
Abstract
Objective:
Evening-dosed HLD200 is a delayed-release and extended-release methylphenidate (DR/ER-MPH) formulation consisting of uniform, dual-layered microbeads with an inner drug-loaded core. DR/ER-MPH is designed to delay the initial release of drug by 8–10 hours, and thereafter, provide a controlled, extended drug release to target onset of effect upon awakening that lasts into the evening. This phase 3 study evaluated the safety and efficacy of DR/ER-MPH on symptoms and temporal at-home functional impairment in children with attention-deficit/hyperactivity disorder (ADHD).
Methods:
This 3-week, randomized, double-blind, multicenter, placebo-controlled, parallel-group, forced-dose titration trial evaluated DR/ER-MPH (40–80 mg/day) in children aged 6–12 years with ADHD. Primary efficacy endpoint was the ADHD rating scale–IV (ADHD-RS-IV), and the key secondary endpoints were the Before-School Functioning Questionnaire (BSFQ), and Parent Rating of Evening and Morning Behavior-Revised, morning (PREMB-R AM) and evening (PREMB-R PM). Safety measures included spontaneously reported treatment-emergent adverse events (TEAEs) and two TEAEs of special interest, appetite suppression and insomnia (with direct questioning on sleep disturbance).
Results:
One hundred sixty-one participants were included in the intent-to-treat population (DR/ER-MPH, n = 81; placebo, n = 80). After 3 weeks, DR/ER-MPH achieved significant improvements versus placebo in ADHD symptoms (least-squares [LS] mean ADHD-RS-IV: 24.1 vs. 31.2; p = 0.002), and at-home early morning (LS mean BSFQ: 18.7 vs. 28.4; p < 0.001; LS mean PREMB-R AM: 2.1 vs. 3.6; p < 0.001) and late afternoon/evening (LS mean PREMB-R PM: 9.4 vs. 12.2; p = 0.002) functional impairment. Commonly reported TEAEs (≥10%) were insomnia and decreased appetite.
Conclusions:
DR/ER-MPH was generally well tolerated and demonstrated significant improvements versus placebo in ADHD symptoms and at-home functional impairments in the early morning, late afternoon, and evening in children with ADHD.
Introduction
A
Currently available extended-release (ER) MPH formulations are administered once daily in the morning before school and have been reported to control symptoms of ADHD in children for up to 12 hours (Childress 2016). Unfortunately, no formulation provides all-day coverage of symptoms, and, because of constraints in the various technologies used to deliver MPH, there is often a delay in the initial onset of action of up to 2 hours (Childress 2016). This leaves the patient vulnerable to inadequate symptom control and impaired functioning during the early morning routine (Childress and Tran 2016; Sallee 2015).
Mornings have been identified as a particularly challenging time of day for school-age children with ADHD and their families (Whalen et al. 2006; Sallee 2015; Faraone et al. 2017). In one study using electronic diaries, mothers reported that their own and their family's activities were significantly restricted by their stimulant-treated child with ADHD; in addition, they had a lower quality of life and less adequate parenting effectiveness (Whalen et al. 2006).
A recent survey of 201 primary caregivers of youth with ADHD revealed that, despite routine morning administration of stimulant medication, ∼75.6% of caregivers regarded the early morning as a time associated with moderate-to-severe ADHD-related functional impairment in their children (Sallee 2015). These findings were corroborated by a subsequent survey of 350 primary caregivers of youth with and without ADHD, which also found that early morning functional (EMF) impairment in stimulant-treated youth with ADHD significantly diminished the emotional well-being of caregivers and exerted a pervasive functional burden on the entire family unit (Faraone et al. 2017). These findings suggest that inadequately controlled early morning ADHD symptoms and EMF impairment remain significant burdens on stimulant-treated youth with ADHD and their families.
Despite the documented importance of EMF impairment (Whalen et al. 2006; Sallee 2015; Faraone et al. 2017), clinical trials of stimulant-treated children with ADHD have primarily focused on improving symptoms during the school day and after-school homework time (Faraone and Buitelaar 2010; Coghill et al. 2013; Maldonado 2013; Storebø et al. 2015). To date, only one randomized, placebo-controlled trial has assessed the efficacy of a long-acting stimulant monotherapy on at-home EMF impairment. In that 4-week crossover study of 30 children with ADHD, very early morning administration of an MPH transdermal patch significantly reduced EMF impairment compared with placebo, as measured by the investigator-rated Before-School Functioning Questionnaire (BSFQ) (Wilens et al. 2010). However, very early administration is inconvenient if done before normal awakening time, and is only possible with a patch formulation in which the patient does not have to be awake. In addition, early morning administration may potentially compromise ADHD coverage in the late afternoon/early evening (Wilens et al. 2010).
Evening-dosed HLD200 is a delayed-release (DR) and ER formulation of MPH (DR/ER-MPH) specifically designed to address the unmet need for a once-daily ADHD medication that provides efficacy upon awakening, but not at the expense of efficacy later in the day. DR/ER-MPH capsules utilize the DELEXIS® drug delivery platform containing uniform microbeads that have two functional layers surrounding an inner MPH-loaded core. The outer DR layer is composed of hydrophobic, hygroscopic, and pH-dependent polymers designed to provide a prolonged, predictable delay in the timing of initial MPH release, which is intended to offer a therapeutic effect upon awakening. The inner ER layer is composed of hydrophobic and soluble polymers designed to provide a controlled, sustained release of MPH with the goal of maintaining efficacy throughout the day and lasting into the evening. Based on the properties of the DR and ER layers, the initial dissolution and subsequent absorption of MPH are not dependent on any single factor, such as a pH trigger, normal variations in gastrointestinal transit, or site of release.
Indeed, the pharmacokinetic profile of evening-dosed DR/ER-MPH shows a consistent, predictable delay in the initial release of MPH until the early morning (i.e., ∼8–10 hours after ingestion), followed by a period of extended, controlled release across the day in healthy adults, and in children and adolescents with ADHD (Childress et al. 2015).
The objectives of this pivotal phase 3 trial were to (1) assess whether DR/ER-MPH improves control of ADHD symptoms throughout the day and reduces at-home functional impairments in the early morning, late afternoon, and evening versus placebo in children with ADHD and (2) evaluate the short-term safety and tolerability of DR/ER-MPH.
Methods
Study conduct
This randomized, double-blind, multicenter, placebo-controlled, parallel-group, forced-dose titration, phase 3 trial of DR/ER-MPH in children aged 6–12 years with ADHD was conducted at 22 sites in the United States, in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All participants and parents/legal guardians provided informed assent and consent, respectively, under procedures approved by each site's institutional review board.
Inclusion/exclusion criteria
Primary diagnosis of ADHD, according to Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) criteria (American Psychiatric Association 2013), was required and confirmed using the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID) (Sheehan et al. 2010). Other inclusion criteria included, but were not limited to, ADHD Rating Scale-IV (ADHD-RS-IV) score ≥90th percentile for age and gender and ≥26 at baseline (DuPaul et al. 1998); Clinical Global Impressions of Severity (CGI-S) score ≥4 at baseline (Guy 1976); Conners' Global Index-Parent (CGI-P) score >10 at baseline (Conners 1989); at least a partial clinical response to MPH as judged by the site investigator; EMF impairment and/or difficulties performing a morning routine of ≥30 minutes in duration occurring between 6:00 and 9:00
Exclusion criteria included, but were not limited to, a history of or current medical condition or laboratory result that could either jeopardize participant safety or interfere with study participation; history of psychosis, bipolar disorder, anorexia nervosa, bulimia, or suicide attempt; current depression, anxiety, conduct disorder, substance use disorder, or other significant psychiatric conditions; history of severe allergic reaction or intolerance to MPH; and use of medication for psychiatric conditions other than ADHD, including antidepressants, mood stabilizers, and antipsychotics.
Study design
The trial was conducted in two phases: a screening/washout phase of up to 2 weeks and a 3-week randomized, placebo-controlled test phase (Fig. 1). During the screening/washout phase, eligible participants were withdrawn from their current ADHD medication. Stimulants, clonidine, and guanfacine required a ≥72-hour washout, and any other medication used to treat ADHD required a ≥7-day washout before randomization. At the start of the treatment phase (Visit 2), participants were randomized in a 1:1 ratio to receive either DR/ER-MPH or placebo once daily each evening for 3 weeks.
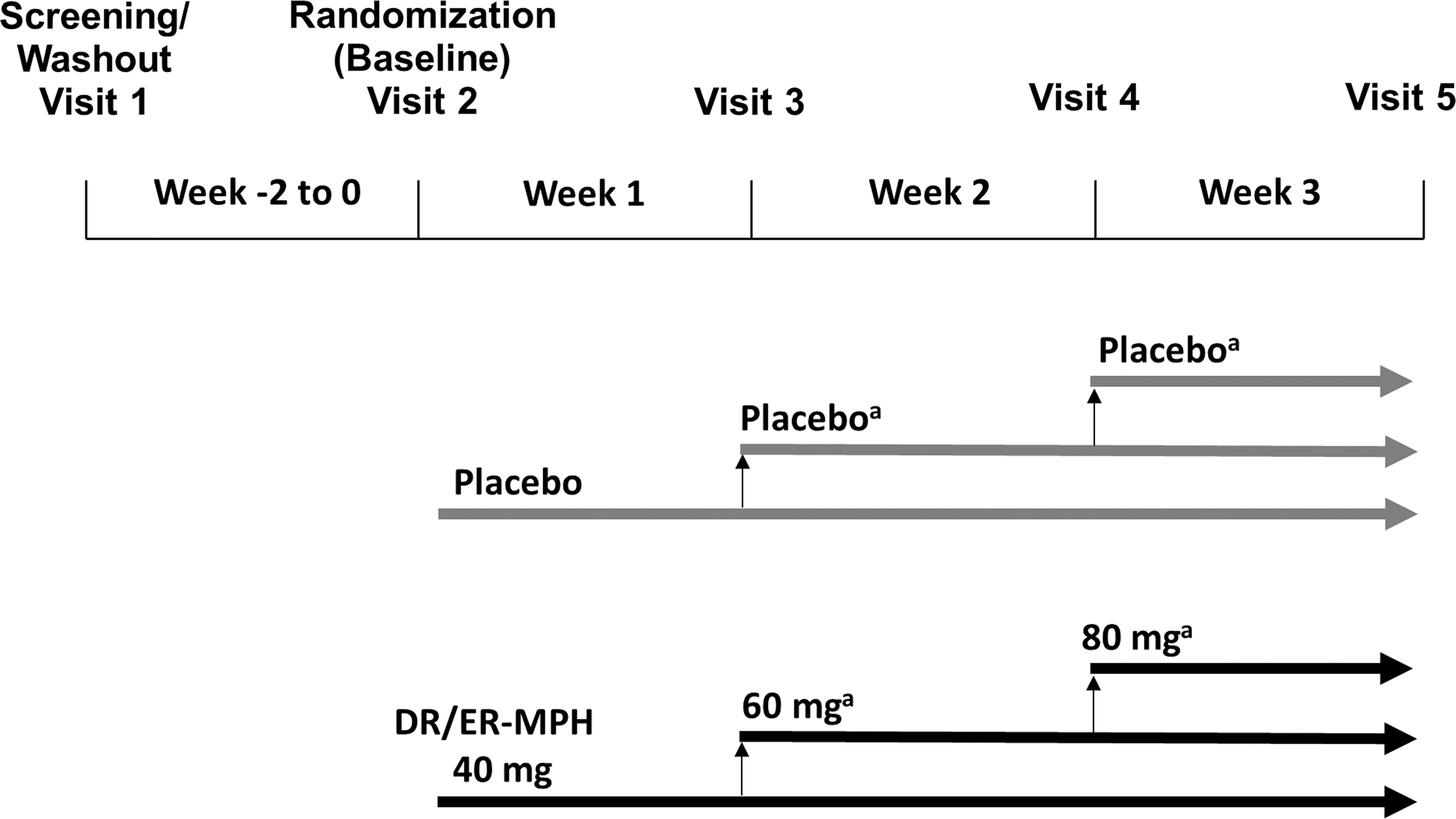
Study design. aOne down-titration to the previous dose strength was permitted during the 3-week randomized test phase, if necessary, for safety or tolerability. DR/ER-MPH, delayed-release and extended-release methylphenidate.
Dosing was initiated at 40 mg/day each evening at 8:00
Efficacy
The primary efficacy endpoint was the mean ADHD-RS-IV total score throughout the day after 3 weeks of treatment. The ADHD-RS-IV evaluates the severity of current ADHD symptoms and consists of 18 items assessing the DSM-IV criteria for ADHD (DuPaul et al. 1998). Each item is scored from 0 (never/rarely) to 3 (very often), with total scores ranging from 0 to 54 and a higher score indicating greater symptom severity. Clinicians administered the ADHD-RS-IV at baseline and at each weekly study visit. Ratings on the ADHD-RS-IV from 6:00 to 9:00
The key secondary efficacy endpoints were the BSFQ total score and the Parent Rating of Evening and Morning Behavior-Revised, morning (PREMB-R AM) and evening (PREMB-R PM) subscales after 3 weeks of treatment. The BSFQ is a validated instrument evaluating EMF impairment in children with ADHD between the time of awakening and before the school day or other morning activities begin (i.e., 6:00 and 9:00
The PREMB-R is a validated, 11-item clinician-rated scale based on a parent interview that assesses at-home functioning (i.e., behaviors that impact activities of daily living, such as getting up and out of bed, doing or completing homework, and falling asleep) during the early morning (PREMB-R AM) and late afternoon/evening (PREMB-R PM) in children with ADHD (Sutton et al. 2003; Faraone et al. 2015b). Each item is rated from 0 (no difficulty) to 3 (a lot of difficulty), with the three-item PREMB-R AM having a maximum score of 9, and the eight-item PREMB-R PM having a maximum score of 24. PREMB-R-reported results were clinician-rated scores derived from structured parent interviews at baseline and at last visit. Parents were instructed to base their responses on the last 2 weekdays before their child's study visit.
Safety
Safety endpoints included spontaneously reported treatment-emergent adverse events (TEAEs); TEAEs of special interest, specifically appetite suppression and insomnia, with sleep disturbances (onset, quality, and quantity) directly queried from participants and parents at each visit; vital signs; electrocardiograms; clinical laboratory tests; physical examination findings; and the Columbia-Suicide Severity Rating Scale for Children.
Statistical analyses
It was estimated that a minimum of 70 participants per treatment arm were required for 90% power at the statistical significance level of p < 0.05, using a two-sided, two-sample t-test. Assuming a dropout rate of 20% to 25%, an estimated sample size of 180 participants was required to randomize at least 140 participants. Analyses were conducted on the intent-to-treat (ITT) and safety populations. The ITT population was defined as all randomized participants who received at least one dose of study drug and had at least one postbaseline evaluation of the primary efficacy assessment; the ITT population was the basis for the primary and secondary efficacy analyses. The safety population was defined as all enrolled participants who received at least one dose of study drug and had at least one postbaseline safety assessment.
Descriptive statistics were presented by visit and treatment group based on the ITT population. The ADHD-RS-IV was analyzed by using a mixed-model repeated measures (MMRM) analysis that included the participant's intercept as a random effect; treatment, study center, and visit-by-treatment interaction as fixed effects; and baseline ADHD-RS-IV total score at Visit 2 as a covariate. The BSFQ and ADHD-AM-RS were also analyzed using the MMRM, as described for the primary efficacy analysis. Treatment differences between DR/ER-MPH and placebo at all postbaseline visits were estimated by using least-squares (LS) means from the MMRM, and treatment comparisons were conducted as a two-sided test at the significance level of p < 0.05. PREMB-R AM and PREMB-R PM were assessed by using an analysis of covariance model with treatment as the main effect, and study center and baseline score at Visit 2 as the covariates.
Results
Participant disposition, demographics, and baseline characteristics
Of the 163 children enrolled, 82 participants were randomized to active treatment with DR/ER-MPH and 81 to placebo (Fig. 2). A total of 161 participants were included in the safety and ITT populations (DR/ER-MPH, n = 81; placebo, n = 80). Seventy-three participants (89%) randomized to DR/ER-MPH and 65 participants (80%) randomized to placebo completed the study.
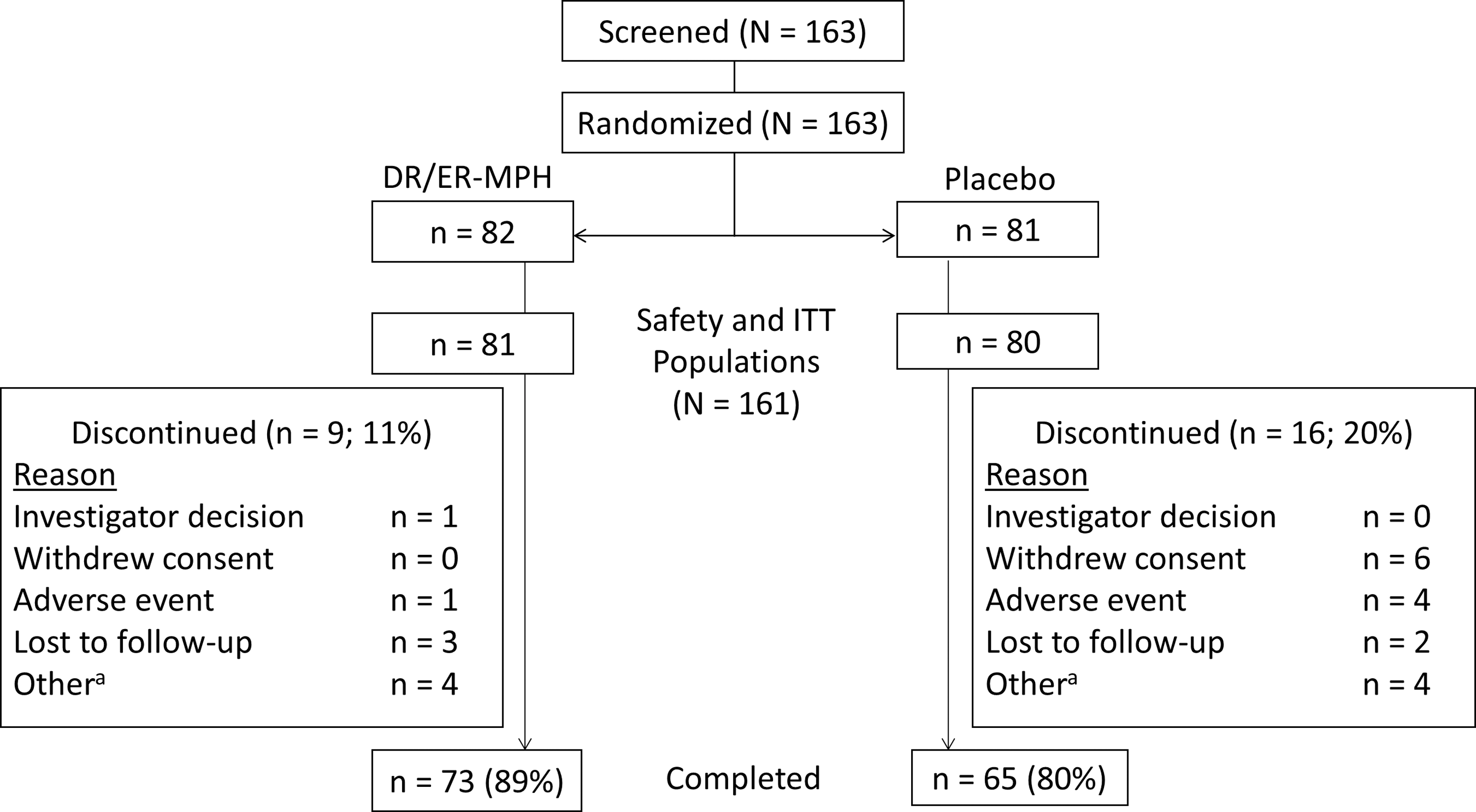
Participant disposition. aReasons for discontinuation consisted of the following: noncompliance with study visits (DR/ER-MPH: n = 1); participant off study drug for 5 days before Visit 5 (DR/ER-MPH: n = 1); Visit 5 out of window and parent discontinued dosing except for the night before Visit 5 (DR/ER-MPH: n = 1); error in the distribution of investigational product (DR/ER-MPH: n = 1); lack of efficacy (placebo: n = 2); participant unable to complete Visit 5 within visit window (placebo: n = 1); noncompliance and lost to follow-up (placebo: n = 1). DR/ER-MPH, delayed-release and extended-release methylphenidate; ITT, intent-to-treat.
The demographic and baseline characteristics of the ITT population are presented in Table 1. Most of these variables were comparable between treatment groups. At baseline, the mean ADHD-RS-IV total score (±SD) was 43.3 ± 7.1 and the majority (65.8%) of participants were markedly ill (CGI-S of 5).
ADHD-RS-IV, Attention-Deficit/Hyperactivity Disorder Rating Scale-IV; CGI-P, Conners' Global Index-Parent; CGI-S, Clinical Global Impressions of Severity; DR/ER-MPH, delayed-release and extended-release methylphenidate; SD, standard deviation.
Primary efficacy: ADHD-RS-IV total score at week 3
The mean DR/ER-MPH dose achieved after 3 weeks of treatment was 68.1 mg, and the most commonly prescribed dosing time (84%) was 8:00
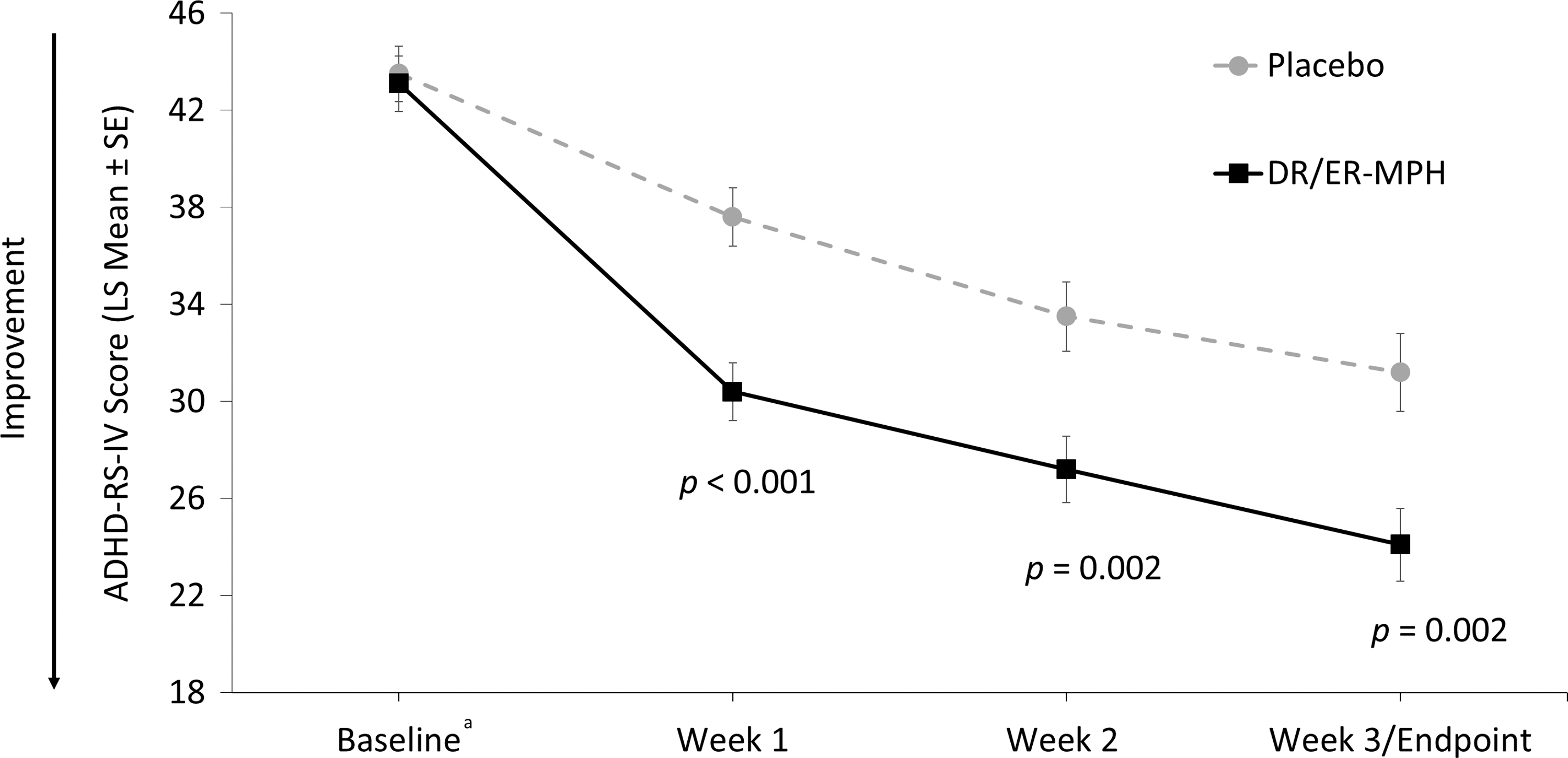
ADHD-RS-IV total scores. The primary efficacy endpoint was the ADHD-RS-IV total score at final visit (week 3). aBaseline scores are represented as arithmetic mean scores with error bars denoting SE for comparison purposes. Mean ADHD-RS-IV scores (±SD) at baseline were 43.1 ± 7.3 for DR/ER-MPH and 43.5 ± 6.8 for placebo. ADHD-RS-IV, Attention-Deficit/Hyperactivity Disorder Rating Scale-IV; DR/ER-MPH, delayed-release and extended-release methylphenidate; LS, least-squares; SD, standard deviation; SE, standard error of the mean.
Key secondary outcomes: BSFQ, PREMB-R AM, and PREMB-R PM at week 3
After 3 weeks of treatment, children treated with DR/ER-MPH versus placebo had a significant improvement in EMF impairment, as measured by BSFQ (LS means [SE]: 18.7 [1.63] vs. 28.4 [1.73]; p < 0.001; Fig. 4). Significant reductions in EMF impairment were also apparent after 1 (p = 0.001) and 2 (p = 0.004) weeks of treatment with DR/ER-MPH (Fig. 4). DR/ER-MPH also demonstrated statistically significant improvements in at-home EMF and late afternoon/evening functional impairment versus placebo after 3 weeks, as measured by the PREMB-R AM (LS mean [SE]: 2.1 [0.26] vs. 3.6 [0.27]; p < 0.001) and PREMB-R PM (LS mean [SE]: 9.4 [0.64] vs. 12.2 [0.66]; p = 0.002), respectively (Fig. 5).
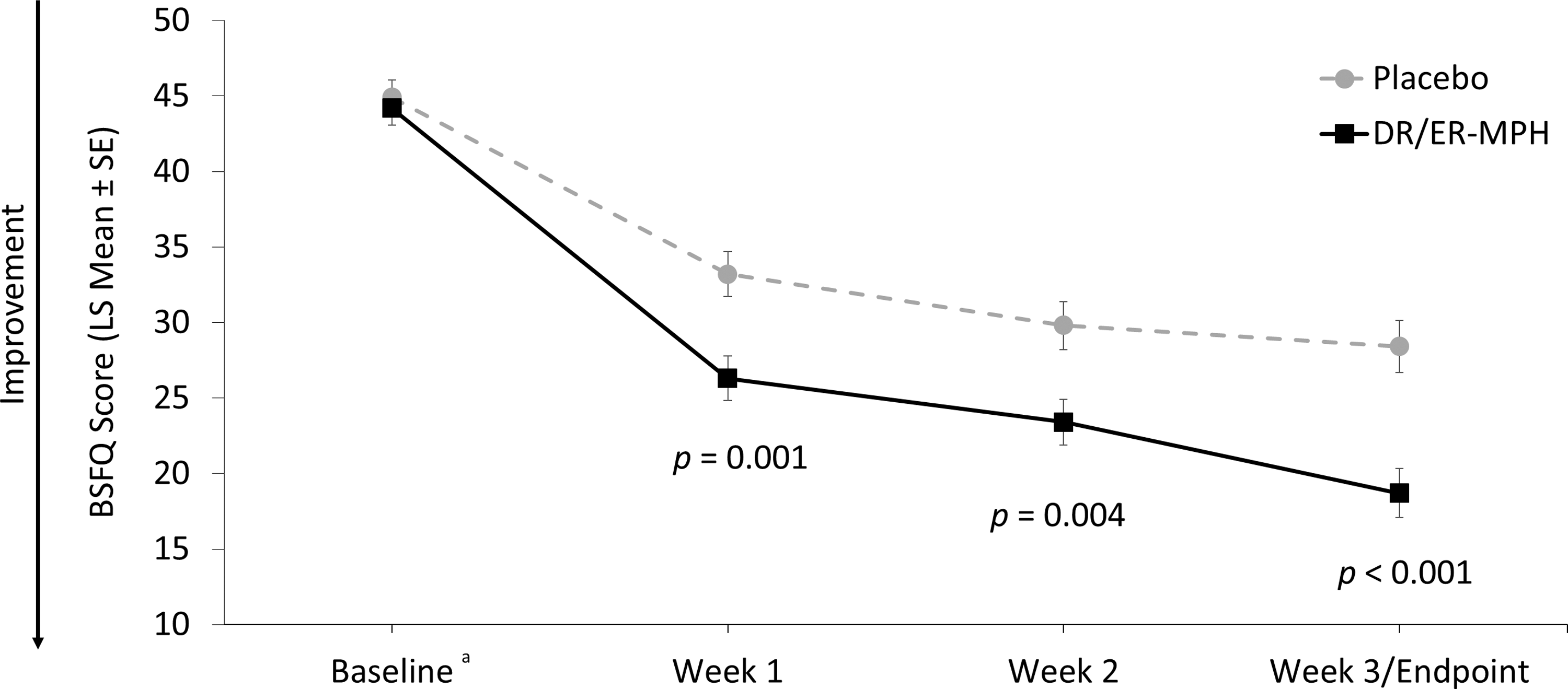
BSFQ total scores by week. The key secondary endpoint was the total BSFQ score at the final visit (week 3). aBaseline scores are represented as arithmetic mean scores with error bars denoting SE for comparison purposes. Mean BSFQ scores (±SD) at baseline were 44.2 ± 10.28 for DR/ER-MPH and 44.9 ± 10.20 for placebo. BSFQ, Before-School Functioning Questionnaire; DR/ER-MPH, delayed-release and extended-release methylphenidate; LS, least-squares; SD, standard deviation; SE, standard error of the mean.
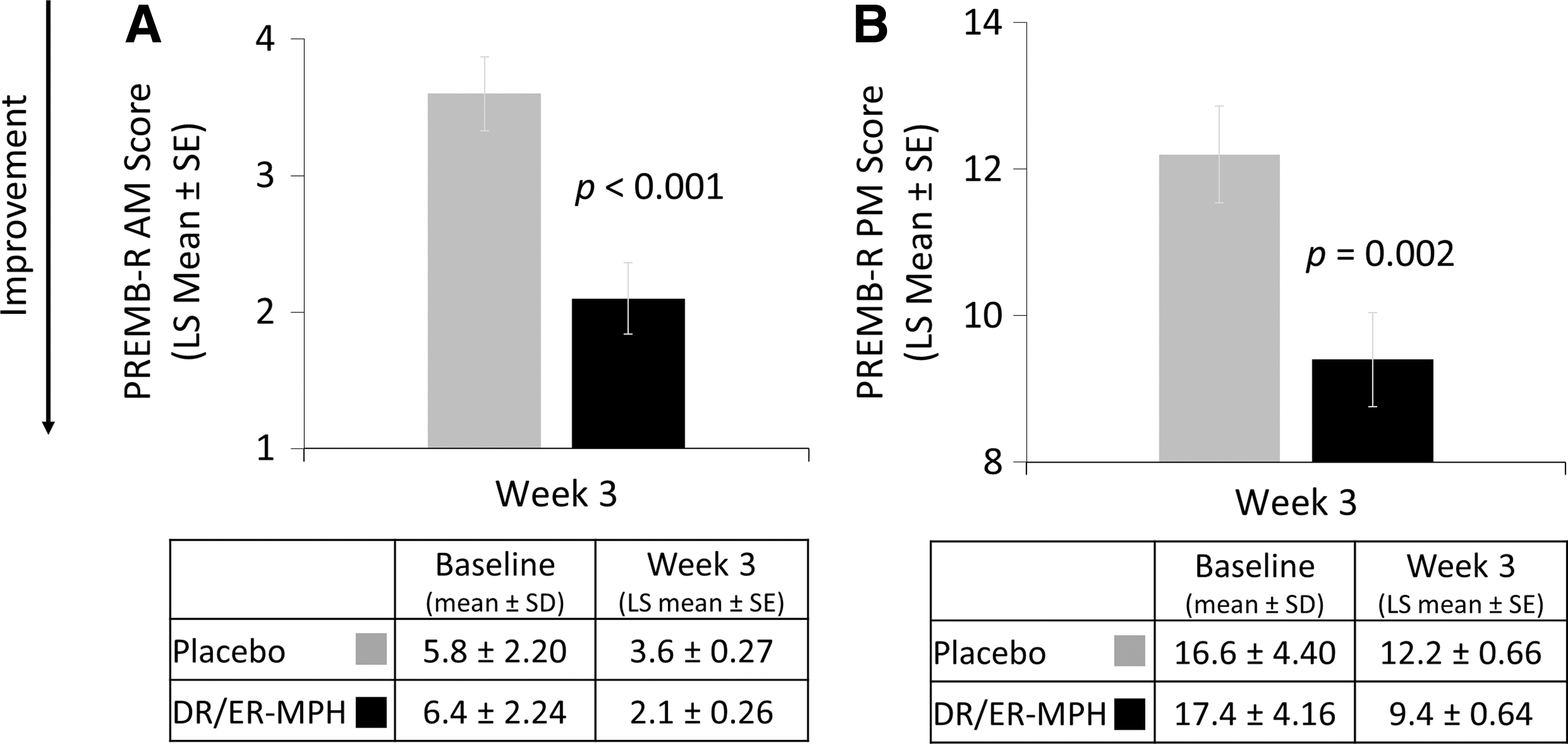
PREMB-R AM and PREMB-R PM scores at week 3. DR/ER-MPH, delayed-release and extended-release methylphenidate; LS, least squares; PREMB-R AM, Parent Rating of Evening and Morning Behavior-Revised, morning subscale; PREMB-R PM, Parent Rating of Evening and Morning Behavior-Revised, evening subscale; SD, standard deviation; SE, standard error of the mean.
Safety
The number of participants reporting any TEAE was 56 (69.1%) on DR/ER-MPH and 39 (48.8%) on placebo. There were no serious TEAEs reported, and one participant on DR/ER-MPH (1.2%; mood swings) and four receiving placebo (5.0%) had TEAEs leading to early study withdrawal. Reasons for premature withdrawal in the placebo-treated group were irritability in two participants; flatulence, regurgitation, and enuresis in one participant; and dizziness, somnolence, tremor, and sleep disorder in one participant. The most common TEAEs (≥10%) reported by children on DR/ER-MPH were insomnia and decreased appetite (Table 2).
Preferred term is based on the Medical Dictionary for Regulatory Activities, Version 18.0 coding dictionary. Participants could have reported >1 adverse event.
Deemed to be related to the study medication by the study investigator.
DR/ER-MPH, delayed-release, and extended-release methylphenidate; SD, standard deviation.
All sleep-related TEAEs were either mild or moderate in severity, with the majority reported as mild (DR/ER-MPH: 82.8%; placebo: 72.7%). In the DR/ER-MPH group, 96.6% of sleep-related AEs were transient and resolved during the course of the study, and no action was taken to reduce the dose in 89.7% of cases. One participant on placebo experienced a sleep-related event that led to discontinuation of the study drug, and four participants on DR/ER-MPH experienced sleep-related events that resulted in a decrease in the dose. All appetite-related TEAEs were mild or moderate in severity, and all but two resolved over the course of the study (i.e., decreased appetite in one participant on placebo and decreased weight in one participant on DR/ER-MPH).
Vital sign changes were consistent with those expected for MPH products (Table 2). After 3 weeks of treatment, mean changes in heart rate and systolic blood pressure from baseline were comparable between treatment groups. The DR/ER-MPH group had a small increase in mean diastolic blood pressure from baseline to week 3, whereas the placebo group had little change.
Discussion
The results of this trial demonstrated that 3 weeks of treatment with evening-dosed DR/ER-MPH is more effective than placebo in improving ADHD symptoms and at-home functional impairments from early morning to evening in children with ADHD. Improvements in both ADHD symptoms and EMF impairment were evident with DR/ER-MPH as early as 1 week of treatment. Evening-dosed DR/ER-MPH was generally well tolerated and demonstrated a safety profile consistent with previously reported studies of MPH (Childress 2016).
In addition to the positive outcome on the primary measure of symptoms (ADHD-RS-IV), the most noteworthy findings of this trial are represented by the positive outcomes on the key secondary measures of functional impairment. Three weeks of treatment with DR/ER-MPH achieved significant reductions in at-home EMF impairment (BSFQ and PREMB-R AM), and these findings were paralleled by reductions in early morning ADHD symptoms, as measured by ADHD-AM-RS. DR/ER-MPH was also associated with a significant reduction in late afternoon/evening functional impairment (PREMB-R PM). Given the specific items that comprise the BSFQ and PREMB-R AM/PM (e.g., “getting up and out of bed” and “falling asleep”), these findings indicate that DR/ER-MPH reduced at-home functional impairment from awakening until bedtime in children with ADHD. To our knowledge, this is the first trial to show improvements in at-home EMF impairment without compromising at-home functioning in the late afternoon/evening with a single dose of a long-acting MPH.
This study emphasized the importance of treatment on functional status as well as symptom severity, and indirectly acknowledges the long-held emphasis on functioning in the DSM diagnostic criteria for ADHD (American Psychiatric Association 2013). Although ADHD symptoms and functional impairment are interrelated, previous studies have shown that improvements in symptoms do not necessarily translate into improvements in functioning (Sasser et al. 2017). Furthermore, functional impairment (i.e., the consequences of symptoms) (Barkley et al. 2006) tend to be the primary reason for referral, and may be a better predictor of long-term outcomes than ADHD symptoms alone (Karpenko et al. 2009; Sasser et al. 2017). Also, the effect of treatment on functional status is critical for parents of youth with ADHD, as EMF impairment creates significant challenges not only for their child but also for the entire family unit (Sallee 2015; Faraone et al. 2017). Therefore, parents of children with ADHD may not consider a treatment to be meaningful unless their child's functioning has improved (Karpenko et al. 2009).
Despite the documented importance of functional impairment, only recently has there been a shift toward defining optimal clinical outcomes for youth with ADHD that go beyond simple symptom reduction to include novel approaches that reliably and validly assess ADHD-related functioning (Rostain et al. 2015; Sasser et al. 2017). The psychometric properties of the BSFQ and PREMB-R AM and PREMB-R PM subscales were recently validated to provide measures of temporal functional impairment for clinical trials in children with ADHD (Faraone et al. 2015a, 2015b). By using a combination of these temporal functional measures, this study provides a unique demonstration of the beneficial effects of a long-acting MPH formulation on both symptoms and at-home temporal functioning.
Evening-dosed DR/ER-MPH was well tolerated at doses ranging from 40 to 80 mg/day. The most common TEAEs reported by children on DR/ER-MPH were insomnia and decreased appetite. Given that this study utilized a forced-dose titration design and sleep was directly queried from participants and their caregivers rather than obtained from spontaneous reports, a higher occurrence of these TEAEs was expected. Nonetheless, 89% of participants treated with DR/ER-MPH completed the study, no serious TEAEs were observed, and only one DR/ER-MPH-treated participant discontinued the study drug because of a TEAE. All TEAEs and vital sign changes associated with DR/ER-MPH were characteristic of those previously reported with MPH formulations (Coghill et al. 2013; Childress 2016).
The positive findings of this study should be considered in light of the several limitations. The short duration of the study limits the ability to extrapolate the findings over the long term. The study included school-age children (aged 6–12 years) only and, therefore, the applicability of these findings to other age groups (i.e., preschool children, adolescents, and adults) is unknown. Lastly, enrolled participants have previously shown at least a partial response to MPH, and, therefore, the response and safety profiles in MPH-naïve patients may be different than those achieved in this study.
Conclusions
After daily evening administration, DR/ER-MPH was generally well tolerated and demonstrated significant improvements in not only ADHD symptoms but also at-home early morning and late afternoon/evening impaired functioning compared with placebo in children with ADHD. The safety profile of DR/ER-MPH is consistent with those of other MPH formulations, with appetite suppression and insomnia being the most commonly reported adverse events. To our knowledge, this is the first study to demonstrate significant improvements in impaired at-home functioning from the early morning until evening with a single dose of a long-acting stimulant in children with ADHD.
Clinical Significance
Despite the availability of multiple long-acting stimulants, there remains a significant unmet need in the treatment of ADHD to provide control of EMF impairment from inadequately controlled ADHD symptoms. In addition to demonstrating ADHD symptom control, DR/ER-MPH is the first long-acting MPH formulation to show improvements in at-home EMF impairment without compromising at-home functioning in the late afternoon and evening with a single evening dose. For clinicians, patients, and their families, evening-dosed DR/ER-MPH represents a shift in the approach to the timing of MPH delivery and provides a new treatment option specifically designed to address the unmet need for a once-daily ADHD medication that provides efficacy upon awakening, but not at the expense of efficacy later in the day.
Footnotes
Acknowledgments
Under the direction of the authors, Amy Horton Williams, PharmD, of MedLearning, Inc., provided medical writing assistance for this article, funding for which was provided by Ironshore Pharmaceuticals & Development, Inc. Marina Komolova, PhD, Stacey Curtiss, PharmD, and Rick Nullmeier from Ironshore Pharmaceuticals & Development, Inc. also reviewed and edited the article for scientific accuracy. Clinical trial registration identification number: NCT02520388.
Disclosures
S.R.P. is a consultant for and has received research support from Ironshore Pharmaceuticals & Development, Inc., and has served as an expert witness for AstraZeneca. T.E.W. received grant support from NIH (NIDA); is a consultant for Alcobra, Ironshore Pharmaceuticals & Development, Inc., NIH (NIDA), Phoenix House and Bay Cove Human Services (Clinical Services), Neurovance/Otsuka, Sunovion, Tris, U.S. National Football League (ERM Associates), and U.S. Minor/Major League Baseball; has published a book, Straight Talk About Psychiatric Medications for Kids (Guilford Press); coedited books, ADHD in Children and Adults (Cambridge Press), Massachusetts General Hospital Comprehensive Clinical Psychiatry (Elsevier), and Massachusetts General Hospital Psychopharmacology and Neurotherapeutics (Elsevier); and is the co-owner of the Before School Functioning Questionnaire and has a licensing agreement with Ironshore Pharmaceuticals & Development, Inc. S.B. has participated in advisory boards for Curemark and Tris Pharma; and has received research support from Arbor Pharmaceuticals, Lundbeck, Neurim Pharmaceuticals, Pearson, Rhodes, Shire, Sunovion, and Supernus. V.K.A. has participated in advisory boards for Ironshore Pharmaceuticals & Development, Inc., Neos, Rho, and Shire; is a consultant for Ironshore Pharmaceuticals & Development, Inc.; and participates in speakers bureau for Lundbeck/Takeda. A.M. is a consultant for Ironshore Pharmaceuticals & Development, Inc. A.J.C. is a consultant and has received research support from Aevi Genomics, Akili Interactive, Alkermes, Allergan, Arbor Pharmaceuticals, Ironshore Pharmaceuticals & Development, Inc., Janssen, Kempharm, Lilly, Lundbeck, Neos Therapeutics, Neurovance, Novartis, Noven, Purdue, Rhodes Pharmaceuticals, Shire, Sunovion, Supernus, and Takeda; and is a speaker for Alkermes, Allergan, Arbor Pharmaceuticals, Janssen, Lilly, Lundbeck, Novartis, Shire, Sunovion, and Takeda. F.A.L. is a consultant or has participated in advisory boards for Ironshore Pharmaceuticals & Development, Inc., Neos, Novartis, Noven, Rhodes, Shionogi, Inc., Shire Canada, Shire Global, Shire U.S., Supernus, and Tris Pharma; and has received research support from Bristol-Myers Squibb, Cephalon, Ironshore Pharmaceuticals & Development, Inc., New River Pharmaceuticals, Novartis, Noven, Shire U.S., Pfizer, and Celltech Medeva. N.J.D. and B.I. are employees of Ironshore Pharmaceuticals & Development, Inc. F.R.S. is an employee of Ironshore Pharmaceuticals & Development, Inc.; and is a member of the Board of Directors for P2D Bioscience. J.H.N. is advisor/consultant for Akili Interactive, Alcobra, Arbor Pharmaceuticals, Cerecor, Enzymotec, Ironshore Pharmaceuticals & Development, Inc., KemPharm, Lundbeck, Medice, Neos, U.S. National Football League, NLS, Pearson, Rhodes, Shire, Sunovion, and Supernus; is a DSMB member of Sunovion; has received research grants from Enzymotec, Lundbeck, and Shire; and has received speaking honoraria (nonpromotional) from Shire and Teva.