly associated with methylphenidate use.
Footnotes
Disclosures
A.C.C. has received research support from Akili, Allergan, Alcobra, Arbor, Emalex, Forest, Ironshore, KemPharm, Lilly, Lundbeck, Medgenics, Neos, Neurovance, NextWave, NLS, Noven, Otsuka, Pearson, Pfizer, Purdue Pharma (Canada), Rhodes Pharmaceuticals L.P., Servier, Shire, Sunovion, Supernus, Theravance, and Tris; been a consultant for and received honoraria from Kempharm, Ironshore, Neos, Pfizer, Rhodes Pharmaceuticals L.P., Shire, Sunovion, Supernus and Tris; received travel support from Adlon, Ironshore, NextWave, Pfizer, and Shire; has received writing assistance on projects from Adlon, Arbor, Ironshore, Neos, NextWave, Pfizer, Purdue, Rhodes Pharmaceuticals L.P., Shire and Tris; received payment for lectures from Arbor, Neos, Pfizer, Shire and Tris; has been an advisory board member for Arbor, Ironshore, Neos, Neurovance, NextWave, Noven, Pfizer, Purdue Pharma L.P. and Rhodes Pharmaceuticals L.P.; and has participated in speakers bureaus for Takeda. A.J.C. is a consultant to Adlon Therapeutics, Aevi Genomics, AiCure, Akili Interactive, Arbor Pharmaceuticals, Ironshore, KemPharm, Lundbeck, MedAvante-ProPhase, Neos Therapeutics, NLS Pharma, Otsuka, Purdue, Shire, Sunovion, Supernus, Takeda, Tris Pharma; has received speaker/promotional honoraria from Adlon Therapeutics, Arbor Pharmaceuticals, Ironshore, Lundbeck, Neos Therapeutics, Otsuka, Shire, Sunovion, Takeda, Tris Pharma; has research grants from Aevi Genomics, Akili Interactive, Arbor Pharmaceuticals, Ironshore, KemPharm, Lundbeck, Neos Therapeutics, Otsuka, Purdue, Shire, Sunovion, Supernus, Takeda, Tris Pharma; and is an employee and board member of the Neuroscience Education Institute. M.N.B. has served on advisory boards for Neos Therapeutics, Shire, and Tris Pharma, Inc., and has participated in speakers bureaus for Neos Therapeutics, Lundbeck, Otsuka, Sunovion Pharmaceuticals, Inc., Takeda, and Tris Pharma, Inc. G.A.E.D and S.B. are employees of Purdue Pharma (Canada). Medical writing services were provided by Susan Bartko-Winters, PhD, of SBW Medical Writing Inc., which was funded by Purdue Pharma (Canada).