
Abstract
Background:
Evidence suggests that monoacylglycerol lipase (MAGL) inhibitors can potentially treat HIV symptoms by increasing the concentration of 2-arachidonoylglycerol (2-AG). We examined a selective MAGL inhibitor ABX1431 in the context of neuroHIV.
Methods:
To assess the effects of ABX1431, we conducted in vitro and in vivo studies. In vitro calcium imaging on frontal cortex neuronal cultures was performed to evaluate the role of ABX1431 (10, 30, 100 nM) on transactivator of transcription (Tat)-induced neuronal hyperexcitability. Following in vitro experiments, in vivo experiments were performed using Tat transgenic male mice. Mice were treated with 4 mg/kg ABX1431 and assessed for antinociception using tail-flick and hot plate assays followed by locomotor activity. After the behavioral experiments, their brains were harvested to quantify endocannabinoids (eCB) and related lipids through mass spectrometry, and cannabinoid type-1 and -2 receptors (CB1R and CB2R) were quantified through western blot.
Results:
In vitro studies revealed that adding Tat directly to the neuronal cultures significantly increased intracellular calcium concentration, which ABX1431 completely reversed at all concentrations. Preincubating the cultures with CB1R and CB2R antagonists showed that ABX1431 exhibited its effects partially through CB1R. In vivo studies demonstrated that acute ABX1431 increased overall total distance traveled and speed of mice regardless of their genotype. Mass spectrometry and western blot analyses revealed differential effects on the eCB system based on Tat expression. The 2-AG levels were significantly upregulated following ABX1431 treatment in the striatum and spinal cord. Arachidonic acid (AA) was also upregulated in the striatum of vehicle-treated Tat(+) mice. No changes were noted in CB1R expression levels; however, CB2R levels were increased in ABX1431-treated Tat(−) mice only.
Conclusion:
Findings indicate that ABX1431 has potential neuroprotective effects in vitro partially mediated through CB1R. Acute treatment of ABX1431 in vivo shows antinociceptive effects, and seems to alter locomotor activity, with upregulating 2-AG levels in the striatum and spinal cord.
Introduction
Approximately 39 million people were living with human immunodeficiency virus type-1 (HIV-1) globally, with 1.3 million newly infected in 2022. 1 The advancement of combined antiretroviral therapies (cART) suppresses HIV-1 replication to undetectable levels 2 but does not eradicate the virus entirely due to its low brain penetration. 3 Low levels of viral replication and chronic immune activation still linger, leading to HIV-1-associated neurocognitive disorders (HAND).4–6 About 50% of cART-treated people living with HIV-1 (PLWH) develop some form of HAND, which includes problems with memory consolidation, attention,7–9 mood,10,11 reduced physical activity levels, 12 and increased pain sensitivity. 13 HAND is associated with synaptodendritic injury caused by inflammatory factors and viral proteins, including transactivator of transcription (Tat), released from infected and/or activated microglia/macrophages. 14
The prevalence of HAND in the cART era raises questions about treating HIV-1-related brain disorders. The endocannabinoid (eCB) system presents promising therapeutic targets for neurocognitive disorders, such as HAND, due to its role in neuroprotection and anti-inflammatory processes.15,16 Endogenous cannabinoid ligands, N-arachidonoylethanolamine (anandamide; AEA) and 2-arachidonoylglycerol (2-AG), show neuroprotective effects in several preclinical models of neurodegenerative disease, including Parkinson's disease, Alzheimer's disease, and multiple sclerosis.17–20 In the context of neuroHIV, reviews have discussed the potential neuroprotective effects of eCB ligands.21,22 AEA and 2-AG mainly target cannabinoid type-1 and -2 receptors (CB1R and CB2R) and are rapidly degraded by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively. As direct application of AEA and 2-AG have short durations of action, 23 the strategy of inhibiting their hydrolytic enzymes possesses considerable promise.24,25
MAGL not only serves as the major 2-AG degradative enzyme but also is a major rate-limiting enzyme for producing arachidonic acid (AA). 26 Accordingly, the MAGL inhibitor, JZL184, dampens inflammation in the central 27 and peripheral 28 nervous system by decreasing 2-AG metabolism and concomitantly reducing the production of its primary metabolite AA, as well as the further downstream proinflammatory prostaglandins.29,30 Moreover, JZL184 prevents gp-120-induced synapse loss through CB2R activation and downregulates production of inflammatory cytokines. 29
The highly selective MAGL inhibitor, MJN110, displays neuroprotective effects in vitro, by reducing neuronal hyperexcitability and restoring dendritic arborization complexity, and in vivo, by mitigating Tat-induced neurocognitive alterations. 31 These findings indicate that MAGL inhibitors represent a viable target for treating HAND. ABX1431 (renamed AG06466) is another highly selective MAGL inhibitor, which has undergone ∼16 clinical trials that have been terminated or completed32,33 and is considered safe to use.
ABX1431 significantly increases 2-AG concentration in the brain and produces antinociceptive effects in rats 34 and also showed anticonvulsant effects in a mouse model of Dravet Syndrome by increasing hippocampal 2-AG concentration. 35
Given that ABX1431 is a highly selective MAGL inhibitor with demonstrated safety in clinical trials, we examined its effects in the context of neuroHIV. Accordingly, the present study aims to determine the potential neuroprotective effects of ABX1431 against HIV Tat-induced neurotoxicity in vitro and its acute effects on various behavioral outcomes and the eCB system using the HIV Tat transgenic mouse model.
Materials and Methods
Experiments and procedures described below were approved by the University of North Carolina at Chapel Hill and conducted following the National Institute of Health Guide and Use of Laboratory Animals.
Primary neuronal culture and microglia conditioned media
Primary neuronal cultures (Fig. 1) were obtained from the frontal cortex (FC) of embryonic day 17 C57BL/6J mice (Charles River, Raleigh, NC) as previously described.36,37 Cultures were incubated in neurobasal media supplemented with appropriate nutrients at 37°C in a humidified atmosphere containing 5% CO2. All experiments were conducted at days in vitro (DIV) 21.

Overall outline of the study.
Primary microglial cultures were derived from whole brain of C57BL/6J (Charles River) pups at 0–1 postnatal days as described. 38 After the treatments on DIV14, microglia conditioned media (MCM) were collected and frozen at −20°C. The MCM dilution used for the calcium (Ca2+) imaging experiments was 1:9.
Live cell calcium imaging
Live fluorescence cell imaging was conducted on the cultured neurons using Zeiss Axio Observer Z.1 inverted microscope at 20×s objective (Carl Zeiss). Intracellular calcium ([Ca2+]i) was measured by treating neurons with a cell-permeant calcium indicator fura-2AM (kd=145 nM; 2.5 μM; Molecular Probes) as previously described. 37 Neurons were incubated with appropriate treatments for 30 min prior imaging. Tat (100 nM) was bath applied to the cultures at the 1-, 3-, and 5-min mark and the excitation pattern was recorded for a total of 15 min. For the MCM experiments, 1 mL of diluted MCM was applied to the FC neuronal cultures at 1 min. The [Ca2+]i production was calculated as previously described. 39 At least three independent experiments were performed for each treatment. For quantitative analysis of ([Ca2+]i levels, 12–15 neurons were randomly selected per treatment per experiment.
Treatments
For in vitro experiments, primary FC neuronal and microglial cultures were treated with HIV-1 Tat1–86 (100 nM; ImmunoDiagnostic; clade 267B). Tat 100 nM concentration was used based on previous data from our laboratory.31,37,38 The concentrations of ABX1431 were based on previous activity-based protein profile studies. 34 At DIV14 microglial cultures were incubated with ABX1431 for 1 h before Tat treatment. Treatment with CB1R antagonist rimonabant (100 nM; Tocris) and CB2R antagonist SR144528 (100 nM; Tocris) was done 30 min before the ABX1431 treatment. Tat was added to cultures and was allowed to incubate for 24 h before media collection.
For behavioral experiments ABX1431 (No. 26023; Cayman Chemical, Ann Arbor, MI) was dissolved in 1:1:18 mixture of ethanol:kolliphor:saline to obtain a 4 mg/kg solution. Based on previous studies,34,40 it has been determined that oral gavage of ABX1431 at 4 mg/kg significantly increased the 2-AG concentration in the mouse brain; therefore, ABX1431 dose of 4 mg/kg was selected for all the in vivo studies. Administration of vehicle or ABX1431 was done through oral gavage acutely 4 h before behavioral testing, and was randomized in all experiments.
Animals
Doxycycline (DOX)-inducible, brain-restricted HIV-1IIIB Tat1–86 transgenic male mice (∼7–9 months of age, N=28) were used and developed on a hybrid C57BL/6J background.41,42 Transgenic Tat(+) and control Tat(−) mice were fed a special DOX chow containing 6 mg/g DOX (Envigo) for 4 months before the experiment and were housed on a reversed 12-h light/12-h dark cycle.
Behavioral procedure
All the behavioral experiments were blinded for treatment and genotype.
Tail-flick and hot plate assay
ABX1431 4 mg/kg or vehicle were administered to the mice and spontaneous nociception was assessed 4 h posttreatment as previously described 43 (Fig. 1). For tail-flick, the distal 1/3 of the tail was placed in a water bath maintained at 56°C±0.1°C. The latency to remove the tail from the bath was recorded. Hot plate was conducted immediately following the tail-flick assay. Mice were placed on the hot plate maintained at 55°C±0.1°C and removed immediately after withdrawing or licking a paw and the time was recorded. A maximum cutoff latency of 10 sec and 15 sec was used for tail-flick and hot plate, respectively, to prevent tissue damage.
Locomotor activity
Mice were habituated to the locomotor activity chambers 24 h before the experiment and then placed into the experimental chambers as previously described 44 (ENC-307W; 22×18 cm floor; MED Associates). Locomotion was recorded for 10 min using night vision cameras. Videos were analyzed using AnyMaze software.
eCB and eicosanoid analysis
Following locomotor activity, animals were sacrificed by isoflurane-induced anesthesia followed by rapid decapitation. Prefrontal cortex (PFC), striatum, and spinal cord were dissected and snap frozen in liquid nitrogen. Endogenous cannabinoid ligands, including AEA, 2-AG, N-oleoylethanolamide (OEA), N-palmitoylethanolamide (PEA), and AA were quantified from samples of the right hemisphere using ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) as previously described.31,45
Western blot analysis
Western blot analysis was carried out on the same three central nervous system (CNS) regions from the left hemisphere as previously described.46,47 Briefly, tissue samples were homogenized on ice in Pierce™ RIPA lysis and extraction buffer (Thermo Scientific) with Halt™ protease and phosphatase inhibitor cocktail. BCA assay was performed on the lysates to determine the protein concentration. Proteins were denatured at 85°C for 10 min and equal amount of protein (20 μg/lane) was resolved at 120 volts for 1.5 h and transferred to nitrocellulose membrane at 1–4°C and 100 volts for 1 h. The membranes were incubated with Intercept blocking buffer at room temperature for 1 h followed by primary antibodies overnight at 4°C and secondary antibody for 1 h the next day.
The membranes were imaged and analyzed in Empiria studio software (LI-COR Biosciences). The following antibodies were used, primary antibodies: anti-CB1R (rabbit polyclonal; Proteintech), anti-CB2R (rabbit polyclonal; AbClonal), and anti-GAPDH antibody (mouse monoclonal; Abcam) as a housekeeping protein. Dilutions for primary are 1:1000, except GAPDH and secondary antibodies are 1:10,000 dilution.
Data represent the fold-change with respect to a control sample represented on all blots and normalized to the housekeeping gene GAPDH.
Statistical analysis
All data are presented as mean±the standard error of the mean. In vitro data were analyzed using analyses of variance (ANOVAs). Animal data sets were analyzed by two-/three-way ANOVAs with time (1, 3, 8, 10 min) as a within-subjects factor and/or treatment (two levels: Vehicle, ABX1431) and genotype [two levels: Tat(−), Tat(+)] as between-subjects factors. All ANOVAs were followed up with Tukey's post hoc test when appropriate. An alpha level of p≤0.05 was considered significant for all statistical tests (Table 1). SPSS Statistics 28 and Prism GraphPad 8.0 were used for data analysis and data graphing, respectively.
Statistical Data for All the Studies Conducted
Bolded values denote significance at p≤0.05.
2-AG, 2-arachidonoylglycerol; AA, arachidonic acid; AEA, N-arachidonoylethanolamine; ANOVA, analysis of variance; CB1R, cannabinoid type-1 receptor; CB2R, cannabinoid type-2 receptor; MCM, microglia conditioned media; NA, not applicable; PFC, prefrontal cortex; SC, spinal cord; Str, striatum.
Results
In vitro studies
To study the role of ABX1431 in the context of neuroHIV, we conducted in vitro Ca2+ imaging studies using primary FC neuronal cultures at DIV21. Tat (100 nM) application onto neuronal cultures significantly increased [Ca2+]i concentration, which was significantly reduced by ABX1431 pretreatment at all concentrations (10, 30, 100 nM; p<0.0001; Fig. 2A). Similarly, to understand the role of MAGL inhibition and Tat in mediating microglial neurotoxicity, we assessed [Ca2+]i responses of FC neuronal cultures to MCM derived from ABX1431 and/or Tat-treated microglia in the presence and absence of CB1R (rimonabant) and CB2R (SR144528) antagonists.
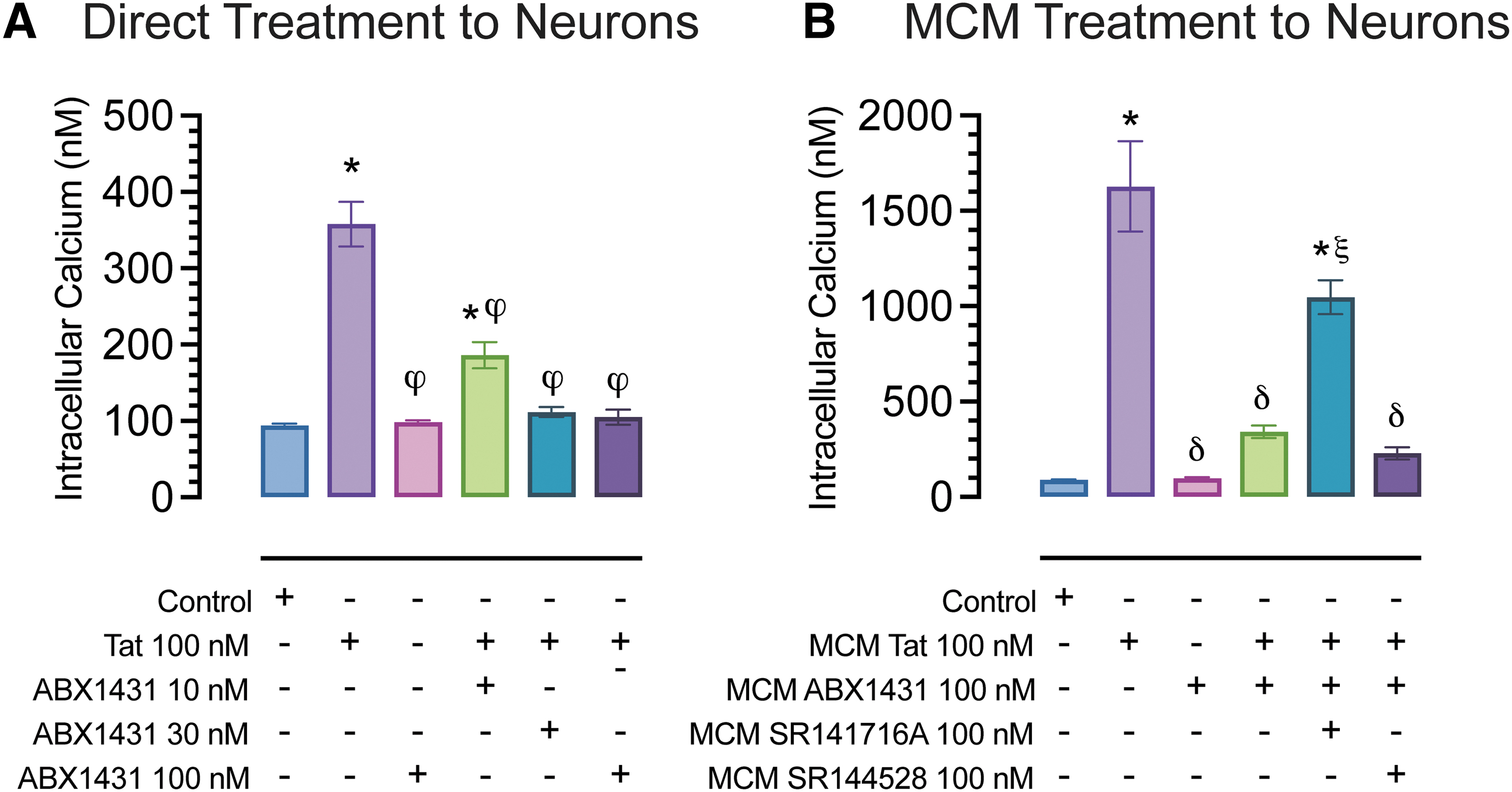
Calcium changes in cultured FC neurons at DIV21.
Application of MCM Tat onto neurons resulted in a significant increase in [Ca2+]i, (p<0.001), which was blocked completely when applying MCM cotreated with ABX1431 and Tat (p<0.0001). MCM derived from cotreatment of ABX1431, Tat, and rimonabant only showed partial neuroprotective effects. Whereas MCM derived from cotreatment of ABX1431, Tat, and SR144528 showed protective effects similar to that of MCM derived from Tat and ABX1431 treatment, which suggests the protective effects of ABX1431 are partially CB1R mediated only and not by CB2R (Fig. 2B).
In vivo studies
Tat(−) and Tat(+) male mice were randomly assigned to vehicle or ABX1431-treated groups. No differences were observed in body mass between groups (Fig. 3A). The hot plate and tail-flick assays were conducted 4 h after ABX1431 oral gavage (4 mg/kg) to access heat-evoked nociception. Hot plate assesses supraspinal-related nociception, and no drug or genotype-dependent effects were observed (Fig. 3B). For tail-flick, spinal-related nociception, acute ABX1431 significantly increased pain latencies in Tat(+) male mice only (p=0.002; Fig. 3C).

Effect of acute ABX1431 at 4 mg/kg on body mass and spontaneous evoked nociception and locomotor activity.
Furthermore, to evaluate effects of acute ABX1431 (4 mg/kg) on motor function, we assessed locomotor activity (Fig. 3D) following the hot plate assay. Over the period of 10 min, there was a significant decrease in locomotion (p<0.001); however, when treatment effect was considered, ABX1431 significantly altered this decrease in locomotor activity over the 10 min (p=0.008). Interestingly, ABX1431 increased the overall locomotor activity (p=0.041). Similarly, there were significant differences in average speed (Fig. 3E), with a significant decrease in speed over time (p<0.001), which was altered by acute ABX1431 (p=0.047). Furthermore, acute ABX1431 exposure significantly increased overall speed (p=0.009). Finally, there were no changes in the overall immobility due to ABX1431 (Fig. 3F), but increased immobility over time (p<0.001) was significantly altered by ABX1431 (p<0.001), which was specifically noted for the 3-min time point, where ABX1431 increased the immobility in both the genotypes.
CNS levels of eCB and receptors
Following behavioral experiments, CNS regions (PFC, striatum, spinal cord) were taken, quantified, and analyzed by two-way ANOVA for eCB and eicosanoid levels through UPLC-MS/MS, including AEA, 2-AG, PEA, OEA, and AA (Fig. 4 and Supplementary Table S1). Lipid concentrations (nmol/g) significantly varied between CNS regions (Fig. 4). The order of 2-AG levels was PFC < striatum < spinal cord. Importantly, acute ABX1431 significantly upregulated 2-AG levels in the striatum and spinal cord regardless of genotype (p<0.05). For AEA, no significant drug or genotype effect was noted with the order of AEA levels being PFC > striatum > spinal cord. Finally, AA levels were comparable between CNS regions, with only the striatum showing a drug×genotype interaction (p=0.031). AA was significantly upregulated in vehicle-treated Tat(+) males compared with Tat(−) males (p=0.009).

Endocannabinoid levels (nmol/g) in prefrontal cortex, striatum, and spinal cord.
Regions from the left hemisphere were used for western blot analysis (Fig. 5). No alterations were found for CB1R protein expression levels (Fig. 5B). A significant treatment×genotype effect was seen for CB2R expression levels in the striatum (Fig. 5C).

Cannabinoid receptors 1 and 2 expression levels in prefrontal cortex, striatum, and spinal cord. CB1R and CB2R expression levels were assessed in PFC, striatum, and spinal cord of vehicle and acute ABX1431-treated Tat transgenic mice through western blot.
CB2R was upregulated in ABX1431-treated Tat(−) mice (p=0.004) and no changes were seen in Tat(+) mice. Raw unedited western blot images have been added to Supplementary Figure S1.
Discussion
HIV-1 Tat activates the glutamatergic N-methyl-D-aspartate (NMDA) receptors36,48–50 and interacts with the lipoprotein receptor-related protein, 51 causing an increase in neuronal intracellular calcium, dendritic damage, and synapse loss.49,52–54 2-AG and AEA have been previously reported to counter these effects of Tat by downregulating intracellular calcium concentrations. 47 In contrast to AEA, 2-AG is present at higher concentrations in the brain and acts as a full agonist at both CB1R and CB2R.55–58 The 2-AG is primarily hydrolyzed by MAGL into AA and glycerol, the primary source for cyclooxygenase-2 mediated production of proinflammatory prostaglandins.26,59 Therefore, the neuroprotective effects of MAGL inhibitors may be due to reduced neuroinflammatory responses because of decreased production of AA and its prostaglandin metabolites, while simultaneously elevating 2-AG to enhance eCB receptor signaling. 59
Most in vivo studies have used shorter Tat exposure times of ≤2 weeks, but here we chose 16-week exposure to model the chronic elevation reflected human disease. While some studies have reported Tat-induced allodynia, hyperalgesia and damaged nerve fibers,60–62 other studies have found either no effects 63 or even decreased pain sensitivity. 13 The effect of Tat on nociception is complex and not clearly understood. 13 Pain sensitivity also depends on the length of Tat exposure, with studies showing hyposensitivity following 3 weeks of Tat induction 13 and hypersensitivity after over a month of Tat exposure.60,64
The hyposensitivity may be due to neuronal dysfunction with initial Tat exposure, which is later reversed into hypersensitivity after prolonged exposure. 43 It is plausible that von Frey filaments (i.e., mechanical allodynia) may be a more sensitive assay to detect Tat-induced hypernociception compared with the hot plate or tail-flick assays. Various MAGL inhibitors have been shown to attenuate pain primarily through CB1R activation.65–69 The present study also showed that an acute ABX1431 (4 mg/kg) dose increased pain latency in Tat(+) male mice.
The mechanisms by which MAGL inhibitors exert their antinociceptive effects include the involvement of CB1R67,70,71 or CB2R 72 while others suggest the involvement of both CB1R and CB2R.71,73,74 Moreover, another study claims that the therapeutic effects of MAGL inhibitor JZL184 is anti-inflammatory and does not depend on cannabinoid receptors. 75 We did not see any alterations in CB1R levels, although 2-AG was upregulated in the striatum and spinal cord.
There is a possibility that the CB1R functional activity may be altered, which cannot be detected by western blot. On the contrary, it is also possible that acute drug treatment was insufficient for substantial 2-AG-induced CB1R receptor desensitization and subsequent internalization or loss of surface receptors as loss of CB1R is demonstrated after prolonged use or chronic exposure to MAGL inhibitors. 76 These differences in the mechanism of action of MAGL inhibitors could be explained by the transient role of CB1Rs resulting from 2-AG overload leading to desensitization of cannabinoid receptors following excessive MAGL inhibition.76,77
Motor deficits, including impaired gait, motor strength, and motor coordination, are comorbidities PLWH face78,79 leading to physical ailments and inadequate daily activity levels, even lower than most other chronic diseases. 12 Preclinical studies have revealed that the presence of Tat significantly decreases locomotion,80–83 which is believed to be due to significant changes in the synaptic organization after Tat exposure. 80 However, we did not find any genotype differences in locomotor activity and speed in our vehicle-treated mice, which is in line with some past studies.84,85
Discrepancies between studies may arise due to shorter open-field experiment duration, which was 10 min in our study and 30 min in a previously reported study, 80 or due to longer Tat exposure (6 months). 81 Perhaps studies such as grip strength may have detected genotype differences. For ABX1431, we found that acute ABX1431 significantly increased overall locomotion and speed of mice regardless of their genotype, which supports previous studies where treatment with MAGL inhibitors, including JZL184 and MJN110, increased locomotor activity.86–88
Differential effects of JZL184 and MJN110 have also been reported, in which JZL184 decreased locomotion, whereas MJN110 increased speed and total distance traveled. 71 These differences in locomotor activity in the presence of various MAGL inhibitors and doses are yet to be understood and require further investigation.
Changes in the eCB system in PLWH have been previously reported.89,90 There is little information about the levels of endogenous ligands such as AEA and 2-AG in neuroHIV. Although we did not see any changes in AEA levels in the CNS regions, ABX1431 significantly increased 2-AG in striatum and spinal cord of both genotypes. The increase in 2-AG levels in the spinal cord may account for the increased latency in the tail-flick assay in ABX1431-treated mice.
Additionally, AA, known to be a key player in inflammatory responses,91–93 was significantly upregulated in vehicle-treated Tat(+) mice, suggesting an increase in proinflammatory mediators in the presence of Tat. 94 This is supported by a recent study where AA cascade and eicosanoid production were upregulated in the brains of HIV-1 gp120 mice. 95 As ABX1431 is a MAGL inhibitor, we showed 2-AG levels to be upregulated, but AA levels did not change. The modest increase of 2-AG and lack of changes in AA may be due to the route of administration of ABX1431, as previous studies have shown that intraperitoneal injection of JZL184 caused a large increase in 2-AG and decrease in AA concentration. 96
Also, the dose of JZL184 used in the previous study 96 (40 mg/kg) was much higher than the present study, which may be responsible for a decrease in AA concentration. It is also important to note that although MAGL is the primary enzyme responsible for the metabolism of 2-AG, serine hydrolases, α/β hydrolase domain 6 and 12 (ABHD6 and 12) are two other enzymes that play a minor but significant role in 2-AG hydrolysis,97,98 which may be responsible for the increase in AA levels. It is also important to consider that lack of changes in AA may be due to the Tat-induced proinflammatory effect, which releases AA from cell membrane phospholipids through mechanism such as tumor necrosis factor and toll-like receptors, which are independent from MAGL. 99 Finally, we did not see any genotype-based changes in the CB1R and CB2R levels, except for CB2R expression in the striatum, which was downregulated in vehicle-treated Tat(+) mice when compared with Tat(−) mice.
The lack of CB1R alterations in neuroHIV has been reported previously90,100,101; however, most studies89,100,102 have shown an upregulation in CB2R in the context of neuroHIV due to its inducible nature upon microglial cell activation leading to its anti-inflammatory function.103–105 Interestingly, we found that acute ABX1431 administration led to upregulated CB2R expression in the striatum of Tat(−) mice without affecting Tat(+) mice, which is the opposite of previous research findings where desensitization of the GPCRs was caused by 2-AG overload.43,76,77 The exact relationship between the effects of MAGL inhibitors on the eCB system and its signaling pathway is yet to be understood, and warrants further investigation.
This study has several limitations; first, we only used male mice in the study. Literature has shown that women are more vulnerable to HAND symptoms.106–111 Therefore, it is critical to understand the role of MAGL inhibitors using female mice along with the consideration of estrous cycle in future investigations. Second, the HIV Tat transgenic model used in this study is a well-established neuroHIV model; however, it only expresses one of many viral proteins in HIV. Therefore, this is an important consideration when generalizing findings to PLWH, as viral proteins may interact, target various signaling pathways, and modify the CNS compared with a single protein. 94
Lastly, the use of DOX to induce Tat expression in the Tat transgenic mouse is another limiting factor as DOX on its own has shown to exert neuroprotective effects and might mask some of the effects seen in this study.112,113 To minimize bias and confound, all animals, including the Tat(−) mice were fed the same DOX chow throughout the study. Furthermore, it would be important to investigate the chronic effects of ABX1431 exerted on nociception, locomotor activity, and eCB levels, along with other comorbidities of HAND, such as anxiety and memory.
Conclusion
In conclusion, the present study demonstrates in vitro neuroprotective effects of a potent MAGL inhibitor on selected behaviors in a neuroHIV mouse model. Specifically, ABX1431 downregulated Tat- and MCM Tat-induced intracellular calcium release through a CB1R-mediated mechanism. Moreover, acute ABX1431 displayed moderate antinociceptive effects and increased locomotor activity in our HIV Tat transgenic mouse model. While ABX1431 caused an increase in 2-AG levels in the striatum and spinal cord, only CB2R was found to be upregulated in the striatum. Mechanistic studies are required to investigate how these alterations in the eCB system by MAGL inhibitors correlate with the behavioral outcomes in the context of neuroHIV. Based on previous clinical studies, ABX1431 is safe to use and therefore repurposing it for the treatment of HAND would facilitate future clinical trials.
Footnotes
Acknowledgment
The authors would also like to acknowledge the work of animal care technician Patrick G. Stutts for maintaining the welfare of our animals through the studies.
Authors' Contributions
Conceptualization: B.J.Y.-S., B.M.I.-J., A.H.L., and S.F.; methodology: B.J.Y.-S., H.P.R., K.M.B., H.D., J.L.P., K.J.R., and S.F.; validation: B.J.Y.-S., H.P.R., and S.F.; formal analysis: B.J.Y.-S. and S.F.; investigation: B.J.Y.-S., H.P.R., and S.F.; resources: S.F.; data curation: B.J.Y.-S., H.P.R., J.L.P., and S.F.; writing—original draft preparation: B.J.Y-S., and S.F.; writing—review and editing: B.J.Y.-S., H.P.R., B.M.I.J., A.H.L., J.L.P., K.J.R., and S.F.; visualization: B.J.Y.-S., H.P.R., and S.F.; supervision: B.J.Y.-S. and S.F.; project administration: B.J.Y.-S., and S.F.; funding acquisition: S.F., A.H.L., and B.M.I.-J.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This research was funded by the National Institute on Drug Abuse (NIDA), R01 DA055523 (S.F.), R21 DA041903 (S.F.), T32 DA007244 (H.P.R.), and P30 DA033934 (A.H.L.). B.M.I.-J. was supported by Japan Society for Promotion of Science (JSPS) Fellowship for Overseas Researchers (P17388), Kakenhi Grant-in-Aid for JSPS Fellows (17F17388), and Kakenhi Grant for Scientific Research (21K06399).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.