
Abstract
Objective:
The study goal was to determine the efficacy and safety of an optimal dose of Evekeo, racemic amphetamine sulfate, 1:1
Methods:
A total of 107 children ages 6–12 years were enrolled in this multicenter, dose-optimized, randomized, double-blind, placebo-controlled crossover study. After 8 weeks of open-label dose optimization, 97 subjects were randomized to 2 weeks of double-blind treatment in the sequence of R-AMPH followed by placebo (n=47) or placebo followed by R-AMPH (n=50). Efficacy measures included the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Rating Scale and Permanent Product Measure of Performance (PERMP) administered predose and at 0.75, 2, 4, 6, 8, and 10 hours postdose on 2 laboratory classroom days. Safety assessments included physical examination, chemistry, hematology, vital signs, and treatment-emergent adverse events (TEAEs).
Results:
Compared to placebo, a single daily dose of R-AMPH significantly improved SKAMP-Combined scores (p<0.0001) at each time point tested throughout the laboratory classroom days, with effect onset 45 minutes postdose and extending through 10 hours. R-AMPH significantly improved PERMP number of problems attempted and correct (p<0.0001) throughout the laboratory classroom days. During the twice-daily dose-optimization open-label phase, improvements were observed with R-AMPH in scores of the ADHD-Rating Scale IV and Clinical Global Impressions Severity and Improvement Scales. TEAEs and changes in vital signs associated with R-AMPH were generally mild and not unexpected. The most common TEAEs in the open-label phase were decreased appetite (27.6%), upper abdominal pain (14.3%), irritability (14.3%), and headache (13.3%).
Conclusions:
Compared to placebo, R-AMPH was effective in treating children aged 6–12 years with ADHD, beginning at 45 minutes and continuing through 10 hours postdose, and was well tolerated.
Trial registration:
Introduction
A
Amphetamines, also known as sympathomimetic amines, increase synaptic levels of the catecholamines dopamine and noradrenaline by inhibiting their reuptake into the neuron and also by releasing them into the synaptic cleft. Amphetamine contains a single chiral center that gives rise to distinct enantiomeric active forms—dextro (
Several amphetamine products for the treatment of ADHD have been developed with different proportions of each isomer. Some consist entirely of
The primary objective of the current study was to establish that an optimal dose of R-AMPH in children 6–12 years old diagnosed with ADHD would result in a significant reduction in ADHD signs and symptoms compared to placebo at 2 hours postdose in a laboratory classroom, while characterizing the onset of effect and duration of action for the medication. A laboratory classroom setting for study of ADHD medications allows for assessment by trained observers over the course of a typical extended school day, with both subjective and objective measures for determining onset and duration of drug effect (Wigal and Wigal 2006).
Methods
Subjects
Males and females between the ages of 6 and 12 years meeting DSM-IV-TR criteria for ADHD combined, inattentive, or hyperactive/impulsive type were eligible for the trial. The diagnosis was confirmed during screening using the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID). Study inclusion required that subjects receive a score of at least 3 (mildly ill) on the clinician-administered Clinical Global Impressions–Severity (CGI-S) scale and have an ADHD-Rating Scale (ADHD-RS-IV) score (DuPaul et al. 1998) at screening or baseline ≥90th percentile normative values for gender and age in at least one of the following categories: Hyperactive/impulsive subscale, inattentiveness subscale, or total score. The ADHD-RS-IV is an 18-item scale based on Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) criteria of ADHD that rates symptoms on a 4-point scale. Each item is scored using a combination of severity and frequency ratings from a range of 0 (reflecting no symptoms or a frequency of never or rarely) to 3 (reflecting severe symptoms or a frequency of very often), so that the total ADHD-RS-IV scores range from 0 to 54. The 18 items can be divided into two 9-item subscales: One for hyperactivity/impulsivity and the other for inattentiveness. Scores were obtained during a clinician-directed interview with the parent/caregiver at each visit. The CGI-S is a clinician-rated scale that evaluates the severity of psychopathology (ADHD symptoms in this trial) on a scale from 1 (not at all ill) to 7 (among the most severely ill) (Busner and Targum 2007). To qualify for inclusion, the subject also had to be in need of pharmacological treatment for ADHD in the judgment of the site principal investigator.
A primary psychiatric diagnosis other than ADHD excluded a subject from participating in the study, as did secondary or co-morbid diagnoses other than ADHD with the exception of simple phobias, oppositional defiant disorders, motor skill disorders, communication disorders, learning disorders, adjustment disorders, and sleep disorders. Subjects with a clinically significant cognitive impairment or a personal history of seizure disorder (except simple febrile seizures), untreated thyroid disease, glaucoma, Gilles de la Tourette's disorder, or chronic tics were excluded from participating in the study. Also excluded were subjects with a personal history of structural cardiac disorders, serious cardiac conditions, or severe hypertension. Those subjects with a family history of sudden cardiac death required review and approval by the medical monitor before being cleared for participation in the study. Taking an investigational drug within 60 days of the screening visit was also exclusionary.
Study design
This multicenter, dose-optimized, double-blind, randomized, placebo-controlled crossover study was conducted to evaluate the safety and efficacy of R-AMPH in pediatric subjects aged 6–12 years with ADHD. A laboratory school protocol, as described by Wigal and Wigal (2006), was used to measure the efficacy, onset of effect, and duration of effect. The study was conducted at seven sites in the United States, in accordance with Good Clinical Practice guidelines. The study was approved by an appropriate institutional review board at each study site.
The study design is outlined in Figure 1. At the screening visit (study visit 1), before the performance of any study-specific procedures, each subject and his or her parent/guardian received an explanation of all study procedures, and the parent/guardian provided voluntary written informed consent for the subject to participate in this study, and the subject gave written assent. Screening assessments were then administered to determine eligibility. Medical history, including medications, was reviewed. A physical examination was performed, with vital signs (sitting blood pressure and pulse, in triplicate via automated machine, along with respiratory rate and temperature), 12-lead electrocardiogram (ECG) (performed at screening only), and laboratory tests, including serum chemistry panel, hematology, urinalysis, and urine screen for commonly abused drugs. Female subjects of childbearing potential were administered a serum pregnancy test.
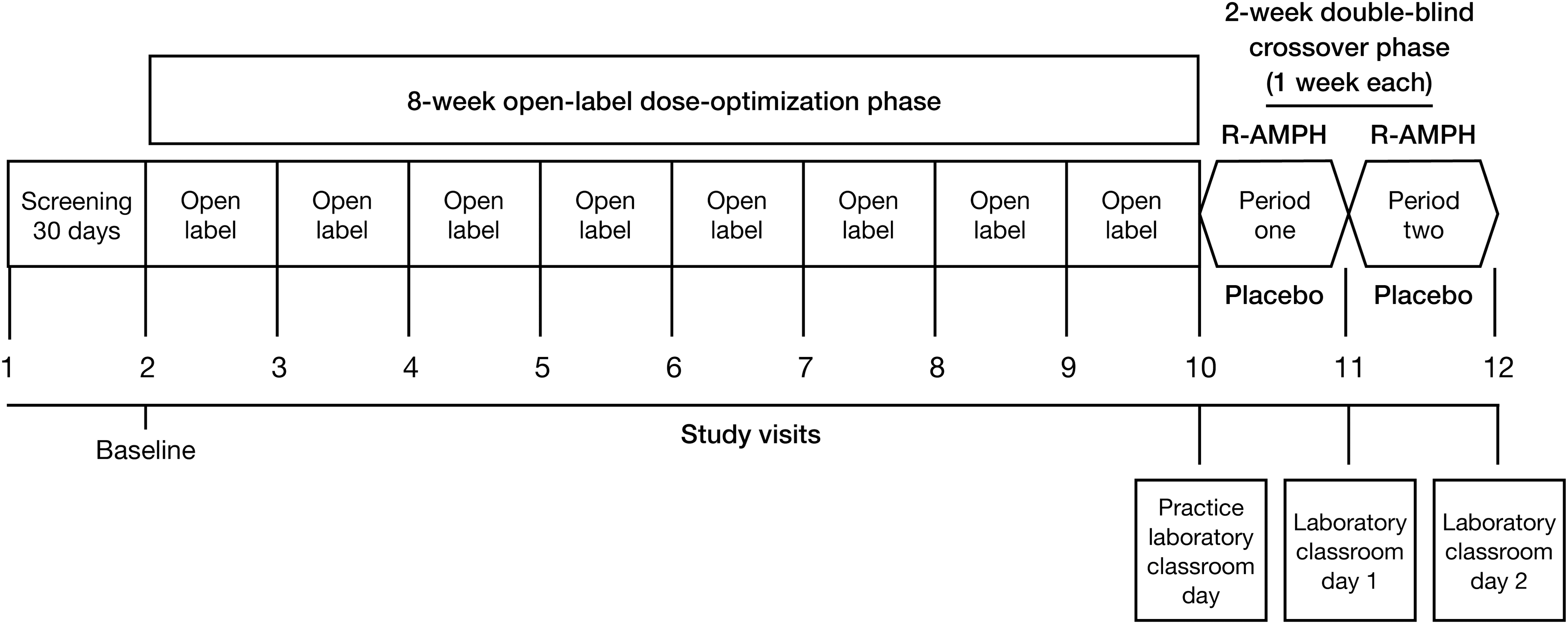
R-AMPH laboratory classroom trial design.
At the baseline visit, the subject's medical history was reviewed again, body weight was obtained, and sitting blood pressure and pulse were again measured in triplicate. A urine pregnancy test was obtained in female subjects of childbearing potential. The ADHD-RS-IV, CGI-S, Conners 3rd Edition Parent Short Form (Conners 3-P), and baseline version Columbia-Suicide Severity Rating Scale (C-SSRS) were also administered. The Conners 3-P is a 45-item parent (or guardian) report that provides evaluation of inattention, hyperactivity/impulsivity, learning problems, executive functioning, aggression, and peer relationships. Subjects meeting all eligibility requirements were prohibited from using any psychotropic medications with the exception of sedative hypnotics prescribed as sleep aids during the trial. Anticonvulsants, antidepressants, antipsychotics, or nonstimulant ADHD medications (atomoxetine, guanfacine, or clonidine) were not allowed within the 30 days before the baseline visit and for the duration of the study. Stimulant medications were allowed until 24 hours before the baseline visit. Other medications allowed during the course of the study included acetaminophen and nonsteroidal anti-inflammatories; nonsedating antihistamines such as cetirizine, loratadine, and fexofenadine; mometasone; and approved courses of prescription and nonprescription medications for the treatment of acute illnesses.
The baseline visit (study visit 2, study day 1) occurred within 30 days following the screening visit. During this visit, subjects were reassessed for eligibility and administered a placement Permanent Product Measure of Performance (PERMP), an objective individualized mathematics examination, to assign the level of difficulty that would be administered throughout the laboratory classroom segment of the study as an objective measurement of efficacy. Subjects meeting all eligibility requirements, including performance at the basic level of the PERMP or higher, were enrolled in the study. Open-label medication was dispensed to the subjects, who were instructed to start taking the medication twice daily (once in the morning with or without food and again 4–6 hours later), beginning on the following morning, study day 2, and continuing through approximately study day 50. The starting dose of R-AMPH for the 8-week open-label dose-titration period of the study was 10 mg per day (5 mg twice daily), which was titrated weekly in 5 mg increments until an optimal dose based on clinical response and tolerability was achieved in the judgment of the site principal investigator. A single down-titration for tolerability reasons was also permitted. To globally assess changes in the severity of ADHD from visit to visit over the course of the open-label phase of the study, clinicians used the ADHD-RS-IV, CGI-S, and Clinical Global Impressions–Improvement (CGI-I). The CGI-I is scored from 1 (very much improved) to 7 (very much worse). The parent/guardian assessed changes in ADHD symptoms via the Conners 3-P performed on study visits 6 (after week 4) and 10 (after week 8). Safety and tolerability were assessed via weekly monitoring of subject blood pressure and pulse and the tracking of spontaneously reported adverse events. The C-SSRS was also used to assess changes from baseline in the occurrence and severity of suicidal ideation and behavior.
At study visit 10, subjects able to tolerate a stable dose of at least 10 mg per day were randomized on a 1:1 basis by an interactive web-based response system (IWRS) to a treatment sequence of 1 week R-AMPH/1 week placebo or 1 week placebo/1 week R-AMPH. The crossover design allowed subjects to serve as their own controls.
At study visit 10, each subject completed an abbreviated practice laboratory classroom session lasting ∼4 hours and was dispensed the first week of double-blind study medication, which was to be taken twice daily, at home and/or school, for the following 6 days. No adjustment of R-AMPH dose was permitted during the double-blind phase. The final dose of the first week of double-blind medication was administered at the laboratory school site by study staff on the morning of the first full-length laboratory classroom day (study visit 11). On the 2 full laboratory classroom days, only the single morning dose was administered (and not the second daily dose), to establish duration of action of the racemic formulation. At the end of study visit 11, double-blind crossover medication, to be taken at home and/or school twice daily for the following 6 days during the second week of the double-blind phase, was dispensed. The final dose of crossover medication was administered at the laboratory school site by study staff on the morning of the second full-length laboratory classroom day (study visit 12).
One week following study visit 12, a final safety assessment was conducted during a postwithdrawal follow-up phone call or visit to the study site.
Outcome measures: Efficacy
The primary objective of the study was to establish that an optimal dose of R-AMPH would result in a significant reduction in ADHD signs and symptoms compared to placebo at 2 hours postdose. The secondary efficacy objectives were to determine the onset and duration of clinical effect for R-AMPH after a single morning dose. Primary and secondary efficacy analysis variables were measured in a laboratory classroom setting that was designed to model a typical school day plus after-school activity totaling 10 hours. The primary efficacy variable was the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP)-Combined score determined at 2 hours postdose on the 2 full-length laboratory classroom days (visits 11 and 12). The SKAMP scale is a validated subjective measure of ADHD symptoms in a laboratory classroom. It comprised 13 items (grouped under the subcategories of attention, deportment, quality of work, and compliance) on which subjects were rated according to a 7-point scale (0=normal to 6=maximal impairment) by trained study personnel (Wigal and Wigal 2006). The SKAMP-Combined score was obtained by summing the rating values for each of the 13 items. Higher SKAMP scores signified greater impairment.
The secondary efficacy variables included SKAMP-Combined scores, SKAMP-Attention subscale scores (four items), SKAMP-Deportment subscale scores (four items), and PERMP scores, each measured before dosing and at 0.75, 2, 4, 6, 8, and 10 hours postdose on the 2 full-length laboratory classroom days. The PERMP is an individualized, five-page mathematics examination consisting of 400 problems. Subjects were instructed by site staff to work at their seats and complete as many problems as possible in 10 minutes. The appropriate level of difficulty for each student was determined based on the results of the PERMP administered at the baseline visit. Performance was evaluated using two scores: The number of problems attempted (PERMP-A) and the number of problems correct (PERMP-C). Additional secondary efficacy variables included the CGI-S, CGI-I, and ADHD-RS-IV ratings obtained during the open-label phase of the study.
Outcome measures: Safety
The secondary safety objective was to investigate the safety and tolerability of R-AMPH compared to placebo in pediatric subjects 6–12 years of age diagnosed with ADHD. The occurrence of treatment-emergent adverse events (TEAEs) was assessed starting at visit 3 (following the first week of the open-label dose-optimization phase of the study) and ending with the postwithdrawal follow-up phone call. Additional safety evaluations included physical examination, with blood pressure and pulse measured in triplicate, as well as height and weight, repeat laboratory tests, and a follow-up C-SSRS administered at each study visit.
Statistical analyses
Primary and secondary efficacy analyses were performed on the intent-to-treat (ITT) population consisting of all randomized subjects who received at least one dose of double-blind study medication and had at least one postbaseline assessment of the primary efficacy variable. The key secondary efficacy analysis was repeated on the clinically evaluable population, defined as all ITT subjects who received the morning dose of double-blind study medication at both laboratory test sessions; completed all laboratory classroom tests; did not miss more than 2 days of therapy during the double-blind treatment period; and did not use prohibited medication during the double-blind treatment period. Descriptive statistics for the SKAMP-Combined, SKAMP-Attention, SKAMP-Deportment, PERMP-A, and PERMP-C scores were calculated for each time point on the laboratory classroom days. The average difference between treatments (R-AMPH vs. placebo) was estimated for each variable using least-square (LS) means from a mixed-effects repeated-measures model, including treatment, sequence, period, and study center as fixed effects, and subject within sequence as a random effect. The treatment comparison was conducted as a two-sided test at the 5% level of significance. Standard errors and 95% confidence intervals (CIs) were calculated. All available data were used; there was no imputation of missing data. Descriptive statistics and changes from baseline were calculated for the additional secondary efficacy variables, including CGI-S, CGI-I, and ADHD-RS-IV, obtained during the open-label phase of the study. Also determined for ADHD-RS-IV was the proportion of responders, defined as subjects who had a ≥50% change from baseline.
Analyses were performed on both the enrolled safety population (defined as all enrolled subjects who received at least one dose of open-label study medication and had at least one postbaseline safety assessment) and the randomized safety population (defined as all randomized subjects who received at least one dose of double-blind study medication and had at least one postbaseline safety assessment). The frequencies of adverse events (AEs), the results of laboratory assessments and physical examinations (including vital signs), and the frequency of suicidal ideation or behavior (assessed using the C-SSRS) were summarized descriptively by the treatment group.
Results
Subject disposition
Subject disposition is summarized in Figure 2. Of 107 subjects enrolled in the 8-week open-label phase of the study, 10 withdrew before randomization, leaving 97 subjects for the ITT population and for the randomized safety population—47 subjects randomized to the treatment sequence of R-AMPH followed by placebo and 50 to the sequence of placebo followed by R-AMPH. Two randomized subjects completed only one of the two double-blind period laboratory classroom days—one subject was unable to come for the second laboratory classroom day, and one was lost to follow-up. Therefore, the clinically evaluable population for repeat analyses of the primary and secondary efficacy variables consisted of 95 subjects (97.94%) from the ITT population. The enrolled safety population for analyzing safety data across the entire study consisted of 105 of the 107 total enrolled subjects. One subject not included withdrew prematurely due to an AE (irritability) after receiving the study drug but before having any postbaseline safety assessments; the other subject was discontinued at the discretion of the investigator, also without having any postbaseline safety assessments.
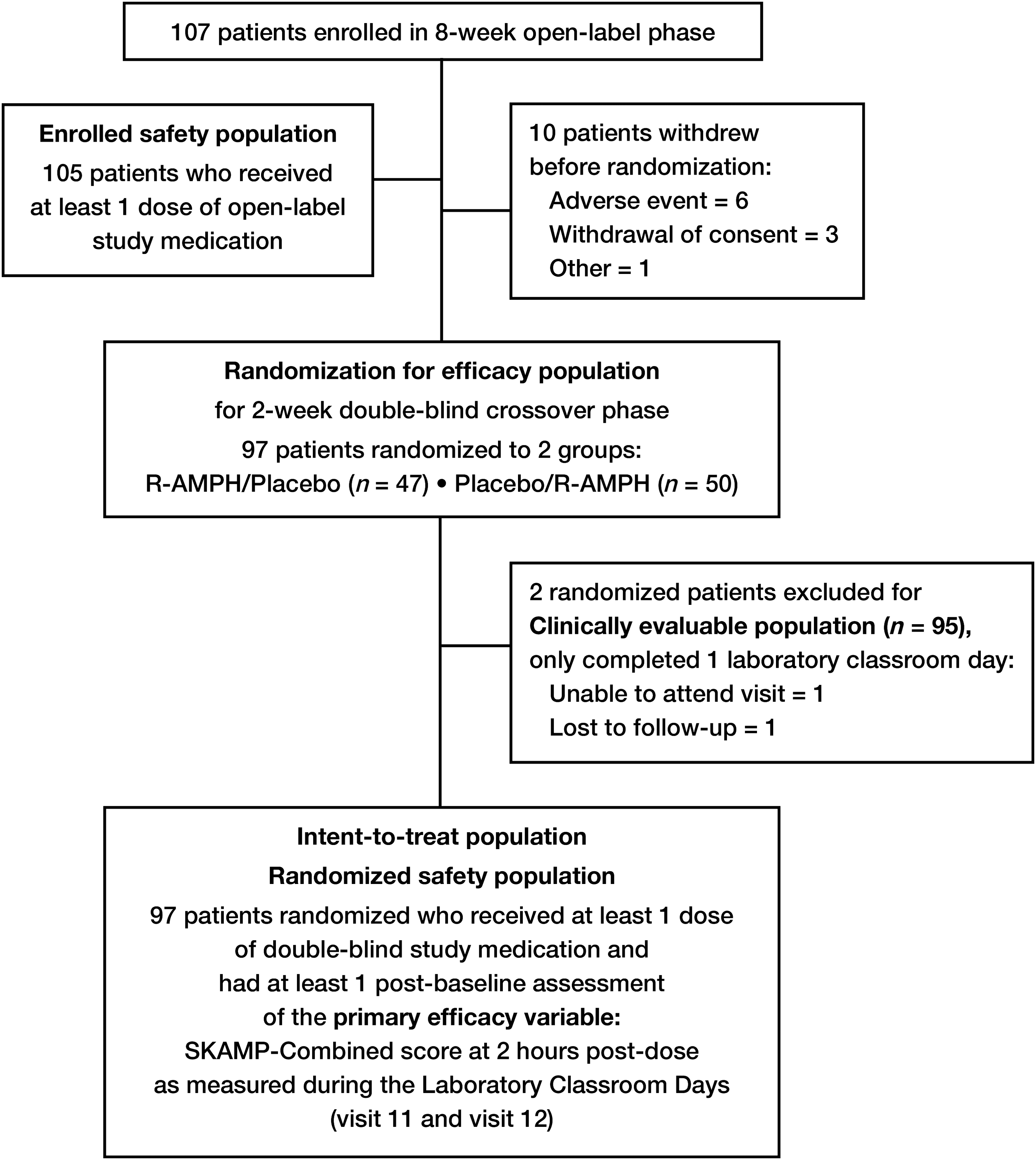
Subject disposition in the R-AMPH laboratory classroom study.
The demographic and baseline characteristics of the ITT population are shown in Table 1. The majority of subjects were male (60.8%) and white (59.8%). Mean subject age was 9.6±1.86 years, with 16.5% aged 6–7 years, 45.4% aged 8–10 years, and 38.1% aged 11–12 years. Most subjects (81.4%) were diagnosed with combined type ADHD, while 18.6% were diagnosed with the inattentive type, and no subjects with strictly hyperactive/impulsive type. All 97 subjects had symptoms≥ADHD-RS-IV 90th percentile at baseline. At screening, 91 subjects (86.7%) had normal ECG findings and 14 subjects (13.3%) had abnormal ECG findings, none of which was recorded as being clinically significant. Medical history and prior medication usage were similar between the R-AMPH/placebo and placebo/R-AMPH groups, the most frequent findings being asthma (21.9%), seasonal allergy (10.5%), drug hypersensitivity (8.6%), and oppositional defiant disorder (8.6%). The most frequently reported prior medications were centrally acting sympathomimetics (48.6%). During the study, for the enrolled safety population, the most frequently administered concomitant medications were centrally acting sympathomimetics (35.2%), selective beta-2-adrenoreceptor agonists (17.1%), propionic acid derivatives (15.2%), and other antihistamines for systemic use (12.4%).
ADHD, attention-deficit/hyperactivity disorder; SD, standard deviation.
Efficacy assessments: Primary measure
The primary efficacy variable was the SKAMP-Combined score determined at 2 hours postdose on the 2 full-length laboratory classroom days (visits 11 and 12). R-AMPH demonstrated significant improvement on the SKAMP-Combined scores at 2 hours postdose compared to placebo at visits 11 and 12 for the ITT population (Table 2). The LS mean SKAMP-Combined score was 10.3 in subjects receiving R-AMPH compared with 18.1 in those receiving placebo. The LS mean treatment difference (R-AMPH vs. placebo) was −7.9 (p<0.0001). A difference in LS means for the change from predose SKAMP-Combined scores also favored R-AMPH treatment compared to placebo for the primary outcome: −10.5 points (95% CI−13.2, −7.8; p<0.0001). When the primary efficacy analysis was repeated on data from the clinically evaluable population (n=95), the results were similar to those observed for the ITT population. In a series of subgroup analyses on the primary efficacy endpoint, statistically significant differences in LS means favoring R-AMPH treatment over placebo were observed regardless of the final daily dose (10–15, 20–25, 30–35, 40 mg), subject age, gender, race, or ADHD type.
CI, confidence interval; ITT, intent-to-treat; LS, least squares; Q, quartile; SE, standard error; SKAMP, Swanson, Kotkin, Agler, M-Flynn, and Pelham Rating Scale.
Efficacy assessments: Secondary measures
Figure 3 shows the SKAMP-Combined scores over the course of the 2 laboratory classroom days—during which, overall, each subject received R-AMPH and placebo. Significant separation from placebo occurred at each time point tested (0.75, 2, 4, 6, 8, and 10 hours) (p<0.0001 for all time points), with onset of R-AMPH effect by 45 minutes postdose (difference in LS means of −5.5 points) and with the greatest R-AMPH effect seen at 4 hours postdose (difference in LS means of −8.3 points). R-AMPH was still effective at 10 hours postdose (difference in LS means of −4.3 points), the last time point measured. The duration of the R-AMPH effect was therefore at least 9.25 hours (from 0.75 hours to the end of the study at 10 hours). Statistically significant differences (p<0.0001) in LS means for the change from predose SKAMP-Combined scores also favored R-AMPH treatment compared to placebo at each postdose time point, with the greatest change from baseline in R-AMPH treatment occurring at 2 hours postdose. When postdose SKAMP-Combined scores were analyzed by clinical site for the ITT population, the differences in LS means favored R-AMPH treatment at all time points for all sites. When SKAMP-Combined scores were examined across different subgroups (final dose, subject age, gender, race, ADHD type), statistically significant differences in LS means favoring R-AMPH treatment over placebo were observed at each time point through 10 hours postdose, with results similar to those for the ITT population as a whole.
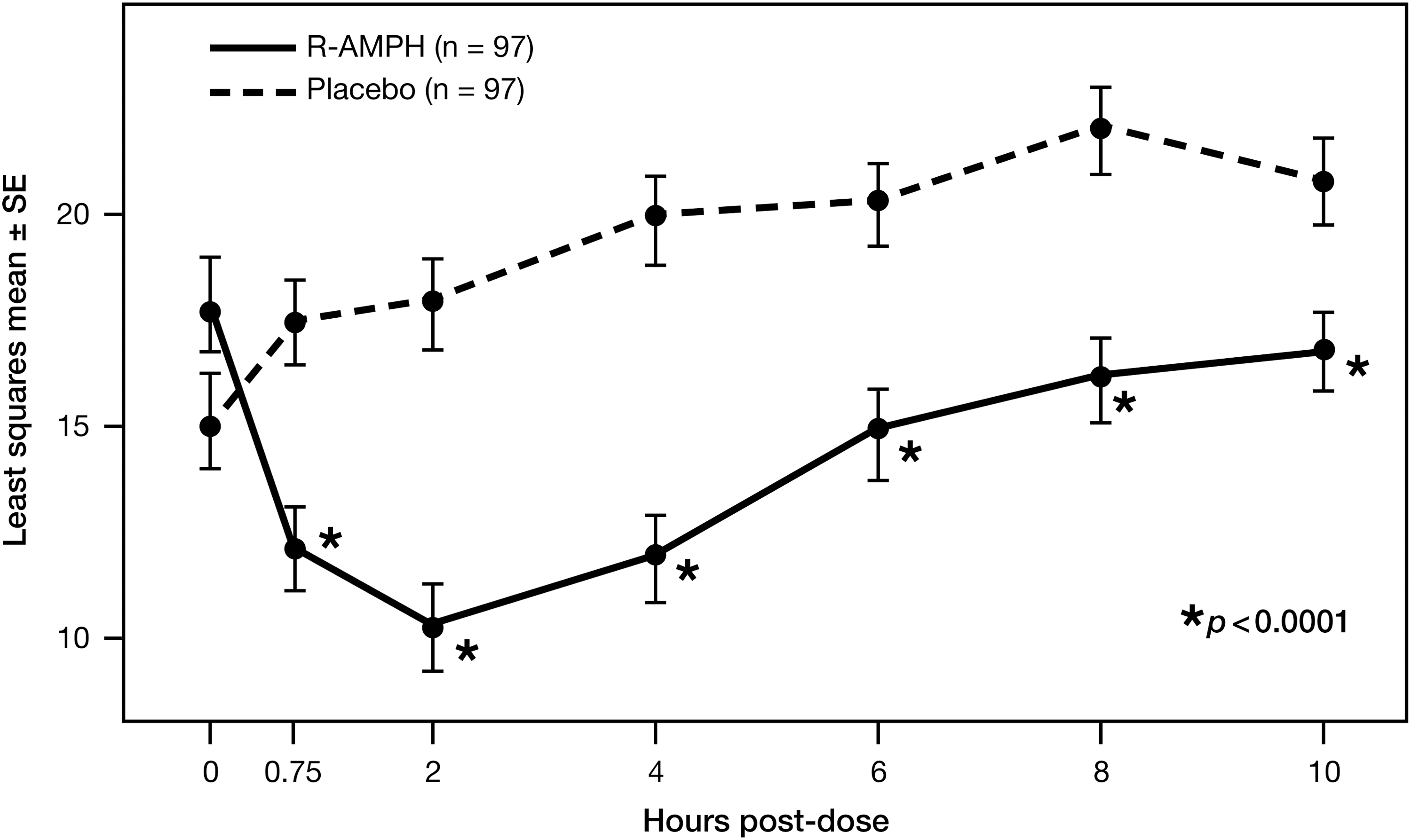
Laboratory classroom SKAMP-Combined scores. SKAMP, Swanson, Kotkin, Agler, M-Flynn, and Pelham.
Results for the SKAMP-Attention and SKAMP-Deportment subscale scores were similar to those for the SKAMP-Combined scores, with 2-hour postdose differences in LS means favoring R-AMPH treatment of −1.6 points on the SKAMP-Attention subscale and −2.5 points on the SKAMP-Deportment subscale. At each time point tested, significant separation of R-AMPH from placebo was observed for both the SKAMP-Attention and SKAMP-Deportment subscales, again with the greatest R-AMPH effect seen at 4 hours postdose (−1.8 points for the SKAMP-Attention subscale and −2.6 points for the SKAMP-Deportment subscale) and the greatest change from predose score seen at 2 hours postdose (differences in LS means of −1.4 points for the SKAMP-Attention subscale and −1.9 points for the SKAMP-Deportment subscale). Statistical significance was robust (p<0.0001) for both subscales for all but two of the time point observations (p=0.0188 at 10 hours for attention and p=0.0018 at 6 hours for deportment).
When subjects received R-AMPH, they attempted more problems on the PERMP and answered more of those questions correctly than when they received placebo. At each time point tested in the laboratory classroom setting (0.75, 2, 4, 6, 8, and 10 hours), both the number of problems attempted (Fig. 4) and the number of problems answered correctly on the PERMP (Fig. 5) were significantly higher (p<0.0001 in all cases) after subjects received R-AMPH. While the greatest R-AMPH effect was seen at 4 hours postdose for the number of problems attempted (difference in LS means of 27.9 problems attempted) and at 2 hours postdose for the number of problems correct (difference in LS means of 28.0 problems correct), similar effect levels were seen between 2 and 6 hours for both categories (range of differences in LS means of 26.3 to 27.9 problems attempted and 24.9 to 28.0 problems correct). When PERMP scores were examined across the different subgroups (final dose, subject age, gender, race, ADHD type) with regard to the number of problems attempted and the number of problems correct, statistically significant differences in LS means favoring R-AMPH treatment over placebo were observed at each time point through 10 hours postdose, with trends similar to those for the ITT population as a whole.
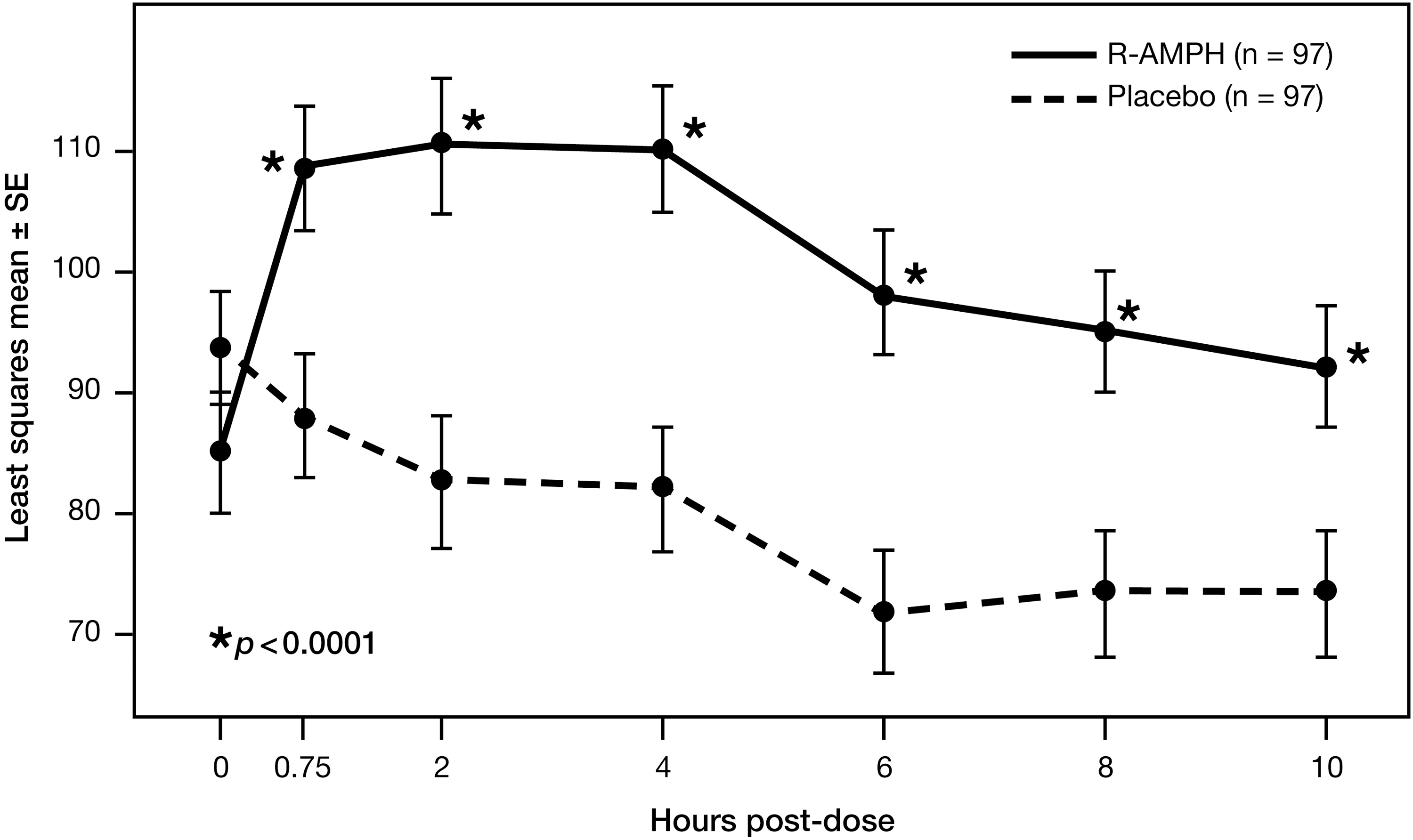
PERMP number of problems attempted over time by treatment group. PERMP, Permanent Product Measure of Performance.
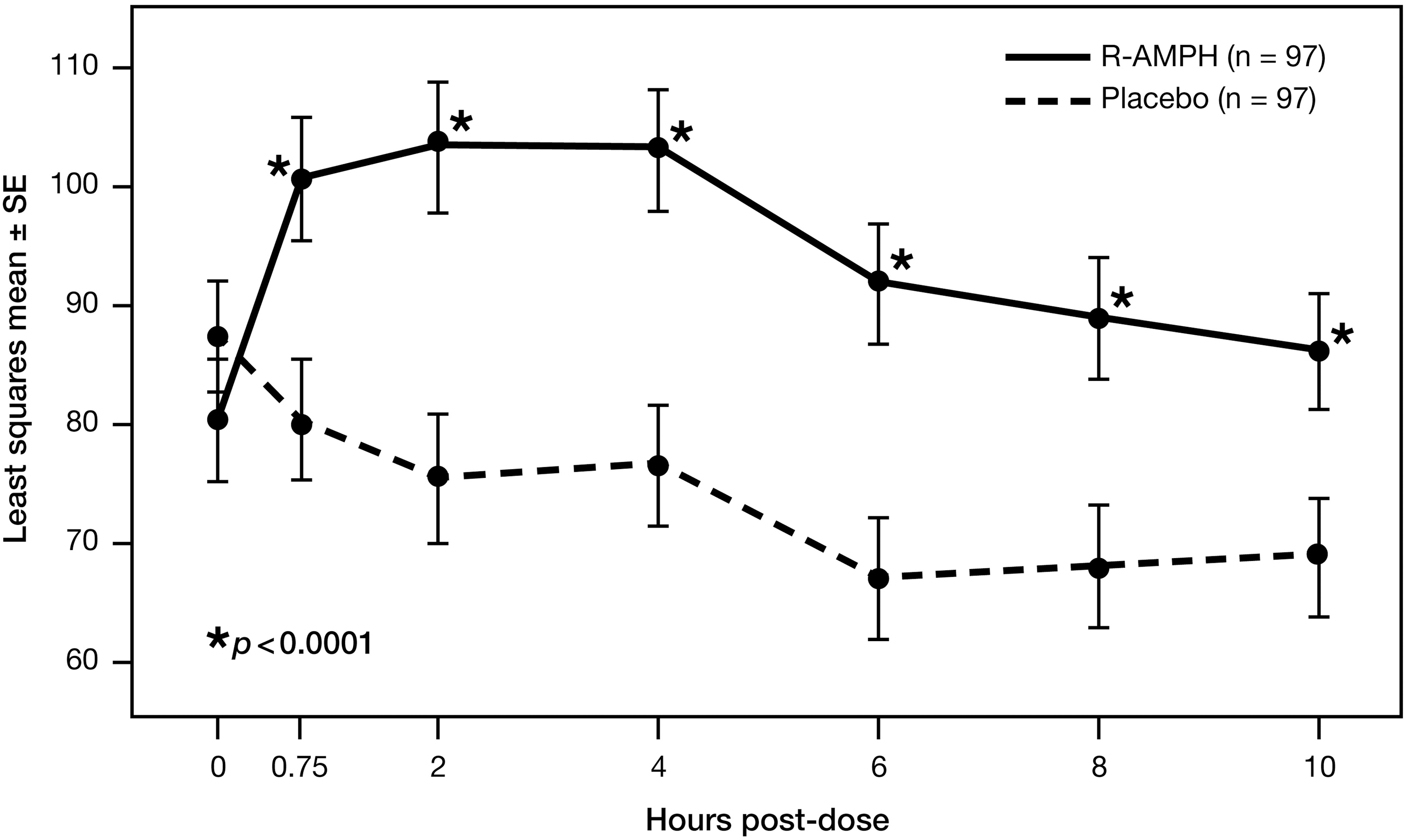
PERMP number of problems correct over time by treatment group. PERMP, Permanent Product Measure of Performance.
Efficacy assessments: Secondary measures during the open-label phase
During the open-label dose-titration period, mean (standard deviation [SD]) ADHD-RS-IV total scores decreased (improved) from 40.8 (7.64) at baseline to 13.0 (7.32) at the end of the 8-week study period (Fig. 6). Mean (SD) changes from baseline showed improvement of ADHD symptoms at each visit through the 8 weeks: From −7.4 (8.91) at week 1 to −27.8 (8.95) at week 8 for total score, from −3.8 (4.93) to −14.1 (5.54) for hyperactivity/impulsivity score, and from −3.5 (4.73) to −13.7 (5.06) for inattentiveness score. Eighty-five subjects (87.6%) were considered responders, defined as ≥50% improvement in the ADHD-RS-IV total score by that time point. When ADHD-RS-IV total scores were analyzed by subject age, gender, and race, mean changes from baseline showed improvement of ADHD symptoms over time through at least week 7 of the open-label phase, with results similar to those for the ITT population as a whole.
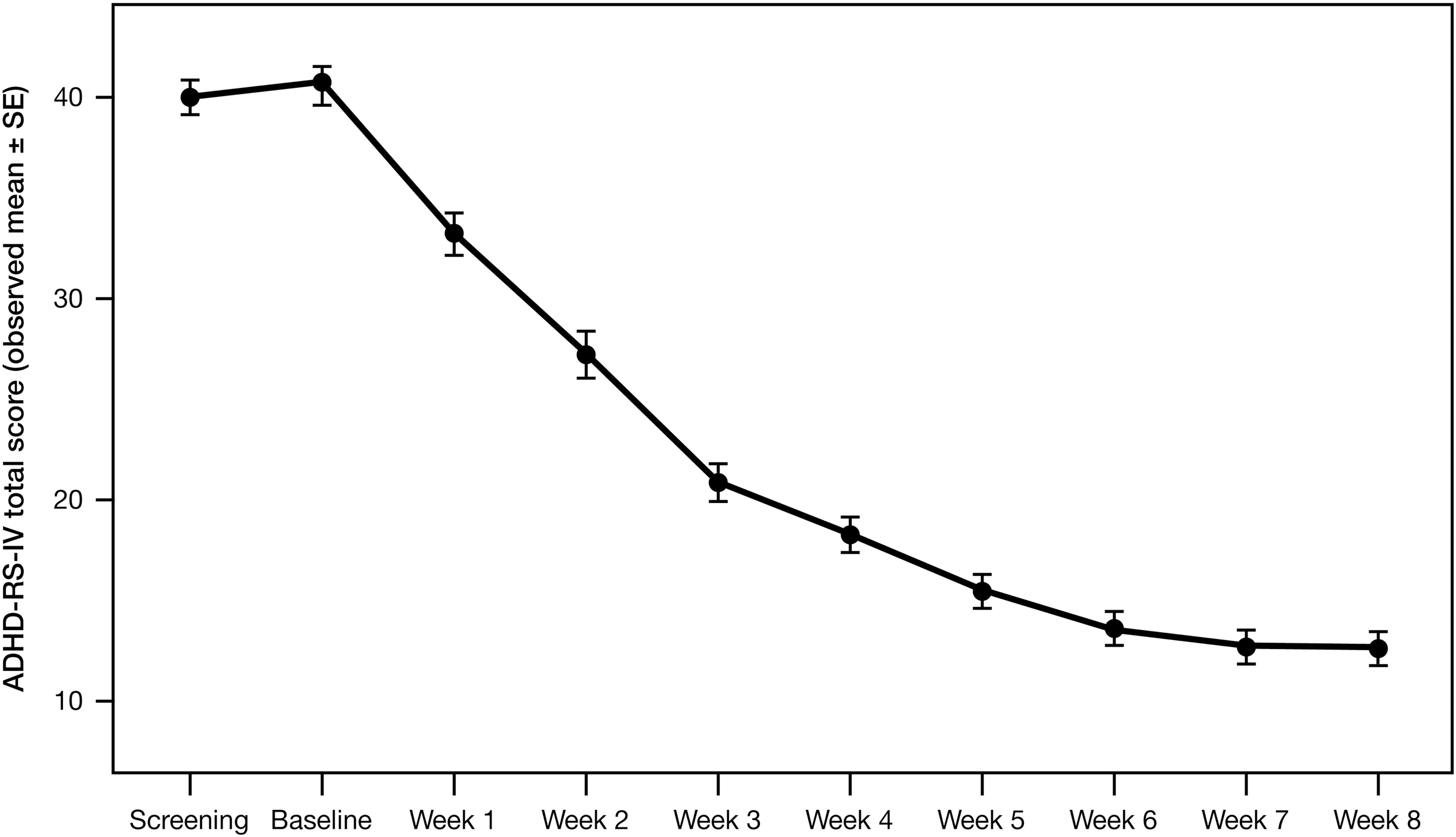
Mean (SE) ADHD-RS-IV total scores in the open-label phase of the study. ADHD-RS-IV, ADHD-Rating Scale IV.
At the screening visit, 49.5% of subjects were considered moderately ill, as rated by the CGI-S score of 4; 37.1% were considered markedly ill (CGI-S score of 5) and 10.3% were considered severely ill (CGI-S score of 6). Only 3.1% of subjects were considered mildly ill (CGI-S score of 3). By the end of the 8-week open-label phase, 33.0% of subjects were considered normal, not at all ill (CGI-S score of 1), 26.8% were borderline ill (CGI-S score of 2), 37.1% were mildly ill, 3.1% were moderately ill, and no subjects were classified as markedly or severely ill. Mean (SD) CGI-S scores decreased from 4.6 (0.70) at baseline to 2.1 (0.91) by the end of the open-label phase (Fig. 7). Based on the CGI-I measure of disease improvement relative to baseline, subjects continued to improve throughout the 8-week open-label phase. By week 4, 100% of subjects showed improvement (42.3% very much improved, 42.3% much improved, and 15.5% minimally improved). By week 8, 71.1% were very much improved, 23.7% were much improved, and 5.2% were minimally improved (Fig. 8). Mean (SD) CGI-I scores improved from 3.3 (0.94) at the end of open-label week 1 to 1.3 (0.58) at the end of open-label week 8.
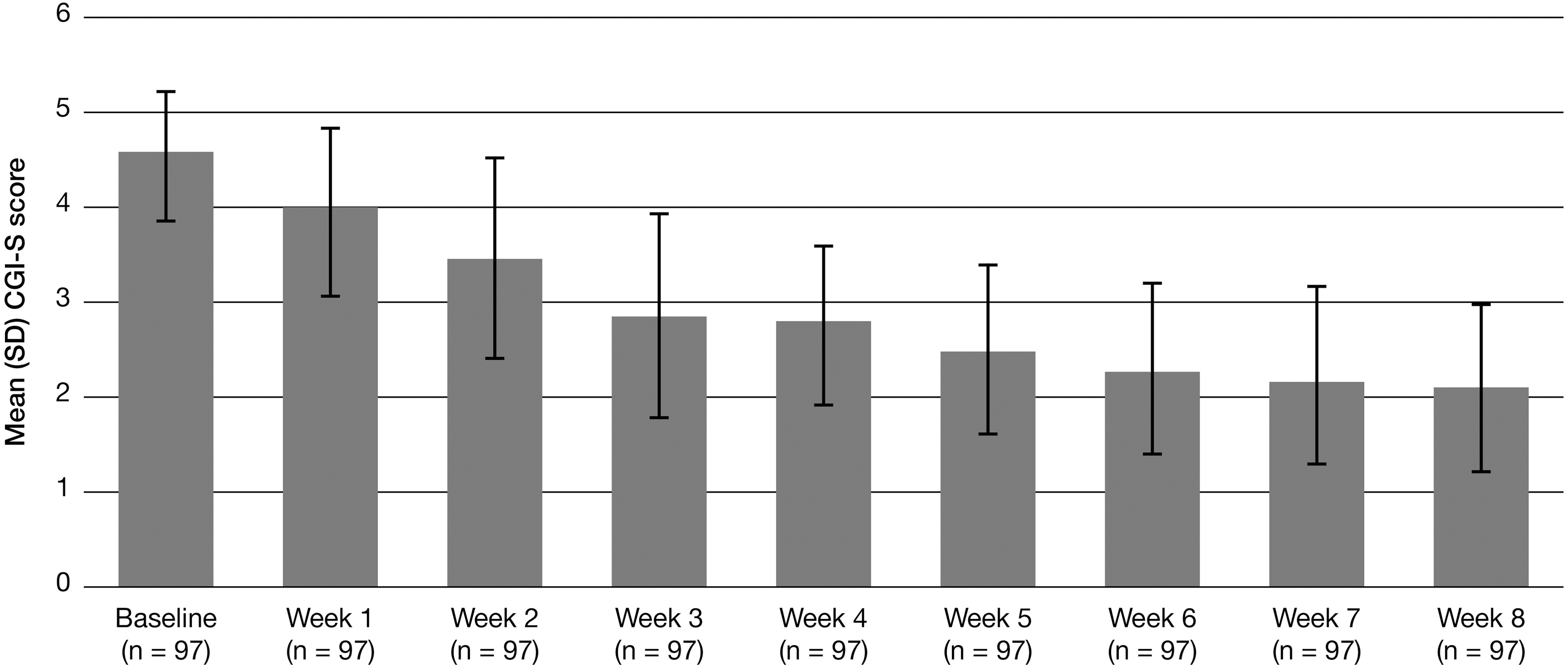
Mean (SD) CGI-S scores in the open-label phase of the study. The CGI-S classifies current disease state as follows: 1=normal, not at all ill; 2=borderline ill; 3=mildly ill; 4=moderately ill; 5=markedly ill; 6=severely ill; 7=among the most extremely ill subjects; CGI-S, Clinical Global Impressions–Severity.
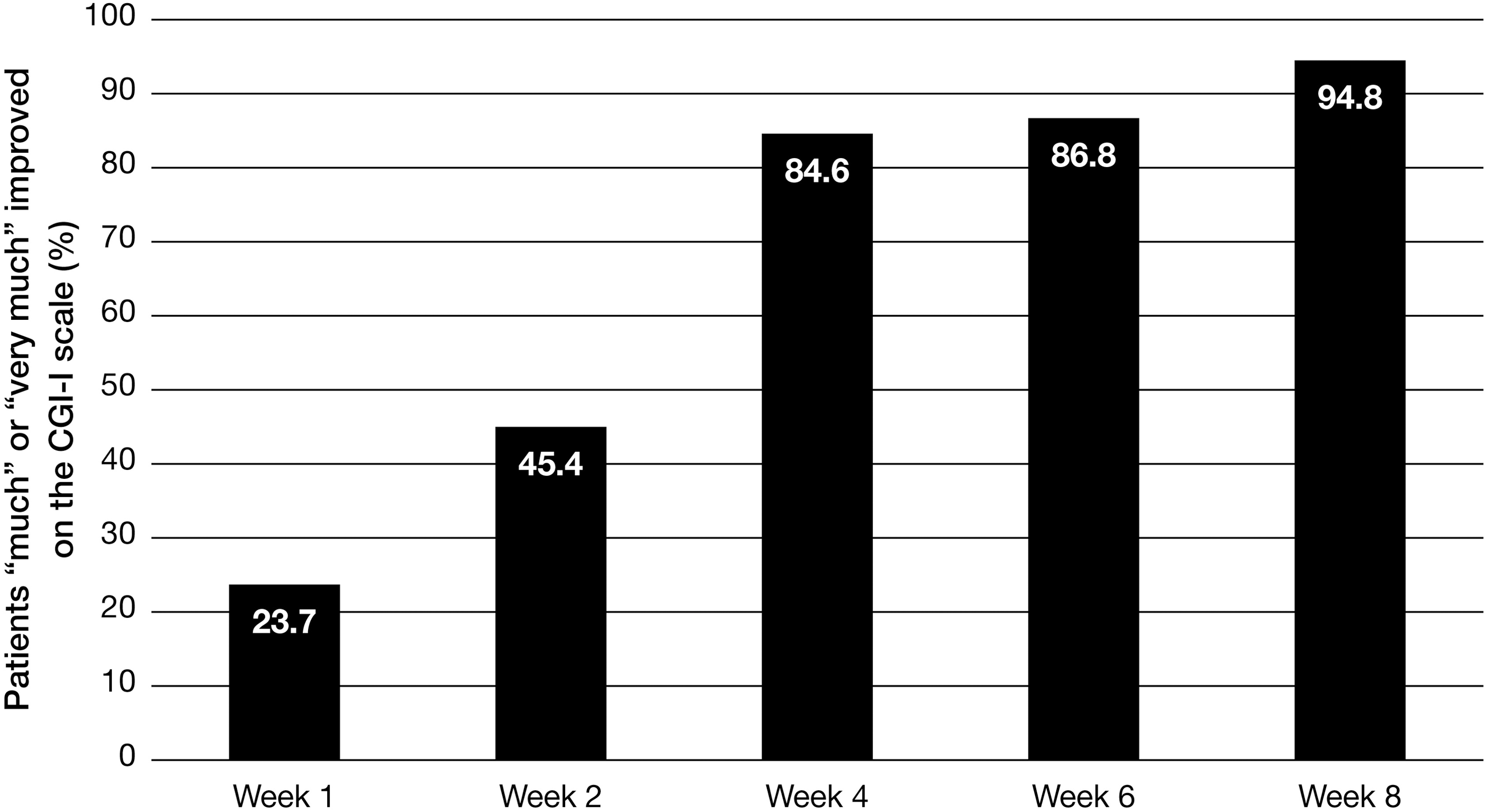
Percentage of subjects either “much” or “very much” improved based on the CGI-I scale in the open-label phase of the study. The CGI-I classifies subject improvement as follows: 1=very much improved; 2=much improved; 3=minimally improved; 4=no change; 5=minimally worse; 6=much worse; 7=very much worse; CGI-I, Clinical Global Impressions–Improvement.
Using the Conners 3-P for parent/guardian assessment, baseline scores were above average (worse) for the categories of inattention, hyperactivity/impulsivity, learning problems, executive functioning, aggression, and peer relationships. Mean (SD) improvements from baseline were observed at week 4 for each of the individual Conners 3-P categories and these improvements continued through week 8: Inattention −16.6 (14.75) at week 4 and −22.0 (16.72) at week 8, hyperactivity/impulsivity −15.6 (14.54) and −23.3 (15.63), learning problems −8.3 (9.51) and −13.7 (12.39), executive function −16.4 (14.03) and −21.4 (14.84), aggression −7.6 (13.81) and −12.1 (16.15), and peer relationships −7.5 (18.23) and −11.9 (19.87).
Safety assessment
Ninety-six (91.4%) of the 105 subjects in the enrolled safety population were exposed to R-AMPH for >56 days during the study. The mean (SD) length of exposure to the study medication was 60.2 (11.73) days, with 53.9 (10.03) days during the open-label phase and 7.0 (0.23) days during the double-blind crossover phase. The mean (SD) daily dose of R-AMPH was 16.7 (4.77) mg for the enrolled safety population and 17.2 (4.55) mg for the randomized safety population (double-blind phase). The mean (SD) and median final dose for the randomized safety population were 23.4 (8.18) mg and 20.0 mg, respectively.
Of the 105 subjects in the enrolled safety population, 69 (65.7%) experienced a total of 253 TEAEs during the open-label phase of the study (Table 3) and 59 (56.2%) experienced treatment-related AEs. The most commonly experienced TEAE was decreased appetite (27.6% of subjects, all judged by the investigator to be treatment related), followed by upper abdominal pain (14.3%, 13.3% treatment related) and irritability (14.3%, 11.4% treatment related). Severe TEAEs were reported for two subjects (1.9%) during the open-label phase: Initial severe insomnia, considered by the investigator to be related to the study drug, which lasted for 10 days and resulted in premature discontinuation; and severe insomnia, considered related to the study drug, which lasted for 1 day. No serious AEs or deaths were reported during the study. In the enrolled safety population, five subjects (4.8%) had TEAEs that led to premature discontinuation of study drug (irritability, affect lability, initial insomnia, acarodermatitis, and rash, each experienced by a different one of the five subjects). An additional enrolled subject withdrew from the study before randomization due to an adverse event (irritability), but this subject was not included in the enrolled safety population due to the absence of any postbaseline safety assessment.
TEAE, treatment-emergent adverse event.
The occurrence of TEAEs in the randomized safety population during the double-blind crossover treatment phase is summarized in Table 4. The most frequently reported TEAEs were similar to those observed during the open-label treatment for the enrolled safety population, but all TEAEs occurred at frequencies of ≤4.1% within each treatment group. Psychiatric disorders (including insomnia in 3.1% and dysphoria in 2.1%) occurred in 7.2% of subjects when receiving R-AMPH during the double-blind phase and in none of the subjects when receiving placebo. During the double-blind treatment phase, no severe TEAEs or serious adverse events were reported, and no subject prematurely discontinued study drug due to a TEAE.
TEAEs, treatment-emergent adverse events.
All subjects had clinically insignificant or normal findings at screening on hematology and chemistry parameters and physical examination. Evaluation of vital signs for the enrolled safety population during the open-label phase of the study showed a mean (SD) increase in systolic blood pressure from baseline to the final open-label visit of the study of 2.9 (8.98) mmHg, a mean (SD) increase in diastolic blood pressure of 4.5 (8.23) mmHg, and a mean (SD) increase in pulse rate of 6.7 (12.72) bpm. During the open-label phase, TEAEs related to vital signs were reported for two subjects—elevated heart rate in one instance and increased blood pressure in the other instance—with resolution in both cases. During the double-blind phase of the study, mean changes from baseline in systolic and diastolic blood pressure were similar when subjects were taking R-AMPH and when they were taking placebo. Eight subjects (8.2%) while taking R-AMPH and three subject (3.1%) while taking placebo had one systolic blood pressure measurement >95th percentile; two subjects (2.1%) while taking R-AMPH and two subjects (2.1%) while taking placebo had one diastolic blood pressure measurement >95th percentile. Mean changes in the pulse rate were minimal and highly variable (0.3 [13.23] bpm on R-AMPH and −0.1 [14.15] bpm on placebo).
During the open-label phase for the enrolled safety population, three subjects scored positive at only one visit on at least one item in the “Suicidal Ideation” section of the C-SSRS. The first of these cases occurred with the subject's mother before the baseline visit and before the study medication was started, in a context of some continuing reaction to the previous death of the subject's dog; the investigator judged this incident as not representing suicidal ideation and not related to study medication. The second case occurred at the week 1 visit due to one fleeting reactive episode in the setting of the subject being upset for being disciplined for misbehavior at school; the investigator judged this incident as being not related to the study medication and not representing an ongoing suicide risk that would prevent the subject from completing the study. In the third case, registered at the week 3 visit, the subject reported that he had said that he “wished that he were dead” for a few minutes after his girlfriend broke up with him. Upon further query, he reported that he did not want to die—he was just very upset. Another subject reported “nonsuicidal self-injurious behavior” at the week 4 visit. He hit himself in the face causing his nose to bleed briefly because he did not want to do homework. None of these occurrences was thought to be related to the study drug, and none resulted in subject discontinuation. The episodes were brief, had resolved before the visit during which they were reported, and were judged as not likely to recur. No occurrences of suicidal ideation or behavior were reported during double-blind treatment for the randomized safety population.
Discussion
In this first formal classroom study of racemic amphetamine (1:1
SKAMP-Combined scores, SKAMP-Attention subscale scores, and SKAMP-Deportment subscale scores were significantly improved at each time point tested when subjects were taking R-AMPH versus placebo, with onset of action by the first time point tested, 0.75 hour postdose, and with efficacy persisting to the last time point measured (10 hours). The greatest R-AMPH effect compared to placebo was seen at 4 hours postdose, and the greatest change from the predose score was seen at 2 hours postdose. When subjects were taking R-AMPH, they also had significantly improved (p<0.0001) PERMP scores compared to placebo at each time point tested through 10 hours, attempting more problems and answering more questions correctly.
The findings regarding the time to onset and duration of effect for a single dose of immediate-release R-AMPH can be contextualized with reference to an earlier pharmacokinetic/pharmacodynamic study of Adderall immediate-release mixed amphetamine salts (MAS, 3:1
During the 8-week open-label dose-optimization phase, disease status steadily improved, as measured by the CGI-S scale for disease severity and the CGI-I scale for disease improvement. By the end of the open-label treatment, 33.0% of subjects were “normal, not at all ill” and only 3.1% remained “moderately ill,” while 94.8% were “much” or “very much” improved. Mean changes from baseline in the ADHD-RS-IV scale showed improvement of ADHD symptoms at each visit through the 8 weeks, and at the week 8 visit, 87.6% of subjects were considered “responders” in having an improvement from baseline of ≥50% on this scale.
The TEAEs in both the open-label and double-blind phases of the trial were similar to those reported in other studies of stimulant use in the school-age population (Biederman et al. 2002; Robb et al. 2014). Overall, there were small mean increases in blood pressure and pulse. During the double-blind phase, one systolic blood pressure measurement>95th percentile was reported for 8.2% of subjects while taking R-AMPH and 3.1% while taking placebo; one diastolic blood pressure measurement >95th percentile was reported for 2.1% while taking R-AMPH and 2.1% while taking placebo.
This racemic mixture delivers
Limitations
This study excluded children with significant psychiatric and medical co-morbidities, which limits the generalizability of the results.
During the 8-week open-label dose-optimization phase of the trial, the dosing regimen was twice daily, with the first dose to be taken in the morning and the second dose to be taken 4–6 hours later. During the double-blind period, the dosing regimen was also twice daily, except for the 2 laboratory classroom days, on which only the single morning dose was administered (and not the second daily dose). The regimen of administering only the single morning dose on the laboratory classroom days was deliberately chosen to test the duration of action of R-AMPH. Consequently, the results reported for the laboratory classroom days might underrepresent the level of symptom control achieved during the open-label phase.
Trials that employ a crossover design, such as laboratory classroom studies, can be subject to both carryover and period effects. Differential carryover effects between treatments were analyzed by the statistical team, and none was reported that might affect the robustness of the statistical conclusions. A period effect could occur when a subject's performance in the second period of the crossover is affected by a change in mental or health status that has arisen independently of treatment. Because no treatment-by-period interaction appeared to be present in the current study, separate analyses of the different periods of data were not conducted to confirm the results of the combined period data.
Conclusions
This placebo-controlled study demonstrated that an optimal dose of R-AMPH effected a significant reduction in the signs and symptoms of ADHD in children aged 6–12 years in a laboratory classroom setting. When receiving R-AMPH, compared to placebo, the children had a significant (p<0.0001) mean reduction (−7.9 points) in the primary endpoint of SKAMP-Combined scores at 2 hours postdose and a significantly greater mean decrease (by 10.5 points, p<0.0001) from their predose scores. R-AMPH significantly improved (p<0.0001) SKAMP-Combined scores at each time point tested throughout the 10-hour laboratory classroom days, with effect onset 45 minutes postdose and extending to the last time point measured (10 hours). Also, the PERMP-A and PERMP-C throughout the 2 laboratory classroom days significantly improved (p<0.0001) while subjects were treated with R-AMPH. During the open-label phase, improvements were observed with R-AMPH in scores of the ADHD-RS-IV and the CGI-S and CGI-I scales. R-AMPH was generally well tolerated, with an adverse-event profile and effects on heart rate and blood pressure similar to those observed with other amphetamine products in the pediatric ADHD population.
Clinical Significance
Results from this trial indicate that R-AMPH is effective in the treatment of ADHD in subjects 6–12 years of age when dosed once or twice daily. R-AMPH received FDA approval for treatment of ADHD in children ≥3 years of age in September 2014.
Footnotes
Disclosures
Dr. Childress has been a consultant, speaker, and research support for Ironshore Pharmaceuticals, Novartis Pharmaceuticals, Pfizer, Shionogi Pharma, and Shire Pharmaceuticals; a speaker and research support for Bristol-Myers Squibb; a consultant and research support for NextWave Pharmaceuticals, Neos Therapeutics, and Rhodes Pharmaceuticals; and a research support for Arbor Pharmaceuticals, Eli Lilly, Forest Research Institute, Johnson & Johnson, Noven Pharmaceuticals, Neurovance, Otsuka Pharmaceutical, Purdue Pharma, Sunovion Pharmaceuticals, Therovance Biopharma, and Tris Pharma. Dr. Brams has been a speaker and research support for Otsuka Pharmaceuticals and Shire Pharmaceuticals; a speaker for Astra Zeneca and Sunovion Pharmaceuticals; and a research support for Arbor Pharmaceuticals. Dr. Cutler has been a consultant, speaker, and research support for AbbVie, Astra Zeneca, Janssen Pharmaceuticals, Eli Lilly, Novartis Pharmaceuticals, Otsuka Pharmaceutical, NextWave Pharmaceuticals, Shionogi Pharma, Shire Pharmaceuticals, and Sunovion Pharmaceuticals; and a consultant and research support for Akili Interactive, Arbor Pharmaceuticals, Johnson & Johnson, Neos Therapeutics, Neurovance, Noven Pharmaceuticals, Purdue Pharma, Rhodes Pharmaceuticals, Supernus Pharmaceuticals, Targacept, and Therovance Biopharma. Dr. Kollins has been a consultant and/or research support for Akili Interactive, Alcobra Pharmaceuticals, Atentiv, Ironshore Pharmaceuticals, Arbor Pharmaceuticals, NEOS Pharmaceuticals, Purdue Canada, Rhodes Pharmaceuticals, Shire Pharmaceuticals, Sunovion Pharmaceuticals, and Tris Pharma. Dr. Northcutt has been a research support for Akili Interactive, Arbor Pharmaceuticals, Bristol-Myers Squibb, DART Neuroscience, Forest Laboratories, Noven Pharmaceuticals, Shionogi Pharma, Shire Pharmaceuticals, Sunovion Pharmaceuticals, and Supernus Pharamceuticals. Dr. Padilla has been a speaker and research support for Eli Lilly, Forest Laboratories, and Shire Pharmaceuticals; and a research support for Arbor Pharmaceuticals, Alcobra Pharma, Merck, Noven Pharmaceuticals, Pfizer, Purdue Pharma, Sunovion Pharmaceuticals, and Takeda Pharmaceutical. Dr. Turnbow has been a consultant and research support for Sunovion Pharmaceuticals; a consultant for Novartis Pharmaceuticals; and a research support for Arbor Pharmaceuticals, Concordia Pharmaceuticals, Purdue Pharma, Shionogi Pharma, and Shire Pharmaceuticals.