
Abstract
Objective:
The purpose of this study was to evaluate the safety and efficacy of asenapine in adolescents with schizophrenia.
Methods:
In an 8 week, randomized, double-blind placebo-controlled trial, subjects (12–17 years of age) meeting Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) criteria for schizophrenia were randomized 1:1:1 to placebo, asenapine 2.5 mg b.i.d., or asenapine 5 mg b.i.d. Subjects who completed the 8 week acute study could participate in a 26 week flexible-dose asenapine-only open-label extension (OLE).
Results:
A similar percentage of subjects completed treatment on day 56 (2.5 mg b.i.d. (n=98): 83%; 5 mg b.i.d. [n=106]: 79%; placebo [n=102]: 79%). In the mixed model for repeated measures analysis of the primary end-point (with Hochberg correction for multiplicity), least squares (LS) mean differences between asenapine and placebo on the Positive and Negative Syndrome Scale (PANSS) total score at day 56 were not significant (−4.8 for 2.5 mg b.i.d., p=0.070; −5.6 for 5 mg b.i.d., p=0.064). Significant improvement in the Clinical Global Impressions-Severity score was observed in the 5 mg b.i.d. group versus placebo on day 56 (LS mean −0.3, p=0.024). In the acute phase, ≥7% weight gain and the composite event of somnolence, sedation, and hypersomnia were more common in both asenapine groups than in the placebo group. Akathisia, fasting glucose elevation, and extrapyramidal syndrome were more common in the 5 mg b.i.d. group than in the placebo group. There were no unexpected adverse events in the OLE, and PANSS total scores decreased by −16.1 points in the group previously treated with placebo (n=62) and by −11.2 points in the continuous asenapine group (n=131) from OLE baseline to week 26.
Conclusions:
Although improvements in PANSS total score at day 56 of the acute phase were numerically greater for both asenapine 2.5 and 5 mg b.i.d. than for placebo and were maintained in the OLE, the primary end-point did not achieve statistical significance in the acute phase. No new or unexpected safety concerns were detected during the acute phase or after an additional 26 weeks of asenapine treatment in the adolescent population with schizophrenia.
Clinical Trials Registry:
NCT01190254 and NCT1190267 at
Introduction
A
Several of the second-generation atypical antipsychotics used to treat schizophrenia in adults have demonstrated efficacy in adolescents with schizophrenia (Findling et al. 2008; Sikich et al. 2008; Haas et al. 2009a,b Kryzhanovskaya et al. 2009; Singh et al. 2011; Findling et al. 2012; Kumar et al. 2013). The 2013 American Academy of Child and Adolescent Psychiatry (AACAP) Practice Parameter for the Assessment and Treatment of Children and Adolescents With Schizophrenia recommends ongoing use of antipsychotic medications, in combination with psychotherapeutic interventions, for treatment of EOS; however, this guideline does not specifically recommend any particular agent or second-generation atypical agents over first-generation agents, and endorses further study (McClellan et al. 2013). Endocrine and metabolic adverse effects, such as weight gain, are potential side effects of atypical antipsychotics (De Hert et al. 2011), and long-term studies of the safety of second-generation antipsychotics in pediatric populations are needed.
Although schizophrenia is a chronic illness requiring long-term therapy, data from the Treatment of Early Onset Schizophrenia Spectrum disorders (TEOSS) study indicate that many adolescents with schizophrenia discontinue treatment with the currently available therapies because of intolerability or lack of effectiveness (Sikich et al. 2008; Findling et al. 2010); therefore, additional treatment options for the EOS population are needed. Asenapine is a sublingually administered second-generation atypical antipsychotic that is approved for acute and maintenance therapy in adults with schizophrenia (Potkin et al. 2007; Kane et al. 2010, 2011; SAPHRIS™ prescribing information 2014). Safety and efficacy in adults with schizophrenia have been demonstrated in controlled studies using doses of 5 or 10 mg b.i.d. The pharmacologic profile of asenapine, a dibenzo-oxepino pyrrole derivative of the tetracyclic antidepressant mianserin, is characterized by high affinity for and antagonism of serotonergic receptors and potent dopamine, α-adrenergic, and histamine antagonism with low cholinergic activity (McIntyre and Wong 2012). Here, we report results from the first double-blind, randomized, placebo-controlled trial that evaluated the 8 week efficacy and safety of asenapine in adolescents (12–17 years of age) with schizophrenia. The safety profile of asenapine in these adolescents was further evaluated in a 26 week, flexible-dose, asenapine-only open-label extension (OLE) trial.
Methods
The 8 week randomized, placebo-controlled, double-blind, fixed-dose acute trial (
Eight week randomized, double-blind, placebo-controlled, fixed-dose acute phase
Adolescents with a confirmed diagnosis of schizophrenia were randomly assigned in a 1:1:1 distribution to placebo, asenapine 2.5 mg b.i.d., or asenapine 5 mg b.i.d. (Fig. S1) (see online supplementary material at
Inclusion criteria
Subjects ages 12–17 years with a current Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) diagnosis of schizophrenia (paranoid, disorganized, or undifferentiated subtype) were eligible to participate in the 8 week acute phase of the trial (American Psychiatric Association 2000). All psychiatric diagnoses specified in the inclusion/exclusion criteria were made according to DSM-IV-TR criteria by a psychiatrist experienced in child and adolescent psychiatric disorders or by a sponsor-approved qualified clinician. The diagnosis of schizophrenia was confirmed using the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL) by a second psychiatrist or qualified sponsor-approved professional. In addition, each subject was required to have the following scores at screening and baseline: A Positive and Negative Syndrome Scale (PANSS) total score ≥80; a Clinical Global Impressions-Severity (CGI-S) scale score of ≥4 (moderately ill); and a score of ≥4 (moderate) on two or more of the five items on the positive subscale of the PANSS (delusions, conceptual disorganization, hallucinatory behavior, grandiosity, suspiciousness/persecution).
Exclusion criteria
Subjects who had been treated with clozapine for treatment-resistant schizophrenia were to be excluded. Subjects with a primary Axis I diagnosis other than schizophrenia or a comorbid Axis I diagnosis that was primarily responsible for current symptoms and functional impairment were excluded, as were subjects with an uncontrolled or unstable clinically significant general medical condition (eg, renal, endocrine, hepatic, respiratory, cardiovascular, hematologic, immunologic, or cerebrovascular disease, or malignancy) or an abnormal laboratory, vital sign, physical examination, or electrocardiogram (ECG) finding at screening that, in the investigator's opinion, precluded the subject's participation in the trial or could interfere with the interpretation of safety and efficacy evaluations. Subjects with uncontrolled or unstable diabetes or a clinically significant abnormal blood glucose concentration at screening or baseline also were excluded. Any subjects reporting suicidal ideation with intent, with or without a plan, in the past 2 months, or suicidal behavior in the past 6 months, as measured by the Columbia Suicide Severity Rating Scale (C-SSRS), were excluded.
Dosing
During the 8 week double-blind treatment period, subjects randomized to asenapine received fast-dissolving black-cherry–flavored sublingual asenapine tablets; and subjects randomized to placebo received matching black-cherry–flavored sublingual tablets without asenapine. Subjects randomized to asenapine 2.5 mg b.i.d. started that regimen on day 1 and continued it for the duration of the treatment period. Subjects randomized to asenapine 5 mg b.i.d. received 2.5 mg b.i.d. until the visit on day 4, at which time the dose of asenapine was increased to 5 mg b.i.d. for the evening dose; and the 5 mg b.i.d. regimen was continued for the duration of the treatment period. Participants who could not tolerate the dose to which they were randomized were discontinued from study participation. Visits were scheduled on days 1, 4, 7, 14, 21, 28, 42, and 56 during treatment.
Efficacy outcomes
The primary efficacy end-point was the change from baseline to day 56 in the PANSS total score. The PANSS is a 30 item instrument in which total scores range from 30 (all symptoms absent) to 210 (all symptoms extreme) (Kay et al. 1987). The key secondary end-point was the change from baseline to day 56 in the CGI-S of subjects' illness score. Possible CGI-S scores range from 1 (normal, not at all ill) to 7 (among the most extremely ill subjects) (Guy 1976). Decreases from baseline within a group are indicative of improvement for both the PANSS total score and the CGI-S score. Other planned secondary end-points included 30% PANSS responders, defined as a subject with a reduction from baseline of ≥30% in the PANSS total score, and the PANSS positive and negative (items P1–P7, and N1–N7, respectively, both with possible range of 7–49) subscale scores.
Safety outcomes
Selected adverse events (AEs) of interest included akathisia; dizziness; insomnia; somnolence, sedation, and hypersomnia combined; oral hypoesthesia and dysgeusia combined; extrapyramidal syndrome (EPS); and the occurrence of a clinically significant weight gain, defined as in increase of ≥7% from baseline to end-point. EPS was defined using a Standardized Medical Dictionary for Regulatory Activities (MedDRA) Query (SMQ [narrow]) that included
Changes in body weight and body mass index (BMI) were also adjusted for growth (age and sex) and presented as mean and median Z scores (Centers for Disease Control and Prevention 2000). Analysis of safety data also focused on mean changes from baseline and predefined levels for the following lipid and endocrine parameters: Fasting glucose, fasting triglycerides, fasting total cholesterol, prolactin, fasting insulin, and hemoglobin A1c (HbA1c). New-onset diabetes was assessed using the MedDRA hyperglycemia/new onset diabetes mellitus SMQ (broad), which also included the preferred terms
Sample size and statistical analysis
The total target sample size was 300 subjects or 100 subjects per treatment group. With this sample size, the study would have ≥85% power to detect a difference between one or more asenapine doses and placebo of 8 points at day 56 on the change from baseline PANSS total score (ie, primary objective), assuming a standard deviation (SD) of ∼16 at day 56 and a 40% dropout rate after 8 weeks in each group. A two sided significance level of 0.05 with multiplicity correction according to Hochberg procedure was applied as the decision rule. For allocation of subjects, a computer-generated randomization list was used. The randomization list was created by the sponsor using SAS statistical software, and was stratified by site with a 1:1:1 allocation using a block size of 3. Allocation of subjects and medication was done using an Interactive Voice Response System by an independent third-party vendor.
Efficacy end-points were analyzed for all randomized adolescents who received one or more doses of asenapine or placebo, and had both baseline and at least one postbaseline PANSS total score (full analysis set [FAS]). The primary and key secondary efficacy end-points were analyzed with a mixed model for repeated measures (MMRM); the MMRM included terms for (pooled) site, treatment group, visit, treatment by visit interaction with baseline score, and visit by baseline score interaction as covariates; and an unstructured variance-covariance structure. PANSS 30% responders were analyzed using logistic regression with treatment, baseline, and (pooled) site in the model (last observation carried forward). PANSS subscale scores were analyzed using an MMRM model similar to that of the primary and key secondary end-point. A subgroup analysis of PANSS total score at day 56 by region (United States vs. non-United States) also was conducted by including the interaction of treatment by region in the MMRM model. P values for all end-points other than the primary, which was Hochberg corrected, were unadjusted.
Safety data were analyzed for all randomized adolescents who received one or more doses of the study drug (all-subjects-as-treated). Treatment-emergent adverse events (TEAEs) were reported, defined as AEs that occurred for the first time during the 8 week treatment period or were present during the screening period but worsened in severity during the treatment. All TEAEs, including those leading to discontinuation, and serious adverse events (SAEs) were summarized descriptively by MedDRA system organ class and preferred term. For TEAEs of interest, statistical comparisons were made between placebo and asenapine at the two sided significance level of p<0.05 using Miettinen and Nurminen (1985) for proportions with no correction for multiplicity.
Twenty-six week, flexible-dose OLE
Study design
Adolescents who completed the 8 week acute phase with an acceptable degree of adherence to trial medication and compliance with the protocol and who, in the investigator's judgement, could benefit from long-term treatment, were eligible to participate in the 26 week OLE (see Fig. S1). Subjects at imminent risk of self-harm or harm to others, in the investigator's opinion and based on C-SSRS responses, were excluded from the OLE. On day 1 of the OLE, all adolescents started with open-label asenapine at a dose of 2.5 mg b.i.d. At the day 4 visit, the dose of asenapine was increased to 5 mg b.i.d. From day 5, dosing was adjusted to either 2.5 mg b.i.d. or 5 mg b.i.d. based on tolerability and/or symptomatology as determined by the investigator. Visits during the OLE were on days 1, 4, 14, 28 (OLE week 4), 42 (OLE week 6), 70 (OLE week 10), 98 (OLE week 14), 126 (OLE week 18), 154 (OLE week 22), and 182 (OLE week 26) during treatment.
OLE efficacy and safety outcomes
Long-term safety and tolerability of asenapine therapy in adolescents were assessed for up to an additional 26 weeks, and summarized descriptively. The TEAEs and laboratory values of interest in the acute trial were also of special interest in the OLE. Exploratory descriptive analyses of efficacy parameters, including change in PANSS total score change and CGI-S and PANSS 30% responders during the OLE, were conducted. Maintenance of PANSS 30% response in the OLE also was analyzed for those subjects who were 30% responders during the acute phase. PANSS was assessed at OLE baseline and weeks 4, 10, 18, and 26; CGI-S and C-SSRS were assessed at each visit; weight and BMI were assessed at OLE baseline and weeks 2, 4, 10, 18, and 26.
OLE statistical analysis
Safety data were reported for all enrolled adolescents who received one or more doses of the study drug and were <18 years of age at enrolment (all-subjects-as-treated). Efficacy data were reported for the FAS, defined as all enrolled adolescents who received one or more doses of asenapine, had both a baseline and at least one postbaseline PANSS total score, and were <18 years of age at enrollment. During the OLE, the average daily dose for each subject was calculated, and then the population mean, median, and mode of the average daily dose were derived. Efficacy and safety measures for the OLE were summarized separately for subjects previously treated with placebo in the double-blind acute phase (hereafter referred to as the placebo/asenapine group) and those who were previously treated with double-blind asenapine (hereafter referred to as the continuous asenapine group). OLE baseline was used for calculating changes from baseline in efficacy parameters, and data were analyzed using an observed cases (OC) approach. TEAEs during the OLE were those that were newly reported during the OLE or were reported in the blinded phase but worsened in severity during the OLE; hence, this definition implies that an AE preferred term reported as starting and stopping during the acute phase and subsequently reemerging during the OLE with the same severity was not counted in the TEAE summaries for the OLE. Data from eight subjects (two in the placebo/asenapine group and six in the continuous asenapine group) who were 18 years old at the time of enrollment in the OLE were excluded from the main OLE efficacy and safety analyses, but were included in some analyses of change to OLE end-point when the acute phase baseline was the point of reference.
Results
Subjects
Eight week acute phase
A total of 306 subjects were randomized to treatment with placebo (n=102), 2.5 mg b.i.d. asenapine (n=98), or 5 mg b.i.d. asenapine (n=106); all randomized subjects received one or more doses of the study medication. The study population was >60% male, more than half were white, and the average age ranged from 15.2 (asenapine 2.5 mg b.i.d. group) to 15.4 (placebo and asenapine 5 mg b.i.d. groups) years across groups (Table 1). In the 8 week acute phase, similar percentages of adolescents completed the study period across the treatment groups (2.5 mg b.i.d.: 83%; 5 mg b.i.d.: 79%; placebo: 79%) (Fig. 1). Mean age at onset of schizophrenia ranged from 13.2 years (2.5 mg b.i.d. asenapine group) to 13.8 years (placebo group), and most subjects were diagnosed with schizophrenia of the paranoid type (DSM-IV-TR 295.30) (Table 2). Less than 10% of the population had a history of Axis I disorders other than schizophrenia or Axis II disorders, and >60% of subjects had received antipsychotic medication previously (Table 2). The FAS for efficacy analysis excluded 6 of the 306 subjects who were randomized and treated but did not have a postbaseline PANSS assessment (2 from each treatment group; n=100 for placebo, n=96 for 2.5 mg b.i.d. asenapine, and n=104 for 5 mg b.i.d. asenapine).
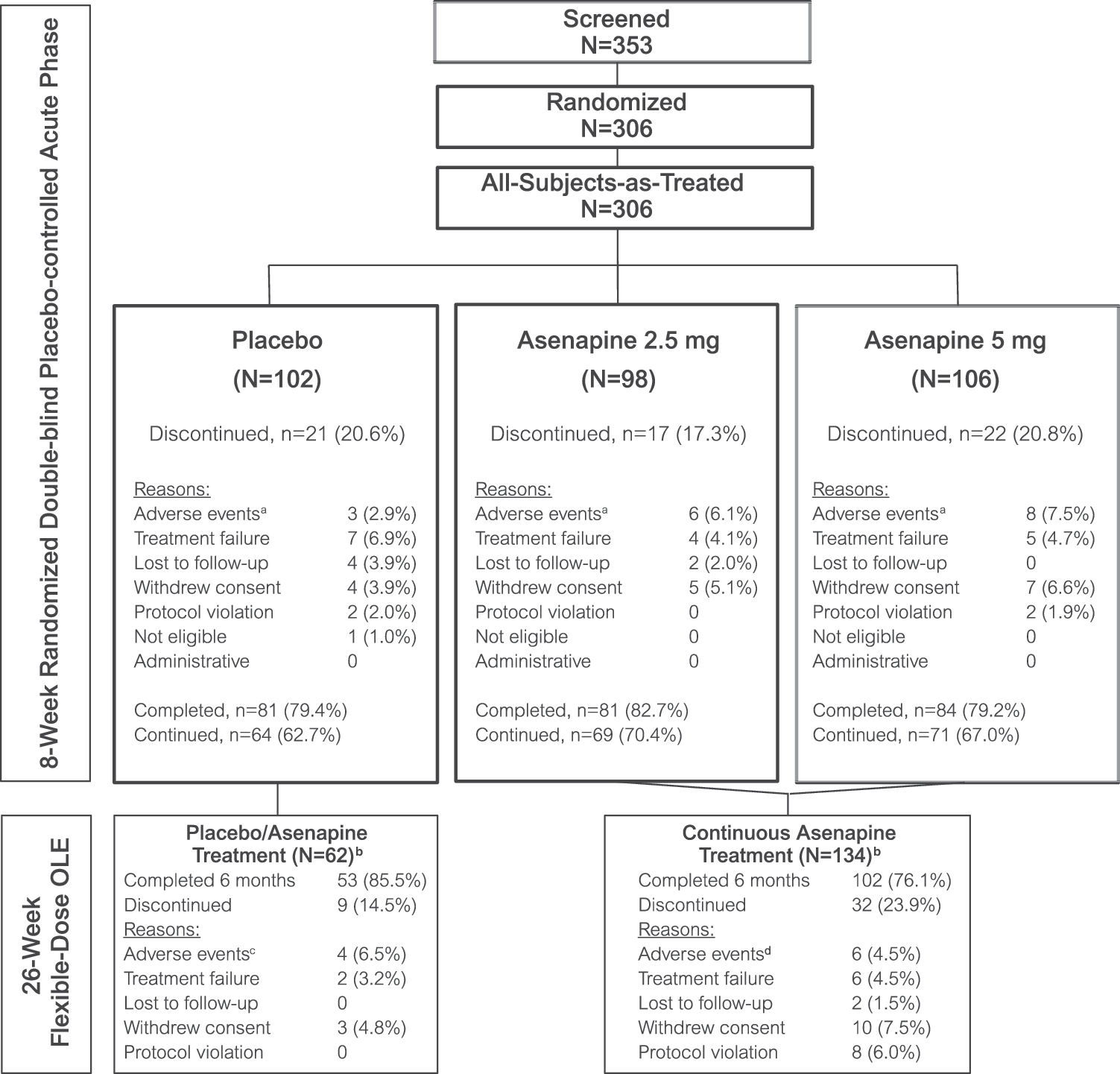
Subject disposition. aOf the AEs in the 8 week acute phase, three in patients randomized to placebo, three in patients randomized to asenapine 2.5 mg, and four in patients randomized to asenapine 5 mg were caused by the disease under study. bThe analysis population included all randomized subjects <18 years of age who received one osr more dose of trial medication; eight subjects (two in the placebo/asenapine group and six in the asenapine/asenapine group) who were 18 year old entered and were treated in the OLE but were not included in the primary analysis. cThree of the AEs in the placebo/asenapine group in the OLE were caused by the disease under study. dFour of the AEs in the continuous asenapine group in the OLE were caused by the disease under study. AE, adverse event; OLE, open-label extension.
Extension-phase characteristics were calculated using the OLE baseline.
BMI, body mass index; OLE, open-label extension.
n=100 for placebo, n=96 for asenapine 2.5 mg b.i.d., n=104 for asenapine 5 mg b.i.d.
CGI-S, Clinical Global Impressions-Severity; DSM-IV-TR, Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision: PANSS, Positive and Negative Syndrome Scale.
Twenty-six week OLE
Most (83%) of the subjects who completed the acute phase enrolled in the OLE. A total of 196 adolescents <18 years of age enrolled and were treated with flexible-dose asenapine in the extension phase. Of these, 62 had been treated with placebo (hereafter referred to as the placebo/asenapine group) and 134 with asenapine (either 2.5 mg b.i.d. or 5 mg b.i.d.; hereafter referred to as the continuous asenapine group) in the antecedent acute phase. The FAS population in the OLE consisted of 193 subjects, excluding 3 additional subjects in the continuous asenapine group who discontinued the OLE before having an open-label PANSS assessment. The demographics of adolescents who participated in the OLE were similar to those who participated in the acute phase (Table 1). A greater percentage of adolescents previously treated with placebo completed the OLE compared with those continuously treated with asenapine (85% vs. 76%, respectively) (Fig. 1). The mean average daily dose of asenapine in the OLE was 8.7 mg/day (median average daily dose 9.8 mg; modal average daily dose 9.9 mg).
Efficacy
Eight week acute phase
In the MMRM analysis of the PANSS total score change from baseline to day 56, the primary objective was not met because the difference from placebo was not statistically significant for either asenapine group. The difference in least squares (LS) mean change from baseline to day 56 in the PANSS total score was −4.8 (unadjusted 95% confidence interval [CI], −9.9 to 0.4; adjusted p=0.070) for 2.5 mg asenapine b.i.d. versus placebo and −5.6 (unadjusted 95% CI, −10.7 to −0.5; adjusted p=0.064) for 5 mg asenapine b.i.d. versus placebo (Fig. 2A). Based on unadjusted p values relative to placebo, the LS mean change from baseline in PANSS total score over time was significantly greater for the 2.5 mg b.i.d. asenapine group on days 21, 28, and 42 and for the 5 mg b.i.d. asenapine group on days 28, 42, and 56 (Fig. 2B). On the key secondary outcome, which can be considered only exploratory because the primary efficacy objective was not met, a significant improvement in the CGI-S score was observed in the 5 mg b.i.d. group when compared with placebo on day 56 (unadjusted LS mean [95% CI] −0.3 [−0.6, −0.0]); (unadjusted p=0.024) (Fig. 2C) but not for the 2.5 mg b.i.d. group.
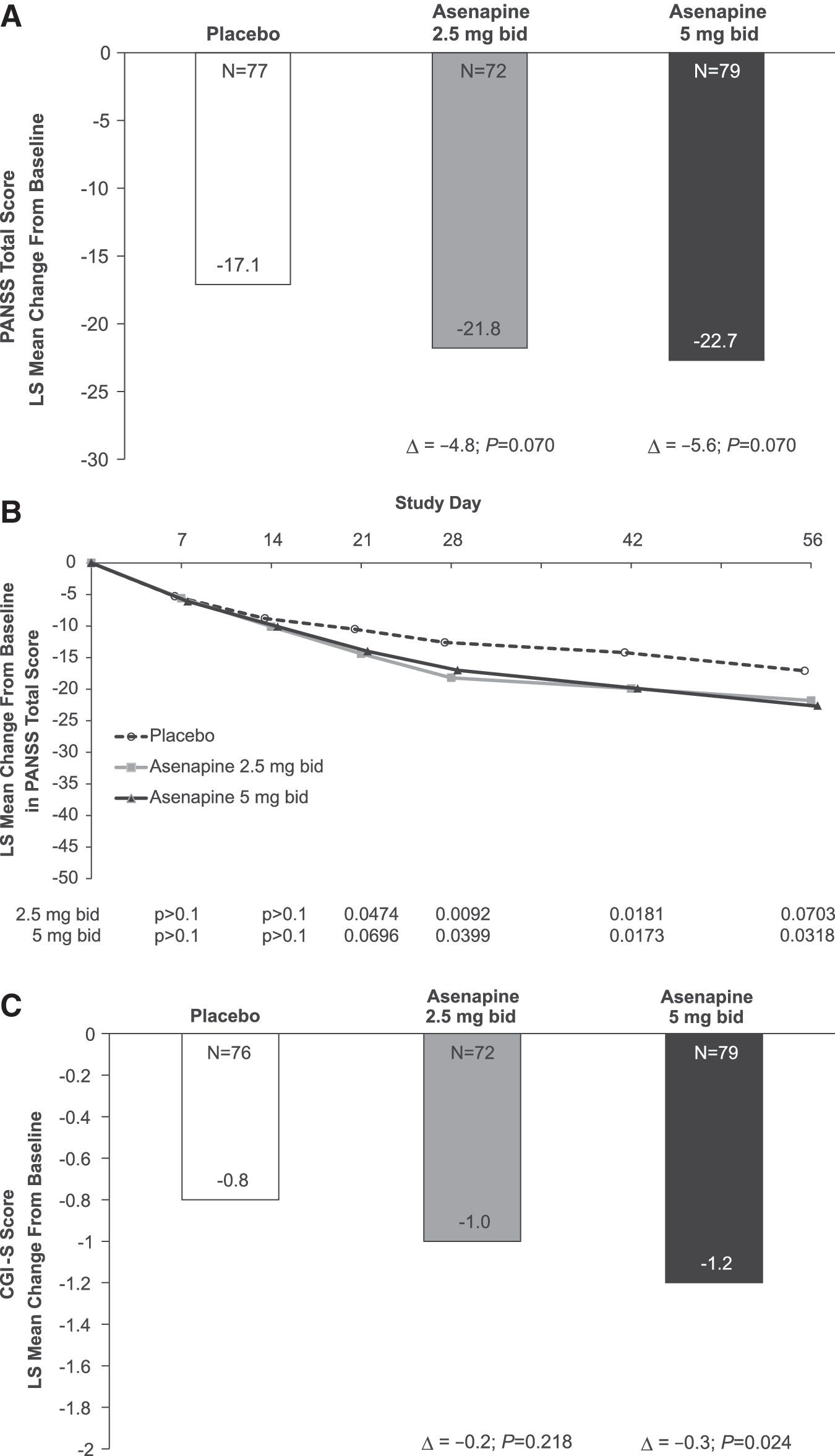
Efficacy during the acute phase (MMRM, full analysis set).
Other planned exploratory end-points of interest included PANSS 30% responders, PANSS subscale scores, and treatment by region interaction. The p value for treatment by region interaction (United States vs. non-United States) in the MMRM for change from baseline in PANSS total score analysis at day 56 was 0.646, suggesting that the treatment effect at day 56 of asenapine compared with placebo was not significantly different for subjects in the United States and not in the United States. In this subgroup analysis, the LS mean changes in PANSS total score on day 56 in the United States (n=48, −3.7 for 2.5 mg b.i.d. vs. placebo and −12.0 for 5 mg b.i.d. vs placebo) and not in the United States (n=252, −5.0 for 2.5 mg b.i.d. vs. placebo and −4.6 for 5 mg b.i.d. vs. placebo) were comparable with the overall treatment differences observed in the primary efficacy analysis, with the exception of United States subjects in the 2.5 mg b.i.d. asenapine group, which could be explained by the imbalance in the sample size.
Based on unadjusted p values, both asenapine dose groups had a higher PANSS 30% responder rate (50% for asenapine 2.5 mg b.i.d., p=0.028; 49% for asenapine 5 mg b.i.d., p=0.048) than placebo (36% of subjects) at day 56. The average change from baseline in the PANSS Positive Subscale Score indicated consistently larger improvement for the asenapine treatment groups relative to placebo starting at day 7. Differences in LS mean changes from baseline to day 56 on the PANSS Positive Subscale were −1.6 for asenapine 2.5 mg b.i.d. (unadjusted p=0.067) and −2.1 for asenapine 5 mg b.i.d. (unadjusted p value of 0.012). Differences in LS mean changes from baseline to day 56 on the PANSS Negative Subscale were −1.2 for both asenapine groups versus placebo (unadjusted p values of 0.097 and 0.099, respectively).
Twenty-six week OLE
At baseline in the OLE, mean PANSS total scores were 78.9 for the placebo/asenapine group (n=62) and 72.8 for the continuous asenapine group (n=131); baseline mean CGI-S scores were 3.7 and 3.5, respectively. On average, numerical improvements for the PANSS total and CGI-S scores in the acute phase were maintained and continued to indicate improvement over the course of 26 weeks of flexible-dose asenapine treatment. Using an OC analysis, the mean change from the OLE baseline to week in the OLE was −16.1 points for the placebo/asenapine group and −11.2 points for the continuous asenapine group (Fig. 3A). Overall, 48% of subjects in the OLE had a 30% decrease from OLE baseline to end-point PANSS total score (56% in the placebo/asenapine group and 43% in the continuous asenapine group). When change in PANSS total score was calculated from the acute phase baseline (including 18-year-olds) to the end of the OLE, representing 34 weeks of asenapine treatment for subjects in the continuous asenapine group, the mean changes were −36.7 points for the continuous asenapine group and −34.4 for the placebo/asenapine group. Of the 103 adolescents who were PANSS 30% responders at the end of the acute phase and who continued in the OLE, a cumulative percentage of 79% (76% in the placebo/asenapine group and 80% in the continuous asenapine group) maintained this effect. The mean change in CGI-S from the OLE baseline to OLE Week 26 was −1.1 points for the placebo/asenapine group and −0.7 for the continuous asenapine group (Fig 3B).
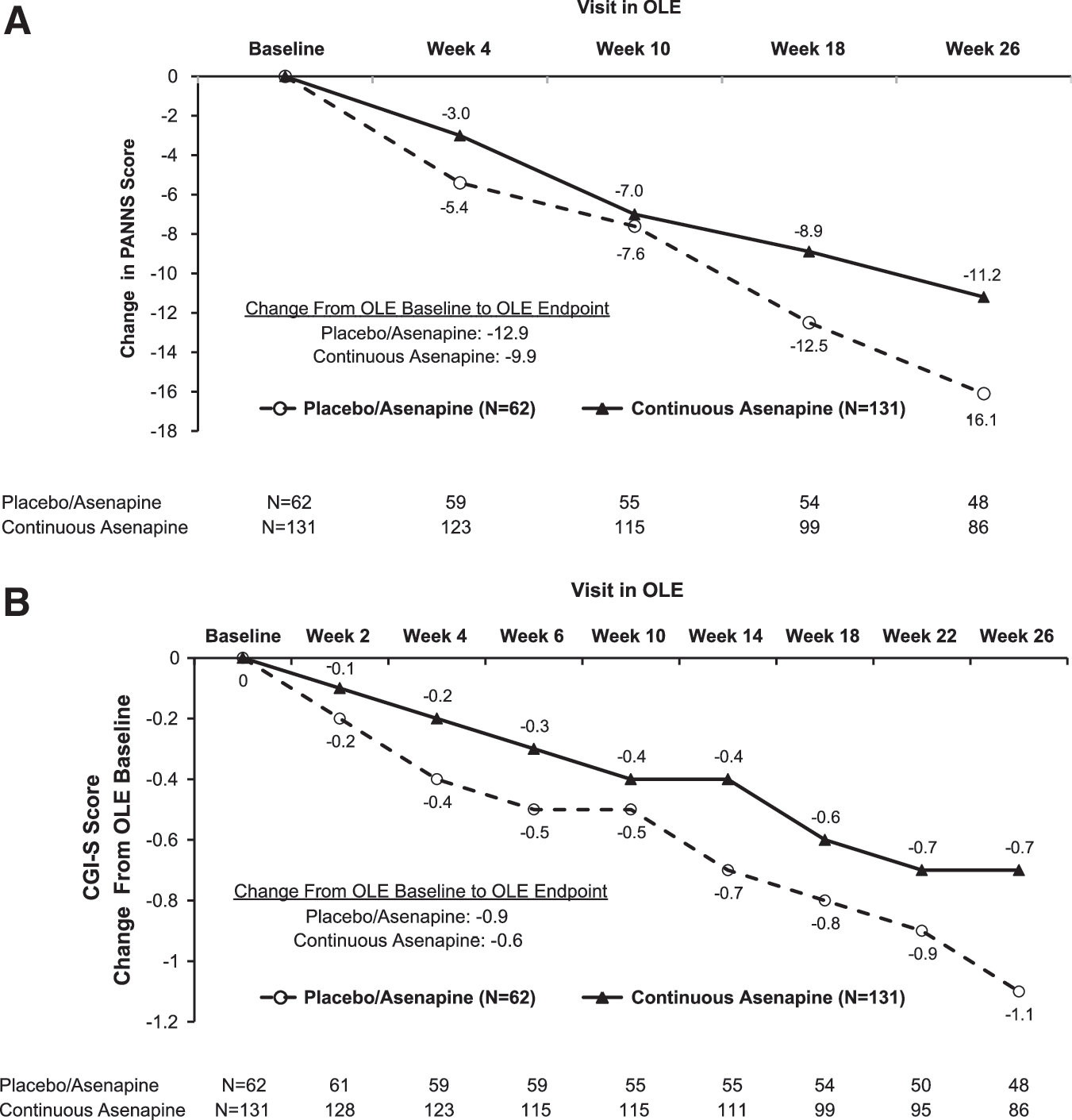
Efficacy during the OLE (OC, full analysis set).
Safety
Eight week acute phase
The mean duration of treatment was 49 days for placebo, 54 days for 2.5 mg b.i.d. asenapine, and 49 days for 5 mg b.i.d. asenapine. No deaths occurred during the acute phase, and no subjects had a TEAE of attempted suicide or C-SSRS scores indicating suicidal intent with a formal plan or actual suicidal behaviors. The percentages of subjects experiencing SAEs were similar across the groups (placebo, 2.9%; asenapine 2.5 mg b.i.d., 3.1%; asenapine 5 mg b.i.d., 2.8%) (Table 3), the most common of which was worsening schizophrenia. The DMC requested that treatment be unblinded for five subjects who experienced serious, unexpected (per the Reference Safety Information in the asenapine Investigator Brochure) AEs that the investigator deemed to be related to study drug or trial procedure. Three of these five subjects were randomized to placebo, and each placebo-treated subject was hospitalized for worsening schizophrenia and study medication was discontinued (days 17, 27, and 56 of treatment). One subject randomized to 5 mg b.i.d. asenapine was hospitalized for worsening of schizophrenia; study medication was continued, and the subject received additional medication and therapeutic procedures. One subject randomized to 2.5 mg b.i.d. asenapine was hospitalized for hallucination (auditory) on day 3, and study medication was discontinued. Of the six (6%) subjects in the asenapine 2.5 mg b.i.d. group who discontinued, one subject each discontinued because of schizophrenia, agitation, depression, auditory hallucination, somnolence, and pneumonia. A total of eight (8%) subjects in the asenapine 5 mg b.i.d. group discontinued; the most common reason was worsening schizophrenia (three subjects) followed by insomnia and akathisia (one subject), dysgeusia (one subject), sedation (one subject), and polycythemia (one subject), and abnormal alanine aminotransferase (one subject). An accidental overdose in a subject who took one extra dose of asenapine 2.5 mg was reported, with no untoward consequences. There were no reports of drug abuse during the acute phase.
For the OLE, TEAEs were counted from the open-label baseline; TEAEs in the OLE were newly reported in the OLE or worsened in severity during the OLE compared with the acute phase.
Every adolescent was counted a single time for each applicable row and column.
Incidence ≥5% in either asenapine group and more than twice that with placebo and not summarized as an AE of interest.
p<0.05 for placebo vs asenapine using Miettinen and Nurminen (1985).
EPS (narrow) indicates that preferred terms in the SMQ (
Adolescents with at least one postbaseline weight value were included; baseline for OLE analysis was the open-label baseline.
AE, adverse event; EPS, extrapyramidal symptoms; OLE, open-label extension; SAE, serious adverse event; SMQ, Standardized Medical Dictionary for Regulatory Activities (MedDRA) Query; TEAE, treatment-emergent adverse event.
The overall incidence of TEAEs was 47% in the placebo group, 62% in the 2.5 mg b.i.d. asenapine group, and 67% in the 5 mg b.i.d. asenapine group (Table 3). Nervous system AEs were the most common overall, experienced by 14% of subjects in the placebo group, 37% in the asenapine 2.5 mg b.i.d. group, and 48% in the asenapine 5 mg b.i.d. group (Table 3). Among the preselected TEAEs of interest, the composite safety end-point of somnolence, sedation, and hypersomnia combined were significantly more frequent in both asenapine groups (2.5 mg b.i.d.: 24%, p<0.05; 5 mg b.i.d.: 29%, p<0.05) compared with placebo (9%) (Table 3). Compared with placebo, dizziness was significantly more frequent in the 2.5 mg b.i.d. group (7% vs. 1%), whereas akathisia (7% vs. 1%) occurred more frequently in the 5 mg b.i.d. group than in the placebo group. The occurrence of EPS, as identified by the MedDRA SMQ (narrow), was numerically greater in both asenapine groups than in the placebo group. More subjects in the asenapine 5 mg b.i.d. group (11%) reported concomitant medication use corresponding to an EPS AE compared with the placebo (3%) and asenapine 2.5 mg b.i.d. groups (2%). Clinically significant weight gain (increase of ≥7% from baseline to end-point) was more frequent in the 2.5 mg (9%; p>0.05) and 5 mg (13%; p<0.05) b.i.d. groups than in the placebo group (3%). Mean weight changes from baseline were 0.1 kg for placebo, 1.3 kg for asenapine 2.5 mg b.i.d., and 1.4 kg for asenapine 5 mg b.i.d. After correcting for growth by age and sex, Z scores for weight and BMI confirmed a clinically meaningful increase in weight for the asenapine 2.5 and 5 mg b.i.d. groups compared with placebo (Table 4).
For the OLE, change from baseline to end-point was calculated using the open-label baseline.
BMI, body mass index; OLE, open-label extension.
With respect to laboratory parameters of interest, the mean change from baseline to end-point in fasting insulin was greater for the asenapine 2.5 mg b.i.d. and 5 mg b.i.d. groups compared with placebo, with an apparent dose-response for asenapine (Table 5). Potential new-onset diabetes per the MedDRA hyperglycemia/new onset diabetes mellitus SMQ (broad) was reported in four (4%) placebo-treated subjects, seven (7%) 2.5 mg b.i.d. asenapine-treated subjects, and seven (7%) 5 mg b.i.d. asenapine-treated subjects; and three asenapine-treated subjects (one in the 2.5 mg bid group and two in the 5 mg bid group) met the IDF criteria for new-onset metabolic syndrome. No notable treatment-group differences were observed for fasting cholesterol, fasting glucose, fasting triglycerides, or HbA1c (Table 5
For the extension phase, mean changes and shifts were calculated using the open-label extension baseline.
Number (%) of adolescents with a shift in hemoglobin A1c outside the predefined limits of change during the treatment phase.
OLE, open-label extension.
Twenty-six week OLE
The mean duration of asenapine treatment in the OLE was 171 days for placebo/asenapine and 163 days for continuous asenapine, with a mean average daily dose of 9.8 mg for both groups. One death occurred during the OLE (placebo/asenapine), secondary to a fall from a sixth floor window. This event was determined by the investigator and the subject's family to be accidental, because the study participant had no history of suicidal ideation, suicide attempt, or deliberate self-harm before or during the trial. SAEs, including the one death, were reported for seven (4%) subjects in the total OLE population (two placebo/asenapine, five continuous asenapine) (Table 3). Hospitalizations related to psychiatric disorders occurred for one subject in the placebo/asenapine group (aggression and anxiety) and five in the continuous asenapine group (three worsening of schizophrenia, one aggression, and one agitation). Ten (5%; four placebo/asenapine, six continuous asenapine) subjects discontinued owing to AEs, four of which were SAEs. Eight subjects (4%) in the total population reported suicidal ideation on the C-SSRS during the OLE phase, none of which included intent with a formal plan or actual suicidal behaviors; four of the eight subjects had a history of suicidal ideation/behavior prior to participation in the acute trial. A TEAE of suicidal ideation was associated with three (1.5%) of these events, but no TEAEs of suicidal behavior or suicide attempt were reported. Parental consent was withdrawn for the six subjects with suicidal ideation. Four accidental overdoses, defined per protocol as any day in which a subject took ≥10 mg of asenapine, were reported during the OLE. One of these overdoses was reported with a TEAE of sedation; this subject's mother gave him a total of four extra tablets of asenapine 5 mg during a six day period (verbatim term for this event was accidental overdose without AE). The DMC had no specific safety concerns during the OLE.
Consistent with the acute phase, the composite end-point of somnolence, sedation, and hypersomnia was the most frequently reported TEAE of interest in the OLE (27% in the placebo/asenapine and 19% in the continuous asenapine group Table 3). With the exception of the TEAE of ≥7% weight gain, which was reported with similar frequency between the OLE groups (13% in the placebo/asenapine group and 15% in the continuous asenapine group), the placebo/asenapine group had a greater frequency of all other TEAEs of interest in the OLE compared with the continuous asenapine group (Table 3). Overall mean weight gain from OLE baseline to end-point was 1.6 kg for the placebo/asenapine group and 1.0 kg for the continuous asenapine group. Z scores for weight and BMI, adjusting for growth based on age and sex and calculated from OLE baseline (Table 4), as well as weight and BMI percentiles overtime (Figs. S2 and S3) (see online supplementary material at
Mean changes from baseline to end-point in metabolic chemistry parameter levels in the total treatment group were generally modest in number (Table 5). Eight (4%) subjects met the criteria for new onset hyperglycemia/diabetes mellitus (SMQ [broad]), of which five in the continuous asenapine group also met the ≥7% weight gain criterion. New-onset metabolic syndrome per IDF criteria was met by seven subjects during the OLE, two in the placebo/asenapine group and five in the continuous asenapine group. Although mean change in prolactin from baseline to end-point was small (3.7 μg/L) during the OLE, prolactin concentrations ≥1.1 times the upper limit of normal were reported for 32% of subjects in the placebo/asenapine group and 31% in the continuous asenapine group. One adolescent had hyperprolactinemia (prolactin concentration of 146 μg/L), breast engorgement, and hyperinsulinemia (insulin 295.86 pmol/L) on day 188. Another adolescent had irregular menstruation (prolactin concentration of 30.3 μg/L at baseline). This TEAE resolved on day 165 (no laboratory data available), and prolactin was normal (19.2 μg/L) on trial Day 129 but again was elevated (40 μg/L) on day 173 without further follow-up available.
Discussion
Although improvements in PANSS total score during the 8 week acute phase were numerically greater for asenapine 2.5 and 5 mg b.i.d. than for placebo, both asenapine groups failed to separate from placebo on the Hochberg-corrected primary analysis of change in PANSS total score from baseline to day 56. The study was designed to have power to detect an 8 point difference, or an effect size of 0.5, in PANSS total score between asenapine and placebo at day 56; however, the anticipated treatment response and effect size may have been overestimated for this largely outpatient population of adolescents, because the actual treatment response was −4.8 for asenapine 2.5 mg b.i.d. and −5.6 for asenapine 5 mg b.i.d., and effect sizes were lower than expected (0.30 for asenapine 2.5 mg b.i.d. and 0.35 for asenapine 5 mg b.i.d.). A change of −0.5 SD is considered to be a clinically significant change in PANSS total score in adults, or typically 8 points on this scale, and the SD in the present study was as anticipated. The assumption of an 8 point difference was derived from the adult asenapine schizophrenia program and publications for other antipsychotic compounds (aripiprazole and quetiapine) that were registered for adolescents (12–17 years of age when randomly assigned to treatment) with schizophrenia (Findling et al. 2008, 2012). In statistical sensitivity analyses (data not shown), differences between asenapine and placebo on the PANSS total score were consistently in the range of −5 points for asenapine 2.5 mg bid and −6 points for asenapine 5 mg b.i.d. Differences in clinical practice between the United States and non-United States sites that recruited adolescents with schizophrenia for this study did not appear to influence the results, as the treatment-by-region interaction was not significant in the MMRM analysis. Likewise, the placebo response on the PANSS total score (−17.1 points) was in the range observed with other agents in both positive and failed studies of schizophrenia in adolescents (Findling et al. 2008, 2012, 2013).
The numerical improvements attained in PANSS total score during the acute phase were maintained or increased over the course of the 26 week flexible-dose OLE, in which the mean average daily dose indicated that most subjects received asenapine 5 mg b.i.d. In addition, the subjects who transitioned to asenapine in the OLE after receiving placebo during the acute phase essentially achieved the same level of response as the continuous asenapine group by the end of the 26 week extension study. The 5 mg b.i.d. dose is the lowest approved dose for adults with schizophrenia (SAPHRIS PI 2014), whereas the 2.5 mg b.i.d. asenapine dose was selected for the present trial based on a pharmacokinetic and tolerability study (Dogterom et al. 2012) with the aim of evaluating whether this lower dose might demonstrate efficacy in this potentially more vulnerable adolescent population. Although the primary end-point was not met for either dose, asenapine 5 mg b.i.d. was associated with improvements in several exploratory secondary efficacy end-points. Statistically significant differences between asenapine 5 mg b.i.d. and placebo at day 56 were observed for the CGI-S score, PANSS positive subscale score, and PANSS 30% responder rate; these findings suggest that subjects receiving the 5 mg b.i.d. dose may have experienced some benefit from asenapine treatment during the acute phase.
With no clear superiority or differential symptom reduction among atypical and typical antipsychotic agents, differential adverse event profiles may be a primary consideration when selecting treatment for an adolescent with EOS (Sikich et al. 2008; Findling et al. 2010; Kumar et al. 2013). The sublingual formulation of asenapine was generally acceptable, because there were no significant differences in the hypoesthesia oral/dysgeusia combined end-point, and only a small increase in this event for the placebo-treated subjects who switched to asenapine in the OLE. As expected, the somnolence/sedation/hypersomnia composite end-point was observed more frequently in the asenapine groups than in the placebo group; however, these events were not a common cause of treatment discontinuation (one subject in the OLE) and were generally considered mild to moderate in severity and transient in nature. Likewise for akathisia, which showed a signal for dose-response in the acute phase, the incidence of this TEAE was lower in the OLE than in the acute phase, and no subjects discontinued because of this event. The numerically greater incidence of EPS in the asenapine 5 mg group, combined with greater use of concomitant medications to treat EPS symptoms, suggests a dose-related signal. Overall, the AE profile in the placebo/asenapine group during the OLE was as expected based on the asenapine AE rates during the acute phase, and the lower rates in the continuous asenapine group during the OLE suggest that the 5 mg b.i.d. dose was well tolerated.
Weight gain occurs in some adults treated with asenapine and has been reported with other atypical antipsychotic medications in adolescents (De Hert et al. 2011). In the present study, weight and BMI were monitored to determine potential clinically significant weight gain and weight gain in excess of normal growth. Because schizophrenia is a chronic illness that may require lifelong treatment, it is important to understand the potential metabolic effects of treatment-related weight gain, particularly because weight gain in adolescents taking atypical antipsychotics might be greater than what is observed in adults. Asenapine treatment was associated with weight gain, increases in fasting insulin, and other metabolic abnormalities in some patients. Overall, the data suggest that asenapine-treated adolescents may experience an initial increase in weight, followed by stabilization and minimal change during up to 26 weeks of additional treatment. Also consistent with the known safety profile of atypical antipsychotic medications with dopamine-antagonistic activity (Peuskens et al. 2014), asenapine treatment was associated with an increase in prolactin concentrations compared with placebo. In the present study, few subjects in the OLE had prolactin-associated AEs, and no dose-response was noted in the acute trial.
Low rates of remission, low effect size of medications, and the potential for adverse events, particularly metabolic abnormalities with long-term treatment, underscore the need for further study of antipsychotic medications in the adolescent population (Masi and Liboni 2011). Although statistically significant efficacy of asenapine in adolescents was not demonstrated in the present study, safety data through 34 weeks of treatment suggest that asenapine was generally well tolerated; and the safety profile in adolescents was consistent with that in adults with schizophrenia. Use of an asenapine dose lower (2.5 mg b.i.d.) than that approved for adults, combined with careful titration to the 5 mg b.i.d. dose, which is the recommended dose in adults, appeared to be successful in potentially mitigating unexpected adverse reactions in this adolescent population.
Conclusions
Although improvements in PANSS total score during the 8 week acute phase were numerically greater for both asenapine 2.5 and 5 mg b.i.d. than for placebo, the differences did not achieve statistical significance. The two asenapine doses had safety and tolerability profiles that, overall, were similar during the acute phase, except for an apparent dose-response for clinically significant weight gain, EPS, and fasting glucose elevation. The numerical improvements attained in PANSS total score and CGI-S score during the acute phase were maintained or increased over the course of the 26 week flexible-dose OLE. No new or unexpected safety concerns were detected during up to 34 weeks of asenapine treatment in this adolescent population with schizophrenia.
Clinical Significance
We report results from the first acute, double-blind, randomized, placebo-controlled trial of asenapine 2.5 mg and 5 mg b.i.d. in adolescents with schizophrenia. These doses were selected based on the results of a previously conducted tolerability and pharmacokinetic study. Although the efficacy of asenapine 5 mg b.i.d. and 10 mg b.i.d. in adult inpatients with schizophrenia has been established, in the present adolescent study the PANSS total scores in the asenapine groups did not separate from placebo after 8 weeks of therapy. Lack of statistical significance may, in part, have resulted from overestimating the expected treatment effect in this predominately outpatient adolescent population. Differences between subjects recruited in the United States versus those recruited in other geographic regions did not appear to influence the results. Exploratory secondary efficacy parameters suggest a potential signal for treatment benefit with 5 mg b.i.d. dose. Asenapine was generally well tolerated in adolescents for up to 34 weeks of treatment, with a safety profile consistent with that in adults with schizophrenia.
Footnotes
Acknowledgments
The authors thank all subjects and investigators who participated in these studies. Medical writing services were provided by Cathryn M. Carter of Arbor Communications, Inc; these services were funded by Forest Research Institute, Inc, an affiliate of Actavis Inc.
Disclosures
Dr. Findling receives or has received research support from, or acted as a consultant and/or served on a speaker's bureau for Alcobra, Alexza Pharmaceuticals, American Academy of Child & Adolescent Psychiatry, American Physician Institute, American Psychiatric Press, AstraZeneca, Bracket, Bristol-Myers Squibb, Clinsys, CogCubed, Cognition Group, Coronado Biosciences, Dana Foundation, Forest, GlaxoSmithKline, Guilford Press, Johns Hopkins University Press, Johnson & Johnson, KemPharm, Lilly, Lundbeck, Merck, NIH, Novartis, Noven, Otsuka, Oxford University Press, Pfizer, Physicians Postgraduate Press, Purdue, Rhodes Pharmaceuticals, Roche, Sage, Seaside Pharmaceuticals, Shire, Stanley Medical Research Institute, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, and WebMD. Drs. Landbloom and Mackle are employees of and own stock in Merck. Dr. Braat was a Merck employee at the time the study was conducted; has no financial disclosures. Dr. Pallozzi was a Merck employee at the time the study was conducted, and owns stock in Merck. Drs. Hundt and Mathews are employees of and own stock in Actavis. Dr. Wamboldt receives research funding from Forest, Pfizer, and Sunovion, as well as royalties from APA Press and Singer.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.