
Abstract
Objectives:
Describe the safety and tolerability of lisdexamfetamine dimesylate (LDX) and provide data on clinical effects for efficacy-related endpoints and pharmacokinetics in preschool-aged children with attention-deficit/hyperactivity disorder (ADHD).
Methods:
This phase 2, multicenter, open-label, dose-optimization study (
Results:
Among 24 participants, the most frequently reported TEAE was decreased appetite (8/24; 33%). At week 8/early termination, mean (standard deviation) systolic and diastolic blood pressure and pulse changes from baseline were −1.1 (7.31) and 1.5 (6.93) mmHg and −0.8 (12.75) bpm, respectively. The mean (95% confidence interval) change from baseline ADHD-RS-IV-PS total score at the final on-treatment assessment was −26.1 (−32.2 to −20.0). Pharmacokinetic parameters of d-amphetamine, a major active metabolite of LDX, were characterized: d-amphetamine exposure increased with LDX dose; mean t max and t 1/2, respectively, ranged from 4.00 to 4.23 hours and 7.18 to 8.46 hours.
Conclusions:
In preschool-aged children with ADHD, LDX was generally well tolerated and reduced ADHD symptoms, consistent with observations in children 6–17 years of age. Based on these findings, a starting LDX dose as low as 5 mg in phase 3 studies in preschool-aged children is supported.
Introduction
Psychostimulants are recommended as first-line pharmacotherapy for treating of attention-deficit/hyperactivity disorder (ADHD) in children and adolescents (Pliszka 2007). Although psychostimulants are used in the treatment of ADHD in preschoolers, few studies have systematically evaluated their effects in this population (Pliszka 2007).
In one literature review, treatment benefits for methylphenidate (MPH) on ADHD symptoms in preschoolers (3–6 years old) with ADHD were reported in eight of nine identified studies (Connor 2002). In a multisite, longitudinal study of preschool-aged children (Preschool ADHD Treatment Study [PATS]) (Kollins et al. 2006), the efficacy and safety of immediate-release MPH in preschoolers (3–5.5 years) was examined (Greenhill et al. 2006; Wigal et al. 2006). During the short-term efficacy phases of the PATS, immediate-release MPH at doses ranging from 2.5 to 7.5 mg three times daily produced significantly greater improvement in ADHD symptoms than did placebo, as measured by reductions in a composite score of parent and teacher ratings on the Conners, Loney, and Milich (CLAM) scale and the Swanson, Kotkin, Atkins, M-Flynn, and Pelham (SKAMP) scale (Greenhill et al. 2006). Across the PATS, adverse events (AEs) were reported more frequently with MPH than placebo and the most commonly reported AEs occurring with MPH versus placebo included emotional outbursts, difficulty falling asleep, and decreased appetite (Wigal et al. 2006). In a small open-label pilot study of the nonstimulant atomoxetine (n = 22), a mean total score decrease from baseline of ∼21 points on the ADHD Rating Scale-IV (ADHD-RS-IV) was reported in children 5–6 years of age after 8 weeks of treatment, with the most frequently reported AEs being emotional lability and decreased appetite (Kratochvil et al. 2007).
There are also data suggesting that the effects of ADHD pharmacotherapy may differ across ages. Teacher-rated effect sizes for MPH on the CLAM scale and the SKAMP scale tended to be smaller in preschool-aged children (Greenhill et al. 2006) than in school-aged children (7–9.9 years) (Greenhill et al. 2001). In terms of pharmacokinetics, preschool-aged (4–6 years) children with ADHD exhibited slower MPH clearance and greater MPH exposure to the same weight-adjusted MPH dose than school-aged (7–8 years) children with ADHD (Wigal et al. 2007). In contrast, reductions in ADHD-RS-IV total score following 8 weeks of open-label atomoxetine tended to be greater in 5-year-old children than in 6-year-old children (Kratochvil et al. 2007).
In a recent review of Medicare claims, it was reported that nearly 40% of preschool-aged children diagnosed with ADHD were treated with a psychostimulant (Davis et al. 2019), despite the fact that in this age group these medications are not approved for use and there is limited evidence for their efficacy and safety. Furthermore, preschool-aged children diagnosed with ADHD can exhibit impaired school readiness compared with age-matched counterparts who do not have an ADHD diagnosis (Perrin et al. 2019). As such, identifying treatment strategies that can extend symptom relief to later portions of the day, especially during extended daycare or preschool hours, would be beneficial. The use of long-acting psychostimulant formulations could also produce more consistent plasma drug concentrations throughout the day compared with immediate-release formulations. Given the importance of identifying safe and effective pharmacotherapies for preschool children with ADHD, further studies are warranted with other approved ADHD pharmacotherapies. Lisdexamfetamine dimesylate (LDX) is approved in the United States and other countries for use in individuals ≥6 years with ADHD (Vyvanse® 2017). This phase 2 study was conducted in the context of a pediatric written request by the U.S. Food and Drug Administration to obtain preliminary safety, tolerability, pharmacokinetic, and clinical response data for LDX in preschool-aged (4–5 years) children with ADHD.
Methods
Study design and treatment
This phase 2, multicenter, open-label, dose-optimization study (
At the start of the screening and washout period, which occurred within 28 days of the first treatment day, eligible participants discontinued their existing ADHD medication (if currently taking a medication that did not control ADHD symptoms with acceptable tolerability) and entered the dose-optimization period, at which time they initiated treatment with 5 mg LDX. On-treatment visits were scheduled every 7 ± 2 days. The LDX dose was titrated weekly (week 1: 5 mg; week 2: 10 mg; week 3: 15 mg; week 4: 20 mg, week 5: 30 mg) until an optimal dose was achieved. This dosing scheme was considered to be conservative with regard to the starting dose, maximum dose, and dose-optimization schedule. The dosing scheme, in particular the relatively low maximum dose, was selected for use based on general safety considerations, as there were concerns that plasma amphetamine concentrations would be high in this population due to their low weight. Determination of the optimal dose was based on achievement of a ≥30% reduction in ADHD-RS-IV Preschool version (ADHD-RS-IV-PS) total score from the baseline visit (visit 0) and a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved) with acceptable safety and tolerability. Reductions to the previous dose level were permitted between weeks 2 and 4 of the dose-optimization period if tolerability was unacceptable. If 5 mg LDX was not tolerated, the participant was discontinued. During dose maintenance (weeks 7–8), participants were maintained on the optimized LDX dose established during the dose-optimization phase. Concomitant medications (i.e., nonstudy medications taken between the dates of the first and last doses of investigational product) were recorded.
Study participants
Participants eligible for inclusion were male or female children 4–5 years of age at the time of consent. Participants were also required to have a primary ADHD diagnosis based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria based on a detailed psychiatric evaluation, baseline ADHD-RS-IV-PS total scores ≥28 for boys and ≥24 for girls, baseline CGI–Severity (CGI-S) score ≥4, and a Peabody Picture Vocabulary Test, Fourth Edition, standard score ≥70 at screening. Additional inclusion requirements were having undergone an adequate course of nonpharmacologic treatment or having symptoms severe enough based on investigator judgment to warrant pharmacologic treatment without prior nonpharmacologic treatment, and having participated in a structured group activity (e.g., preschool, sports) that allowed for assessment of symptoms and impairment outside the home. With regard to nonpharmacologic therapy, there were no protocol-specified methods or treatment durations. However, all treatments had to be described to and approved by the study sponsor or a delegate before a prospective participant could enter the study.
Key exclusion criteria included having well-controlled ADHD symptoms while receiving an ADHD medication with acceptable tolerability; having a concurrent condition that could confound safety assessments, increase risk to the participant, prohibit the participant from completing the study, or make it difficult to adhere to study procedures; or having previously failed to fully respond to amphetamine therapy. Currently being considered at risk for suicide in the opinion of the investigator, having previously made a suicide attempt, or currently demonstrating active suicidal ideation (intermittent passive suicidal ideation was not necessarily exclusionary); having a documented allergy, hypersensitivity, or intolerance to amphetamine or excipients in LDX; having history of serious cardiac problems, including family history of sudden cardiac death or ventricular arrhythmia; having a screening or baseline blood pressure ≥95th percentile for age, sex, and height; having a clinically significant electrocardiogram (ECG); and being ≤5th percentile for age and sex at screening for height or weight, were also exclusionary. Participants reporting investigational drug use or participation clinical study ≤30 days before screening were also excluded.
Assessments
Safety and tolerability
All AEs were collected from the time of informed consent through the follow-up; AEs were categorized based on severity, seriousness, and relatedness to treatment (per the investigator) or study discontinuation. Treatment-emergent AEs (TEAEs) were defined as those starting on or after the first study drug dose or those starting before the first study drug dose but increasing in severity after starting treatment. Vital signs (systolic and diastolic blood pressure [SBP and DBP] and pulse) were assessed at all visits. Blood pressure was initially monitored using automated blood pressure cuffs. However, when it was determined that the automated cuffs did not accurately measure blood pressure in these 4- to 5-year–old children, blood pressure was measured manually. Assessments were made after participants were seated for ≥5 minutes, with the average value being based on three measurements obtained at ∼2-minute intervals. Clinical laboratory measurements were obtained at screening and at week 8/early termination (ET). A single 12-lead ECG (performed after 5 minutes of rest) was assessed at screening and every postbaseline visit; at baseline, average ECG values were based on three measurements obtained at ∼5-minute intervals.
Clinical effects
Clinical effects were assessed with the ADHD-RS-IV-PS and CGI-I. The ADHD-RS-IV-PS was administered at baseline and each study visit through week 8/ET. The 18-item ADHD-RS-IV-PS (McGoey et al. 2007), which is adapted from the ADHD-RS-IV (Dupaul et al. 1998), is a clinician-administered scale that rates ADHD symptom frequency based on DSM-IV-TR criteria using age-appropriate examples for preschool children. Items are scored on a 4-point scale (range: 0 [never or rarely] to 3 [very often]); total score ranges from 0 to 54. The items can be grouped into two 9-item subscales to assess hyperactivity/impulsivity and inattention. Overall ADHD improvement was assessed using the CGI-I (Guy 1976) (range: 1 [very much improved] to 7 [very much worse]) at each postbaseline visit, relative to baseline severity measured by CGI-S (Guy 1976) score (range: 1 [normal, not at all ill] to 7 [among the most extremely ill]).
Pharmacokinetics
Blood samples for pharmacokinetic assessment of LDX and d-amphetamine were taken at various predose times during weeks 1 through 7 in all participants. A subset of participants had additional samples collected at week 7 at prespecified time points (predose and 1, 2, 3, 4, 6, and 8 hours postdose).
The pharmacokinetics of LDX and d-amphetamine were evaluated using noncompartmental analysis (NCA). The pharmacokinetic parameters estimated included maximum plasma concentration (C max), time to C max (t max), area under the plasma concentration curve at steady state (AUCtau), terminal half-life (t 1/2), and total body clearance for extravascular administration at steady state (CLss/F). Plasma LDX and d-amphetamine concentrations were determined by validated bioanalytical methods using liquid chromatography with tandem mass spectrometry in a manner consistent with previous reports (Ermer et al. 2016a,b). Quantification limits for d-amphetamine and LDX were 2.00 and 1.00 ng/mL, respectively.
Data presentation and statistical analysis
A formal sample size calculation was not conducted for the primary safety and tolerability assessments. Sample size for the rich pharmacokinetic NCA was calculated to achieve 80% probability of ensuring that the 95% confidence intervals (CIs) for total body clearance for extravascular administration (CL/F) and the oral volume of distribution (Vz/F) were within 60%–140% of the geometric mean point estimates, with estimates of between-participant variability being 0.128 standard deviations (SDs) for Vz/F and 0.258 SDs for CL/F. Using these criteria, it was determined that at least 6 participants were required for the rich pharmacokinetic sampling NCA.
Safety and tolerability assessments were conducted in the safety analysis set (all participants taking ≥1 study drug dose) and are presented using descriptive statistics. Clinical outcome assessments were conducted in the full analysis set (FAS; all safety analysis set participants having ≥1 postdose ADHD-RS-IV-PS total score assessment); these data are presented using descriptive statistics with two-sided 95% CIs. Baseline for safety/tolerability and for clinical outcome assessments was defined as the last value collected before the first study drug dose; for clinical outcomes, the final on-treatment assessment (FoTA) was defined as the last assessment obtained after baseline while on study drug. For the ADHD-RS-IV-PS, total score change from baseline is presented at each study visit and at the FoTA; changes from baseline for the ADHD-RS-IV-PS subscales are presented at the FoTA. For the CGI-I, dichotomized improvement (improved vs. not improved) at the FoTA is reported, with participants categorized as improved if the CGI-I score was 1 (very much improved) or 2 (much improved) and as not improved if the CGI-I score was 3 (minimally improved) through 7 (very much worse).
Pharmacokinetic NCA was conducted in the full pharmacokinetic set (all safety analysis set participants for whom primary pharmacokinetic data were sufficient and interpretable) using Phoenix® WinNonlin® v.6.3 (Pharsight®, St. Louis, MO).
Results
Participant disposition and demographics
The safety analysis set and FAS included 24 participants; the pharmacokinetic set included 8 participants; 19 participants completed the study (Fig. 1). Reasons for study discontinuation were AEs (n = 2), withdrawal by participant/legally authorized representative (n = 2), and lost to follow-up (n = 1).
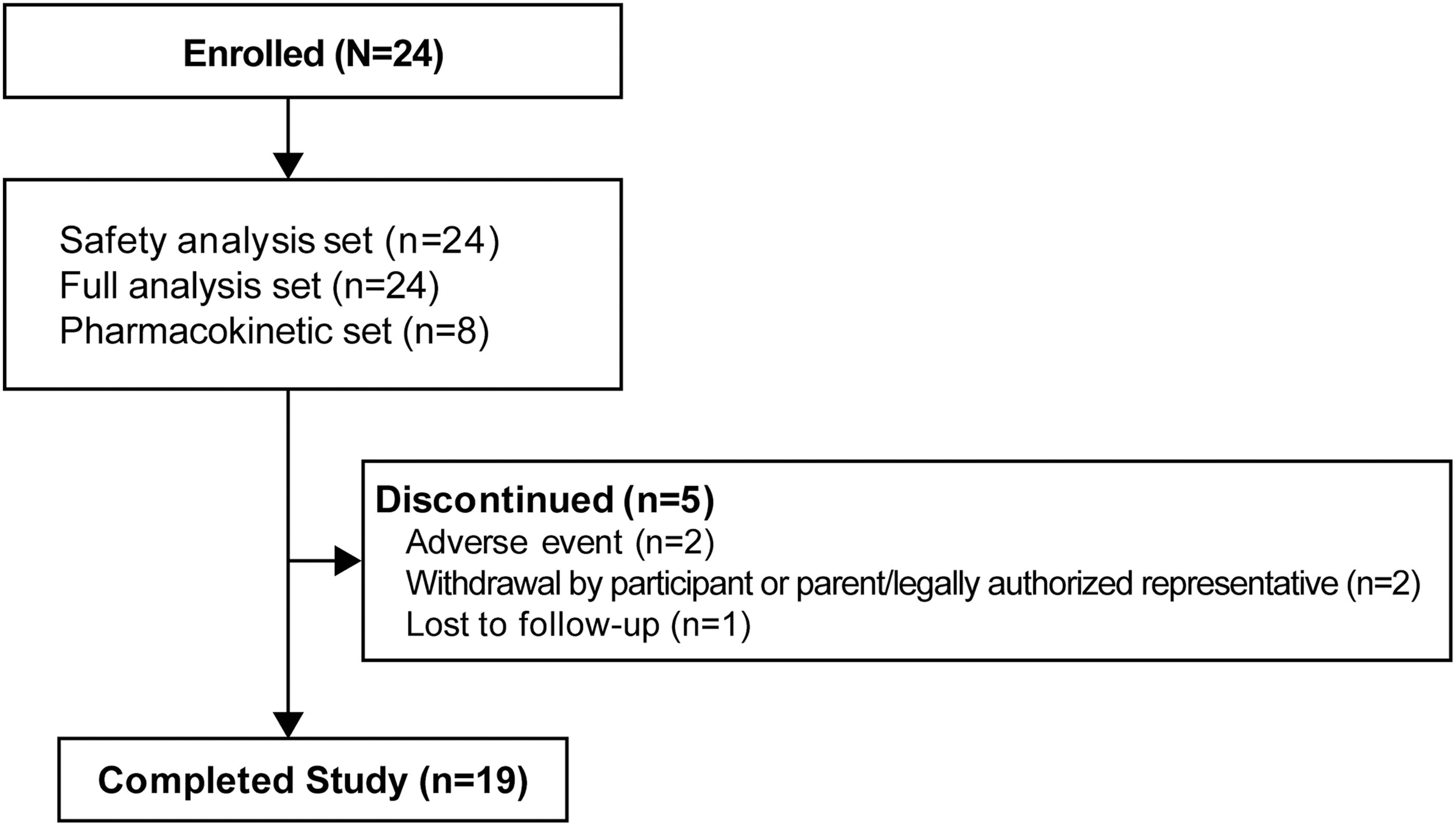
Participant disposition.
Demographic and baseline clinical characteristics are summarized in Table 1. Most participants were male (18/24 [75%]) and white (15/24 [63%]). Based on body mass index (BMI), most (15/24 [63%]) participants were a healthy weight (BMI ≥5th to <85th percentile based on CDC standards). Most participants were diagnosed as having the combined ADHD subtype (21/24 [88%]) and were categorized as being markedly ill based on the CGI-S (10/24 [42%]). The mean ± SD ADHD-RS-IV-PS total score was 43.6 ± 5.54 (median 44; range 32–51) at baseline.
Demographic and Baseline Clinical Characteristics, Safety Analysis Set
BMI based on Centers for Disease Control and Prevention BMI percentiles for children and adolescents (underweight: BMI <5th percentile; healthy weight: BMI ≥5th to <85th percentile; overweight: BMI ≥85th to <95th percentile; obese: BMI ≥95th percentile).
No participants were categorized as “normal, not at all ill,” “borderline mentally ill,” “mildly ill,” or “among the most extremely ill.”
ADHD, attention-deficit/hyperactivity disorder; BMI, body mass index; CGI-S, Clinical Global Impressions–Severity; SD, standard deviation.
Prior and concomitant medications
Before the study, ADHD medications used by study participants were clonidine (n = 2), guanfacine (n = 1), and immediate-release mixed amphetamine salts (n = 1). No concomitant ADHD medications were used during the study.
Lisdexamfetamine dosing
The mean (SD) daily LDX dose at week 8 was 22.9 (7.25) mg/day. The maximum LDX dose was 30 mg in 10 participants, 20 mg in 2 participants, 15 mg in 6 participants, 10 mg in 5 participants, and 5 mg in 1 participant. The optimized LDX dose was 30 mg in nine participants, 20 mg in two participants, 15 mg in six participants, and 10 mg in two participants; the five participants who discontinued did not achieve an optimized dose.
Safety and tolerability
TEAEs were reported by 79% (19/24) of participants (Table 2); 30 mg LDX had the highest TEAE frequency (60%; 6/10). No serious TEAEs, severe TEAEs, or deaths were reported during the study. Two participants discontinued due to TEAEs while on 5 mg LDX (increased hyperactivity and worsened sleep [n = 1]; manic-like symptoms [n = 1]); both instances were considered moderate in severity (except for worsened sleep, which was considered mild), related to study drug, and resolved following discontinuation. TEAEs reported by ≥2 participants across all doses are summarized in Table 2; the most frequently reported TEAEs (occurring in ≥4 participants) were decreased appetite, insomnia, and upper respiratory tract infection. Among participants reporting insomnia-related TEAEs, there were four reports of insomnia, two reports of initial insomnia, and one report of middle insomnia.
Summary of Treatment-Emergent Adverse Events, Safety Analysis Set
LDX, lisdexamfetamine dimesylate; TEAE, treatment-emergent adverse event.
All mean ± SD vital sign changes from baseline at week 8/ET were variable in this small sample (pulse −0.8 ± 12.75 bpm; SBP −1.1 ± 7.31 mmHg; DBP 1.5 ± 6.93 mmHg; heart rate −3.860 ± 10.7809 bpm; Fridericia-corrected QT interval 2.007 ± 13.5858 ms). In two participants, TEAEs related to blood pressure changes (increased DBP: n = 1, 20 mg LDX; increased blood pressure: n = 1, 10 mg LDX) were reported; neither participant withdrew due to these TEAEs. At week 8/ET, mean ± SD weight decreased to 20.67 ± 2.310 kg from 21.03 ± 24.55 kg at baseline (mean ± SD change from baseline: −0.41 ± 0.824 kg). Two participants exhibited a weight decrease ≥7% from baseline.
Clinical effects
ADHD-RS-IV-PS total score decreased over the course of the study (Fig. 2), with the mean (95% CI) change from baseline at the FoTA being −26.1 (−32.2 to −20.0). Score decreases were also observed for the hyperactivity/impulsivity (−12.5 [−15.7 to −9.3]) and inattention (−13.6 [−16.9 to −10.2]) subscales. At FoTA, a majority of participants (20/24 [83%]) were categorized as improved on the dichotomized CGI-I.
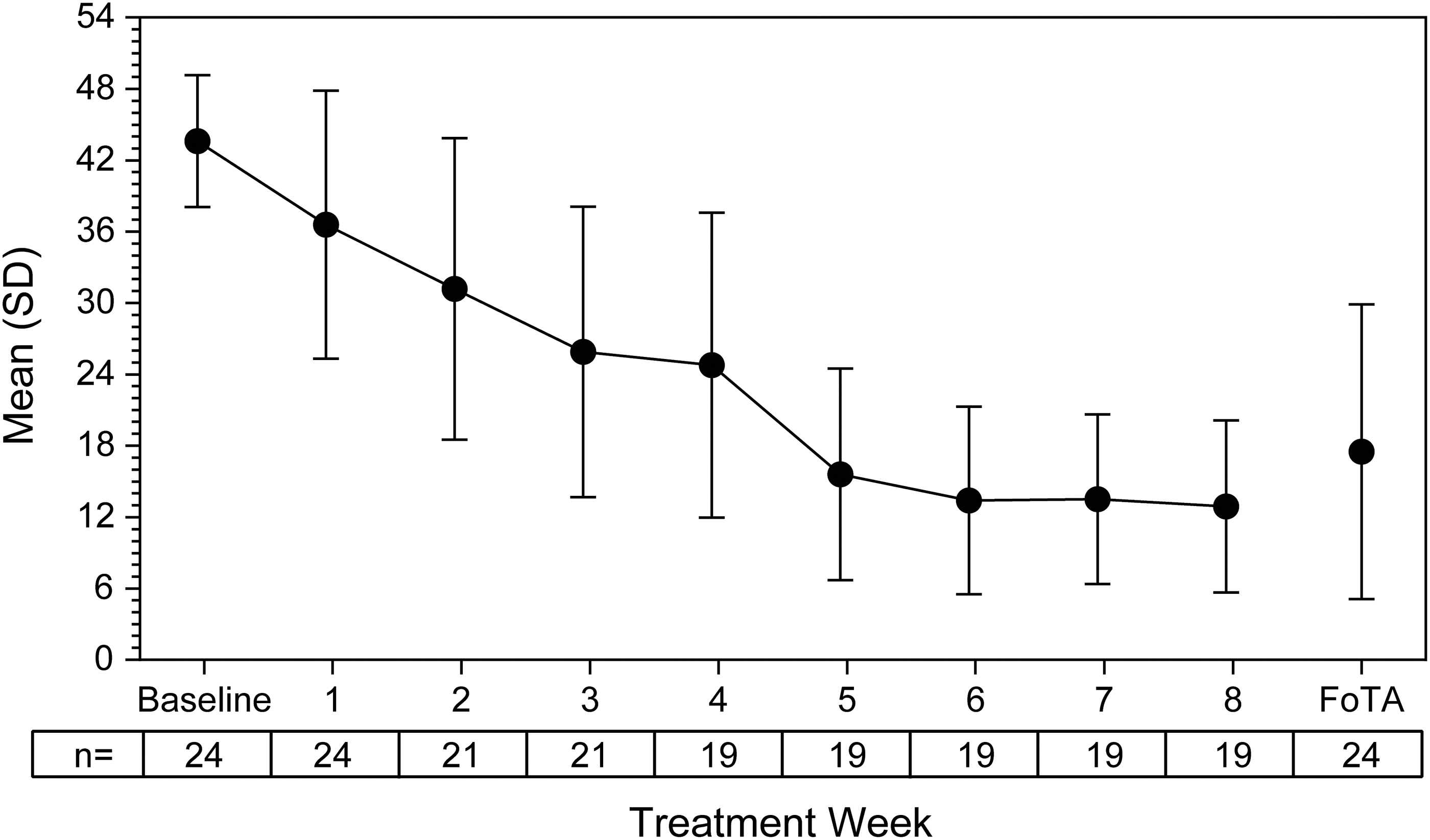
Mean (SD) ADHD-RS-IV-PS total score by treatment week. ADHD, attention-deficit/hyperactivity disorder; ADHD-RS-IV-PS, ADHD Rating Scale-IV Preschool version; FoTA, final on-treatment assessment; SD, standard deviation.
Pharmacokinetics
Plasma concentration-versus-time profiles for LDX and d-amphetamine were consistent across participants (Fig. 3), with LDX concentration peaking at 1 hour postdose and d-amphetamine concentration peaking from 2 to 6 hours postdose. Pharmacokinetic NCA parameters for LDX and d-amphetamine are summarized in Table 3. LDX and d-amphetamine exposure, as measured by C max and AUCtau, was dose proportional. Steady-state LDX clearance (CLss/F) did not change appreciably based on LDX dose, participant weight, or participant age. For d-amphetamine, there was no apparent relationship observed between dose-normalized AUCtau and participant age or weight.
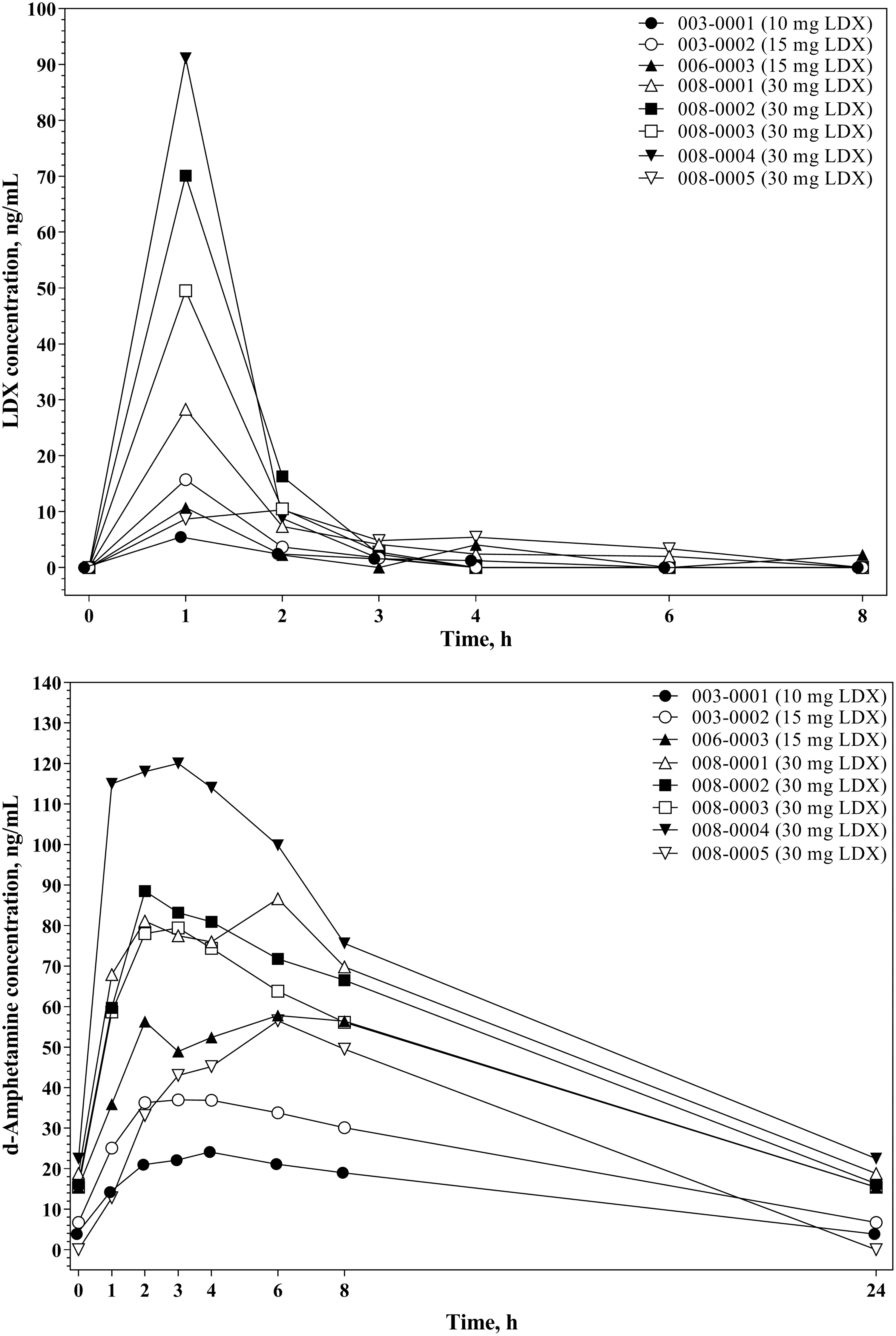
Individual lisdexamfetamine and d-amphetaminea plasma concentrations versus time, full pharmacokinetic subset. aThe reported 24-hour values for d-amphetamine are estimates based on the next predose value (0 hours).
Summary of Pharmacokinetic Parameters by Lisdexamfetamine Dimesylate Dose, Full Pharmacokinetic Set
AUCtau, area under the plasma concentration versus time curve at steady state; AUCtau/dose, dose-normalized AUCtau; CLss/F, total body clearance for extravascular administration divided by the fraction of the absorbed dose at steady state; CLss/F/WT, weight-normalized CLss/F; C max, maximum plasma concentration; LDX, lisdexamfetamine dimesylate; SD, standard deviation; t 1/2, terminal half-life; t max, time to maximum plasma concentration; Vz/F, oral volume of distribution; Vz/F/WT, weight-normalized Vz/F.
Discussion
This study indicates that the safety and tolerability profile of LDX in preschool children with ADHD is generally consistent with its known profile in older children (Biederman et al. 2007b; Findling et al. 2011; Wigal et al. 2012; Coghill et al. 2013); no new or unique AEs were observed. Furthermore, treatment with LDX improved clinical outcome, as measured by reductions in ADHD-RS-IV-PS scores and improvement on the CGI-I, in this population of preschool-aged children.
In the current study, mean ADHD-RS-IV-PS total score at baseline was ∼44, which is substantially higher than the score of 10.85 observed in a normative population of preschool-aged children and places the study population in the 98th percentile for ADHD-RS-IV total scores. Furthermore, 16 of 24 (67%) study participants were considered markedly ill or severely ill based at baseline on CGI-S ratings. This clinical profile is consistent with other study populations of preschool-aged children diagnosed with ADHD (Greenhill et al. 2006; Kratochvil et al. 2007). In Kratochvil et al. (2007), mean ADHD-RS-IV PS total scores were ∼38 at baseline and 19 of 22 (86%) study participants were considered markedly ill or severely ill based on the CGI-S. In the PATS study, rating scale scores also indicated that study participants had severe ADHD symptoms, with mean scores ranging from ∼35 to 40 on the Conners Teacher Rating Scale and Conners Parent Rating Scale at baseline, which represent mean T scores >75 (i.e., >99th percentile) (Posner et al. 2007), and being ∼47 on the Clinical Global Assessment Scale at baseline (Greenhill et al. 2006), which is indicative of a moderate degree of impairment in several areas of function or severe impairment of functioning in one area (Shaffer et al. 1983).
Consistent with other studies of the effects of psychostimulants in preschool-aged children with ADHD (Wigal et al. 2006) and with studies in older children and adolescents with ADHD (Biederman et al. 2007b; Findling et al. 2011, 2013; Wigal et al. 2012; Coghill et al. 2013, 2017; Newcorn et al. 2017), decreased appetite and insomnia were among the most frequently reported TEAEs with LDX in this study. Furthermore, increases from baseline in DBP and decreases from baseline in SBP, pulse, and weight were observed. These findings are partially consistent with findings from the PATS study in preschool children, which reported greater increases from baseline in DBP and SBP with immediate-release MPH relative to placebo but lesser increases from baseline in pulse relative to placebo (Wigal et al. 2006). In contrast, findings in older children and adolescents with ADHD have consistently reported that LDX treatment is associated with increases in pulse (Biederman et al. 2007b; Findling et al. 2011, 2013; Coghill et al. 2013, 2017; Newcorn et al. 2017). These discrepancies may be related to high variability in the vital sign results, which could be related to the small sample size, high baseline vital sign values in a few participants, and the normally high variability of vital sign data. Participants with high baseline vital sign values tended to normalize during the study, resulting in an overall decrease from baseline in pulse and SBP at the FoTA. The initial use of automated blood pressure cuffs, which were not calibrated for this age group (once identified blood pressure measurements were taken manually), did not appear to contribute substantially to vital sign measure variability. No clinically meaningful trends were observed in the ECG results from this study, which is consistent with other studies of LDX in older children and adolescents (Biederman et al. 2007b; Findling et al. 2011, 2013).
LDX treatment was associated with improved clinical effects, as indicated by reductions in ADHD-RS-IV-PS total and subscale scores and improvement on the dichotomized CGI-I. Although these efficacy-related data are limited by the lack of a placebo comparator group, they are consistent with improvements in ADHD-RS-IV total scores observed in open-label safety studies of LDX in older children and adolescents with ADHD (Findling et al. 2013; Coghill et al. 2017), and an open-label study of atomoxetine in 5- and 6-year-old children (Kratochvil et al. 2007). In a long-term open-label safety extension, 52 weeks of dose-optimized LDX (30–70 mg) was associated with a 26-point reduction from baseline ADHD-RS-IV total score and with 87% of participants being categorized as improved on the CGI-I at study endpoint (Findling et al. 2013), which is similar to the 26-point reduction from baseline in ADHD-RS-IV-PS total score and 83% of participants categorized as improved on the CGI-I in the current study. Furthermore, treatment with the nonstimulant atomoxetine was associated with 21-point reduction from baseline on ADHD-RS-IV total score and with 73% of participants being categorized as improved on the CGI-I at study endpoint in a small 8-week open-label pilot study in 5- and 6-year-old children (Kratochvil et al. 2007). Comparisons of the current data with data from the PATS study are complicated by the use of different endpoints (ADHD-RS-IV-PS and CGI-I vs. CLAM and SKAMP), study designs (open label vs. placebo controlled), and medication formulations (once-daily long-acting medication vs. a three-times daily immediate-release medication). However, both studies support the effectiveness of stimulants in the treatment of ADHD in 4- to 5-year-old children.
Results of the pharmacokinetic NCA indicated that the profile of d-amphetamine in this population of preschool-aged children following 30 mg LDX was roughly comparable to previously published findings in older children 6–12 years of age with ADHD (Boellner et al. 2010) in terms of t max (3.41 hours) and t 1/2 (8.90 hours), but was substantially different with regard to C max (53.2 ng/mL in 6- to 12-year-olds vs. 86.20 ng/mL in 4- to 5-year-olds) and AUCtau (745.3 h × ng/mL in 6- to 12-year-olds vs. 1223 h × ng/mL in 4- to 5-year-olds). The increases in d-amphetamine exposure observed in this study compared with the previous study in 6- to 12-year-olds (Boellner et al. 2010) support the use of a starting dose as low as 5 mg LDX in children 4–5 years of age.
These findings should be considered in light of several limitations. First, the sample size was relatively small, so the results should be interpreted cautiously and may not generalize to all preschool-aged children with ADHD. Second, there were numerous investigators involved relative to the small sample size and, in the absence of placebo controls, the study could not account or control for experimenter or reporter bias. Third, there was variability in the vital sign results possibly due to sample size, high baseline vital sign values in a few participants, and the normally high variability of vital sign data. Fourth, the ability to estimate LDX and d-amphetamine pharmacokinetic parameters was limited because of the rapid elimination of LDX with respect to the sampling period used in the study and the relative level of the assay limit of quantitation. Lastly, this was a small, short-term, open-label study, so conclusions regarding the long-term safety and tolerability profile of LDX, the maximum tolerable dose of LDX, and the long-term effects of LDX on clinical outcomes in this population cannot be determined. Future studies, including placebo-controlled studies and long-term safety studies, are required to examine these issues.
Conclusions
In preschool-aged children (4–5 years old), the safety and tolerability of LDX was generally consistent with its known effects in older children and adolescents with ADHD (Biederman et al. 2007a; Findling et al. 2011, 2013; Wigal et al. 2012; Coghill et al. 2013, 2017). Treatment with LDX was also associated with reductions in ADHD-RS-IV-PS scores and improvement on the dichotomized CGI-I. The dosing scheme used in this study was considered to be conservative and was selected for use based on general safety considerations. The study findings indicate that the starting dose of 5 mg, maximum dose of 30 mg, and the titration scheme used was well tolerated and conferred treatment benefits. Taken together, these findings support the conduct of a placebo-controlled study of LDX in 4- to 5-year-old children.
Clinical Significance
Although ADHD is generally diagnosed during elementary school, ADHD symptoms, particularly hyperactivity, are observed in preschool-aged children. Off-label lisdexamfetamine use has been reported in preschool-aged children, but data are limited regarding its safety, efficacy, and pharmacokinetics. We report that in preschool-aged children (4–5 years), lisdexamfetamine has a safety profile consistent with observations in children 6–17 years of age and reduces ADHD symptoms. These pharmacokinetic data support a starting dose of 5 mg lisdexamfetamine in this population.
Footnotes
Acknowledgments
Under the direction of the authors, writing assistance was provided by Madhura Mehta, PhD, Wendy van der Spuy, PhD, and Craig Slawecki, PhD, employees of CHC. Editorial assistance in the form of proofreading, copyediting, and fact checking was also provided by CHC. The authors exercised full control over the content throughout development of the article and had final approval of the article for submission.
Disclosures
A.C.C. has served on advisory boards for Akili, Arbor, Cingulate, Ironshore, Neos, Neurovance, NLS, Noven, Pfizer, Purdue, Rhodes, Supernus, and Tris; has been a consultant for Arbor, Ironshore, Jazz, KemPharm, Neos, Neurovance, Rhodes, Shire, Sunovion, and Supernus; has been a speaker at Arbor, Ironshore, Neos, Pfizer, Shire, and Tris; has received research support from Aevi, Akili, Alcobra, Arbor, Eli Lilly, Forest, Ironshore, KemPharm, Lundbeck, Neos, Neurovance, Noven, Otsuka, Pearson, Pfizer, Purdue, Rhodes, Shire, Sunovion, Supernus, and Tris; and has received writing support from Arbor, Ironshore, Neos, Pfizer, Purdue, Rhodes, Shire, Sunovion, and Tris.
R.L.F. receives or has received research support, acted as a consultant and/or has received honoraria from Acadia, Aevi, Akili, Alcobra, Allergan, Amerex, American Academy of Child & Adolescent Psychiatry, American Psychiatric Press, Arbor, Bracket, Daiichi-Sankyo, Epharma Solutions, Forest, Genentech, Ironshore, KemPharm, Luminopia, Lundbeck, Merck, NIH, Neurim, Noven, Nuvelution, Otsuka, PCORI, Pfizer, Physicians Postgraduate Press, Purinix, Receptor Life Sciences, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, Syneurx, Teva, TouchPoint, Tris, and Validus.
P.M. and Y.W. are employees of Shire, a member of the Takeda group of companies, and hold Takeda stock.
J.W. was an employee of Shire, a member of the Takeda group of companies, at the time this research was conducted and holds Takeda stock; he is currently employed by Ironwood Pharmaceuticals (Boston, MA).
B.R. was an employee of Shire, a member of the Takeda group of companies, at the time this research was conducted and holds Takeda stock; she is currently employed by Yumanity Therapeutics, Inc. (Cambridge, MA).
S.H.K. has received research support and/or consulting fees from Akili Interactive, Bose, Jazz, KemPharm, Medgenics, Neos, Otsuka, Rhodes, Shire, and Sunovion.
The data reported in this report were analyzed by J.W., PhD.