
Abstract
In the striatum, adenosine A2A receptors (A2AR) are mainly expressed within the soma and dendrites of the striatopallidal neuron. A predominant proportion of these striatal postsynaptic A2AR form part of the macromolecular complexes that include A2AR-dopamine D2 receptor (D2R) heteromers, Golf and Gi/o proteins, and the effector adenylyl cyclase (AC), subtype AC5. The A2AR-D2R heteromers have a tetrameric structure, constituted by A2AR and D2R homomers. By means of reciprocal antagonistic allosteric interactions and antagonistic interactions at the effector level between adenosine and dopamine, the A2AR-D2R heterotetramer-AC5 complex acts an integrative molecular device, which determines a switch between the adenosine-facilitated activation and the dopamine-facilitated inhibition of the striatopallidal neuron. Striatal adenosine also plays an important presynaptic modulatory role, driving the function of corticostriatal terminals. This control is mediated by adenosine A1 receptors (A1R) and A2AR, which establish intermolecular interactions forming A1R-A2AR heterotetramers. Here, we review the functional role of both presynaptic and postsynaptic striatal A2AR heterotetramers as well as their possible neuroprotective role. We hypothesize that alterations in the homomer/heteromer stoichiometry (i.e., increase or decrease in the proportion of A2AR forming homomers or heteromers) are pathogenetically involved in neurological disorders, specifically in Parkinson's disease and restless legs syndrome.
Introduction: Precoupling and Oligomerization
Precoupling of G protein-coupled receptors (GPCR) with G proteins and signaling molecules and receptor oligomerization are two concepts that are changing our classical views of GPCR physiology and pharmacology.1,2 The classical view of freely moving molecules of GPCR, G proteins, and effectors in the plasma membrane, which establish ligand-guided associations and dissociations by random collision (collision-coupling mode), is being replaced by a ligand-induced rearrangement of precoupled molecules. 1 Furthermore, the classical view of single GPCR units is being replaced by oligomeric functional units. A large number of experimental data support the view of a common functional building block constituted by two GPCR units (homodimer) and one heterotrimeric G protein (with its preferred α and βγ subunits). 3
A GPCR heteromer is defined as “a macromolecular complex composed of at least two (functional) receptor units (protomers) with biochemical properties that are demonstrably different from those of its individual components.” 4 It is becoming apparent that a common quaternary structure of GPCR heteromers is a heterotetramer, formed by two different GPCR homodimers coupled to their cognate G proteins.1,3 This has been recently reported as the most probable quaternary structure of two striatal adenosine A2A receptor (A2AR) heteromers, the postsynaptic A2AR-dopamine D2 receptor (D2R) heterotetramer and the presynaptic adenosine A1 receptor (A1R)-A2AR heterotetramer.4,5 Here, we review the functional role of both heterotetramers as well as their possible neuroprotective role. Thus, we hypothesize that alterations in the homomer/heteromer stoichiometry (i.e., increase or decrease in the proportion of GPCR protomers forming homomers or heteromers) are pathogenetically involved in neurological disorders, specifically in Parkinson's disease (PD) and restless legs syndrome (RLS).
Functional Role of the Postsynaptic Striatal A2AR-D2R Heterotetramer
In the striatum, A2AR and D2R are mostly expressed within the soma and dendrites of the striatopallidal neuron, one of the two subtypes of GABAergic efferent neurons that constitute more than 95% of the striatal neuronal population. 6 A predominant proportion of these striatal postsynaptic A2AR and D2R form part of the macromolecular complexes that include A2AR-D2R heterotetramers, Gs (more properly Golf subtype, but Gs/olf proteins are referred as Gs proteins, for short) and Gi/o proteins (Gi proteins, for short), and the effector adenylyl cyclase (AC) subtype AC5 (Fig. 1).5,7 These complexes provide the frame for the canonical Gs-Gi antagonistic interaction at the AC level (Fig. 1A). This canonical interaction implies the ability of an activated Gi-coupled receptor to inhibit a Gs-coupled receptor-mediated AC activation, 8 thus requiring the simultaneous respective interaction of the Ras GTPase domains of the α subunits of the Gs and Gi proteins with the C2 and C1 catalytic domains of AC5. 9 For instance, through this interaction, activation of D2R leads to an inhibition of A2AR-mediated activation of AC (Fig. 1A). In addition, the precise quaternary structure of the A2AR-D2R heterotetramer provides the frame for the ability of A2AR ligands to establish allosteric interactions with D2R ligands, not only between agonists but also between antagonists. 10 When either an A2AR agonist or an A2AR antagonist binds to the orthosteric sites of the A2AR homodimer within the A2AR-D2R heterotetramer, they both produce an allosteric decrease in the affinity and efficacy of D2R ligands (Fig. 1B). On the contrary, when an A2AR agonist and an A2AR antagonist bind simultaneously to the two orthosteric sites of the A2AR homodimer, they counteract each other's effects. 10
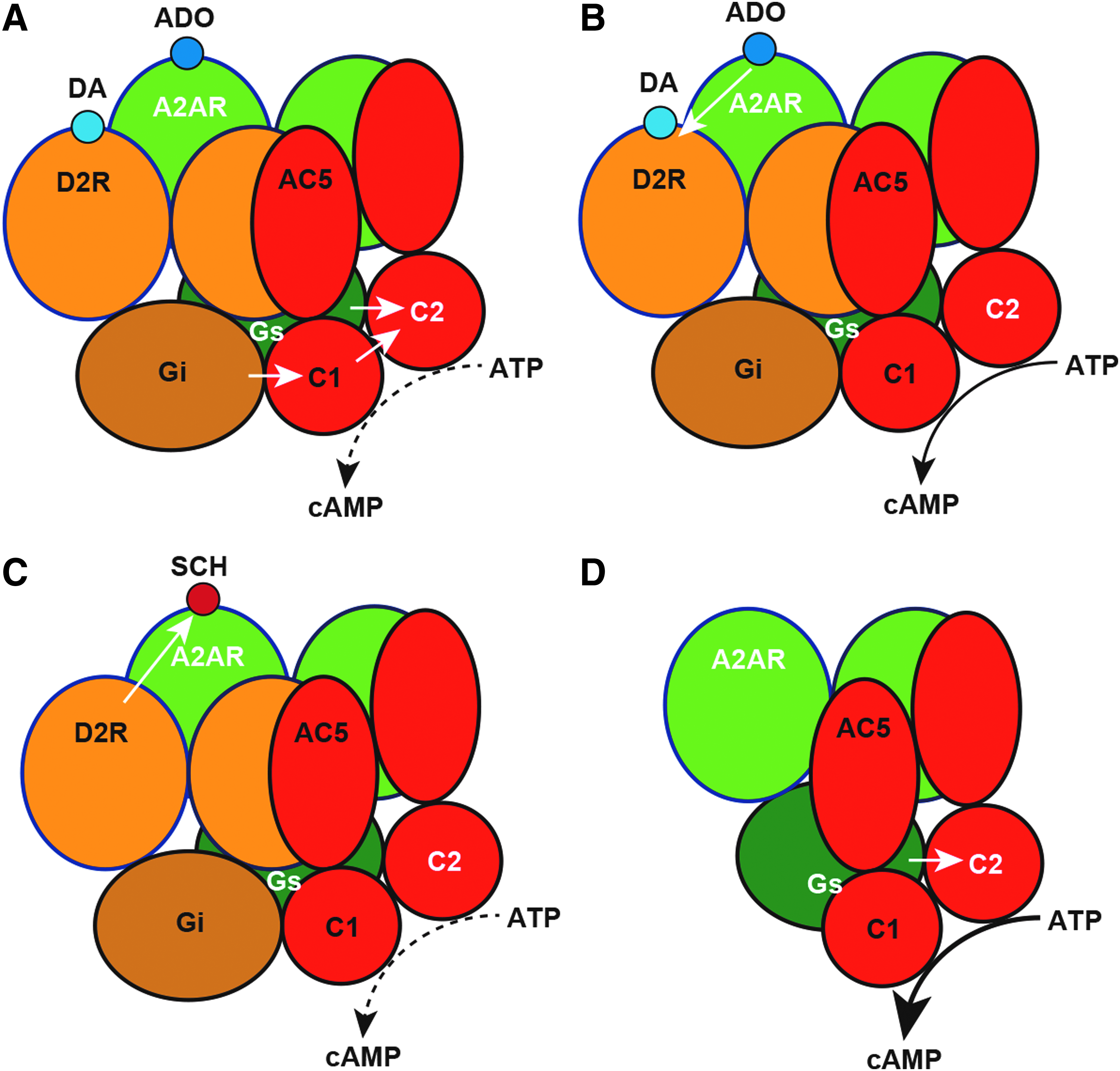
Functional and pharmacological properties of the A2AR-D2R heterotetramer.
Furthermore, there is also evidence for significant reciprocal allosteric interactions, by which D2R agonists negatively influence A2AR agonist binding.11,12 This would be expected by the experimentally supported evidence that indicates that, according to Kenakin, “the allosteric energy flow is reciprocal in nature; that is, if the allosteric modulator changes the affinity of the agonist, then the agonist will also change the affinity of the modulator in a like manner.” 13 Different efficacies where observed when evaluating the negative allosteric modulation of A2AR agonist binding by several D2R agonists, some of them clinically used as antiparkinsonian agents. Apomorphine produced a significantly stronger modulation of A2AR agonist binding than pramipexole and rotigotine. 12
We have recently shown that the canonical Gs-Gi antagonistic interaction, and therefore the functional integrity of the striatal A2AR-D2R heterotetramer-AC5 complexes, depends on the integrity of very specific interactions between transmembrane domains (TMs) of the receptors, which constitute transmembrane heteromeric and homomeric interfaces, as well as between TMs of the receptors and putative TMs of AC5. 2 The methodology involved a peptide-interfering approach based on the use of synthetic peptides with the amino acid sequence of all TMs of both receptors and AC5. The synthetic peptides are fused to the HIV transactivator of transcription (TAT), which determines the orientation of the peptide when inserted in the plasma membrane. The TAT-TM peptides are thus screened for their ability to destabilize oligomerization detected by bimolecular fluorescence complementation (BiFC). In this technique, two complementary halves of a fluorescent molecule are separately fused to two different GPCR protomers or to a GPCR protomer and AC5. This peptide-interfering approach is providing a very powerful tool that allows the characterization of all TM interfaces involved in GPCR oligomerization.2,5,10
The homomeric TM/TM interactions were the same for both the A2AR and the D2R homodimers: A TM6/TM6 interface irrespective of cotransfection with the other molecularly different receptors. 2 The A2AR-D2R heteromeric interface involved a symmetrical TM4–5/TM5–4 interface, but its integrity also depends on a concomitant strong electrostatic interaction between the C-terminal domain of the A2AR (A2AR-CT) and the intracellular end of TM5 of the D2R. Thus, disruption of the interaction between A2AR-CT and D2R-TM5 has been shown to significantly reduce A2AR-D2R heteromerization.2,10,14–16 The allosteric interactions between A2AR and D2R ligands have also been shown to depend on the integrity of the transmembrane and intracellular heteromeric interfaces.10,14–17
With its reciprocal canonical and allosteric interactions, the A2AR-D2R heterotetramer acts as an integrative molecular device, which facilitates the switch between the activation and inhibition of the striatopallidal neuron 7 : A preferential A2AR versus D2R activation leads to an increase in neuronal activity determined by an A2AR-mediated AC5 activation, facilitated by the allosteric counteraction of D2R signaling. 7 Conversely, a preferential D2R over A2AR activation leads to a decrease in neuronal activity determined by a D2R-mediated activation of phospholipase C and facilitated by the canonical and reciprocal allosteric interactions, thus switching off the A2AR-mediated AC5 activation. 7 Indeed, the activation of the striatopallidal neuron leads to withdrawal behaviors and, when intense, to motor arrest and catalepsy. On the contrary, striatopallidal neuron inhibition leads to psychomotor activation.7,18
Functional Role of the Presynaptic Striatal A1R-A2AR Heterotetramer
Adenosine plays an important modulatory role driving the function of corticostriatal terminals. This control is mediated by adenosine A1R and A2AR, which establish intermolecular interactions forming A1R-A2AR heterotetramers.5,19 Interestingly, these heteromers work as an adenosine concentration-dependent switch, which determines the opposite effects of adenosine on glutamate release depending on a predominant A1R or A2AR activation within the heteromer.19,20 Importantly, the affinity of adenosine is higher for A1R than for A2AR. Therefore, low concentrations of adenosine primarily activate A1R, which induces an inhibition of glutamate release. On the contrary, on high concentrations of adenosine, such as those obtained under strong glutamatergic transmission and the consequent neuronal and glial ATP release and conversion of ATP on adenosine, 21 the simultaneous activation of A2AR leads to an allosteric modulation within the heteromer, with a reduction of affinity and efficacy of adenosine for the A1R.5,19 Under these conditions A2AR signaling prevails and an adenosine-mediated facilitatory glutamate release is achieved. Importantly, the molecular mechanisms of these interactions are beginning to be understood and they are related to the specific quaternary structure of the A1R-A2AR heterotetramer. 5 According to the reciprocal nature of allosterism (see the Functional Role of the Postsynaptic Striatal A2AR-D2R Heterotetramer section), 13 we could also expect that A1R ligands allosterically modulate A2AR ligands in the A1R-A2AR heterotetramer, but this needs still to be demonstrated.
The A1R is a classical Gi-coupled receptor. However, different to the A2AR-D2R heterotetramer, the A1R-A2AR heterotetramer does not sustain a canonical Gs-Gi antagonistic interaction at the AC level and the A2AR-mediated AC activation is unopposed by A1R activation. 5 This correlates with the A2AR-mediated striatal glutamate release unopposed by A1R activation. 19 Particularly striking was the finding that deletion of A2AR-CT enables the canonical Gs-Gi antagonistic interaction in cells coexpressing A1R and A2AR. 5 Different from the A2AR-D2R heterotetramer (see the Functional Role of the Postsynaptic Striatal A2AR-D2R Heterotetramer section), A2AR-CT deletion did not disrupt A1R-A2AR heteromerization, indicating its lack of participation on the stabilization of the quaternary structure of the A1R-A2AR heterotetramer. 5 Interestingly, when compared to the A2AR-D2R heterotetramer, different TM/TM homomeric and heteromeric interfaces were observed using TAT-TM peptides in BiFC experiments: TM4–5/TM5–4 homomeric interfaces for both A2AR and A1R homomers and a TM5–6/TM6–5 for the A2AR-A1R heteromeric interface. 5 Molecular dynamics computational analysis already predicts that GPCR homodimers display several possible homodimeric TM interfaces. 22 Our studies indicate that the preferred homomeric interfaces in a GPCR heterotetramer are determined by the heteromeric partner.
An important implication from the study of the role of GPCR heteromers is that they are required for the functional expression of Gi-coupled receptors in the modulation of certain subtypes of AC. More specifically, those AC isoforms can be inhibited by the α subunits of Gi/o proteins, including AC1, AC5, and AC6. 23 Artificially, Gi-coupled receptors can inhibit forskolin-induced AC activation, but we have demonstrated that to inhibit a Gs-coupled receptor-mediated AC5 activation, they need to be part of a GPCR heterotetramer. This has been so far shown for the A2AR-D2R, A1R-D1R, and D1R-D3R heterotetramers, where destabilization of their heteromeric interface leads to the disruption of the canonical Gs-Gi antagonistic interaction at the AC level.2,24,25 The A1R-A2AR heterotetramer represents a particular case, since, because of interference with the A2AR-CT, the Gi-coupled A1R cannot counteract the Gs-coupled A2AR-mediated AC activation. 5 Nevertheless, it is quite well established that the main mechanism involved in the modulation of neurotransmitter release by Gi-coupled receptors, including A1R, is by a decrease in the probability of neurotransmitter release through a direct inhibition of Gi protein βγ subunits of N- and P/Q-type voltage-dependent calcium channels.26,27 On the contrary, Gs protein-coupled receptors can produce and increase in the probability of neurotransmitter release by an AC-cAMP-PKA-dependent mechanism.28,29 Therefore, the A1R-A2AR heterotetramer is particularly designed to inhibit and stimulate glutamate release by an AC-independent and AC-dependent mechanism, respectively.
In addition to providing the frame for allosterical interactions between ligands binding to their orthosteric sites and interactions at the effector level (canonical Gs-Gi antagonistic interaction), heteromerization can lead to changes in the properties of specific ligands for one of the protomers, independent of other ligands binding to the other protomer (Fig. 1C). Indeed, the proof of concept came from experiments in transfected mammalian cells, where the potencies of different selective A2AR antagonists at binding to the A2AR alone or when coexpressed either with D2R or A1R were compared. 30 Interestingly, the most dramatic finding was that the A2AR antagonist SCH442416 showed a selective low affinity for the A2AR when coexpressed with D2R compared with its affinity for the A2AR alone or coexpressed with A1R (Fig. 1C). 30 More specifically, SCH442416 showed a pronounced negative cooperativity of its binding to the A2AR homodimer within the A2AR-D2R heterotetramer.7,30
This was further demonstrated in striatal preparations from mice with conditional striatal D2R-KO, which, in contrast to wild-type mice, did not show the binding negative cooperativity of SCH442416. 7 As expected, SCH442416 showed a dissociation on its ability to produce locomotor activity (which should depend on its binding to the postsynaptic A2AR-D2R heterotetramer) versus its ability to inhibit electrically and optogenetically induced striatal glutamate release (which should depend on its binding to the presynaptic A1R-A2AR heterotetramer).7,30 Both in rats and mice, higher systemic doses were necessary to produce locomotion than those necessary to produce inhibition of glutamate release.7,30 Importantly, the preferential presynaptic profile of SCH442416 was confirmed by further studies by other research groups31,32 and was suggested to provide a therapeutic approach for conditions with increased corticostriatal transmission, such as cannabinoid use disorder. 33
The same mechanism has recently been reported for the selective significant decrease in potency of the μ-opioid receptor agonist methadone, compared with morphine and fentanyl, in the μ-opioid-galanin Gal1 receptor heteromer. 34 Since these heteromers are selectively localized in the ventral tegmental area (localization of cell bodies of dopaminergic cells involved in the processing of natural rewards), this could explain a selective dissociation of the clinical therapeutic versus euphoric/rewarding effects of methadone compared with other opioids. 34 Therefore, changes in the pharmacological properties of ligands induced by receptor heteromerization should constitute a very important strategy in the search for new therapeutic agents with further selectivity for their therapeutic versus side, unwanted effects.
Neuroprotective Role of the Postsynaptic Striatal A2AR-D2R Heterotetramer: Impact on D2R Signaling in PD
PD, the second most common age-related neurodegenerative disorder, is characterized by a progressive loss of nigrostriatal dopaminergic cells, which affects ∼1% of individuals older than 60 years. 35 PD symptoms include resting tremor, rigidity, bradykinesia, or postural instability and, in advanced stages, cognitive dysfunction and dementia. 36 Only 10–15% of PD cases classify as early-onset familial PD (recognized as having a first-degree affected family member), 37 while the remaining cases are idiopathic, thus indicating a key role for nongenetic and environmental factors in PD pathogenesis. Indeed, exposure to environmental toxins (such as pesticides, solvents, heavy metals, and other pollutants) can cause dopaminergic cell death. 38 Furthermore, oxidative stress, mitochondrial dysfunction, and inflammation play key roles in PD pathopsyciology.39,40 Accordingly, PD treatments are mainly based on the use of prodopaminergic drugs, intending to restore neurotransmitter deficiency on the loss of nigrostriatal dopaminergic neurons. 41 However, this pharmacological treatment also presents many undesired effects, specially on chronic consumption. Thus, although L-DOPA has been proved very efficacious in the treatment of PD symptoms, its long-term treatment tends to lose efficacy and also to induce severe collateral motor effects (dyskinesia and rigidity) and psychiatric symptoms. 42 Interestingly, most epidemiological studies support a protective benefit of habitual drinking of caffeinated beverages,43–45 thus suggesting a neuroprotective role of adenosine receptors in PD.
The striatum plays a central role in PD pathophysiology. Indeed, dopamine depletion throughout the course of PD progression is followed by a substantial cellular and molecular striatal remodeling. Postmortem studies using PD necropsies revealed a reduced dendritic length and spine density in striatal GABAergic neurons. 46 The caudal part of the putamen, where PD-associated dopamine deficits are more pronounced, shows the highest reduction in spine density. 47 In addition, some biochemical alterations in postmortem PD brains indicate a protein homeostasis dysregulation, 48 which includes the expected downregulation of the rate-limiting enzyme in dopamine synthesis tyrosine hydroxylase 49 and the existence of the intracytoplasmic α-synuclein fibrillar aggregates that constitute the PD histopathological hallmark, the Lewy body. 48 The levels of several neurotransmitters and the expression of their receptors have been found to be altered, not without some discrepancies. 48 Thus, while the levels of dopamine are reduced in the whole striatum (caudate, nucleus accumbens, and putamen) and globus pallidus, they were not found to be altered in the subthalamic nucleus, thalamus, and substantia nigra. 50 Divergences were found when measuring the striatal D2R density, with some studies reporting that D2R is upregulated, 51 downregulated, 52 or unaltered.53–55 Interestingly, the adenosinergic system has also been found dysregulated in PD, with no changes in adenosine levels, 56 but a significant increase in A2AR density.55,57 Therefore, it is not unreasonable to expect that altered dopamine and adenosine levels and D2R and A2AR densities within the striatum of PD subjects should determine alterations in the A2AR-D2R heteromer composition and function, thus impacting PD pathophysiology.
Indeed, we could demonstrate the existence of A2AR-D2R heteromers in native tissue by using a multimethodological approach (i.e., immunoelectron microscopy, proximity ligation assay, and time-resolved fluorescent resonance energy transfer) in the striatum of control and unilateral 6-OHDA-lesioned rats (a widely used animal model of PD). 58 Interestingly, a significant reduction of the A2AR-D2R heteromer content was observed in the 6-OHDA-denervated striatum. 58 The striatal A2AR-D2R heteromer disruption observed in this PD animal model might then constitute a neuroadaptive response associated with dopaminergic denervation in PD. Future efforts should determine similar changes in the A2AR-D2R heteromer status in postmortem caudate-putamen from PD subjects. Indeed, establishing the A2AR-D2R heteromer status in PD could determine the design of selective combined pharmacotherapeutic strategies restoring the unbalanced A2AR-D2R heteromer function potentially associated with PD.
In the same PD animal model, the A2AR demonstrated a significant constitutive activity and this uncontrolled activity was blocked by caffeine and other prototypic A2AR inverse agonists. 59 Therefore, we assumed that the striatal A2AR-D2R heteromer disruption observed in this PD animal model, and the expected loss of the negative canonical and allosteric control by the D2R was behind the gain of A2AR constitutive activity, which could be involved in the striatal neurodegeneration of PD (Fig. 1D). This provides a rationale for the use of A2AR antagonists in PD and for fostering the research of mechanisms that could restore an unbalanced expression of A2AR homomers versus A2AR-D2R heteromers in this disease.
Neuroprotective Role of the Presynaptic Striatal A1R-A2AR Heterotetramer: Reduction of A1R Signaling in RLS
RLS is a very common neurological disorder, characterized by periodic, rest-induced, mostly nocturnal, movement-responsive urge to move the legs or periodic leg movements during sleep (PLMS) and hyperarousal.60–63 The deficits of sensorimotor integration that promote PLMS and hyperarousal are interrelated, and adenosine seems to be a very important pathogenetic link. Several preclinical and clinical data indicate the existence of a hypoadenosinergic state secondary to brain iron deficiency (BID) as an initial pathogenetic mechanism in RLS.63–65 It has been demonstrated that BID in the experimental animal leads to a generalized downregulation of A1R. 66 A1R downregulation in the cortex and in the areas of origin of the ascending arousal systems could explain the hyperarosusal.63,64 On the contrary, A1R downregulation in the striatum could explain an increased sensitivity of corticostriatal terminals, which has recently proposed to be a main mechanism responsible for the deficits of sensorimotor integration that promote PLMS.65,67 In fact, corticostriatal glutamatergic terminals are targets for the drugs most often prescribed in RLS, the dopamine receptor agonists pramipexole and ropinirole and the α2δ-ligand gabapentin. 67
The possible key role of A1R downregulation in corticostriatal glutamatergic terminals in the sensorimotor symptomatology of RLS was significantly supported by preclinical and clinical experiments. First, we predicted that equilibrative nucleoside transporter inhibitors, by increasing the striatal extracellular levels of adenosine (which would facilitate the binding probability of adenosine to the lower expressed A1R), could provide a new therapeutic approach for RLS. In fact, we recently reported encouraging results with the nonselective ENT1/ENT2 inhibitor dipyridamole in an open trial with RLS patients. 68 At the preclinical level, as predicted, an A1R antagonist produced hypersensitivity of corticostriatal terminals and reduced the frequency of optogenetic stimulation necessary to induce corticostriatal glutamate release. 65 Furthermore, dipyridamole, by increasing the striatal extracellular concentration of adenosine and the activation of presynaptic A1R, was able to counteract optogenetic-induced glutamate release in both naive rats and in rats with BID. 65 Finally, as we also expected, this effect of dipyridamole was counteracted by the A1R antagonist. 65
Similar to what seems to occur with postsynaptic A2AR in PD, the pathogenesis of RLS could involve a change in the stoichiometry of A2AR and A1R forming and not forming heterotetramers. A1R downregulation would mean a relative increase in A2AR/A1R expression ratio, which should lead to a relative decrease of A1R-A2AR heteromers and a relative increase of A2AR not forming heteromers. Unopposed by the A1R signaling in the heterotetramer, the relative increase in A2AR expression and constitutive activity should be indirectly responsible for the A1R downregulation-mediated increased sensitivity of the corticostriatal glutamatergic terminals. This would predict that A2AR antagonists that target A2AR not forming heteromers could also be of therapeutic use in RLS. In fact, we have previously shown that the nonselective pre/postsynaptic A2AR antagonist MSX-3 significantly counteracts optogenetically induced corticostriatal glutamate release. 69
Adenosine A1R-A2AR heteromers have also been demonstrated in cortical astrocytic cultures, where they modulate GABA transport by GAT-1 and GAT-3 transporters. 70 The same as the presynaptic striatal A1R-A2AR heteromer, it acts as an adenosine concentration-dependent switch, which, in this case, determines the opposite effects of adenosine on GABA transport depending on a predominant A1R or A2AR activation within the heteromer. 70 If also functionally present in the striatum, it could be involved in the pathogenesis of RLS. A reduction of A1R density would imply an increased A2AR-mediated GABA transport and, therefore, a reduction in the extracellular levels of the inhibitory neurotransmitter GABA.
Concluding Remarks
GPCR heteromers are changing classical views of GPCR physiology and pharmacology. First, they are providing a better understanding of interactions between different neurotransmitters and exogenous ligands, based on their ability to convey canonical interactions at the effector level and allosteric interactions between endogenous and exogenous ligands. This, for instance, provides the rationale for the use of A2AR antagonists in PD, which increase the therapeutic index of L-DOPA.71,72 Second, GPCR heteromerization determines potential pharmacodynamic differences between exogenous compounds, such as the A2AR antagonist SCH442416, with its preferential binding to the presynaptic A1R-A2AR versus postsynaptic A2AR-D2R heterotetramers. This provides the rationale for the use of SCH442416-like compounds in conditions with an excess of corticostriatal signaling, for instance, in substance use disorders.31–33,73 Finally, we believe this review provides sufficient background that supports a pathogenetic role of postsynaptic striatal A2AR-D2R heterotetramers in PD and presynaptic A1R-A2AR heterotetramers in RLS, based on their decreased expression versus the expression of the Gs-coupled A2AR not forming heteromers, which, free from the antagonistic control of the Gi-coupled D2R and A1R, facilitate an increased neuronal activation and presynaptic glutamate release, respectively. In both cases, A2AR antagonists that could preferentially target A2AR not forming heteromers could constitute a successful therapeutic strategy.
Footnotes
Acknowledgments
This work was supported by the intramural funds of the National Institute on Drug Abuse, FEDER/’Ministerio de Ciencia, Innovación y Universidades–Agencia Estatal de Investigación’ (SAF2017-87349-R) and ISCIII (PIE14/00034), the Catalan government (2017 SGR 1604) and “Fundació la Marató de TV3” (Grant 20152031), FWO (SBO-140028).
Author Disclosure Statement
No competing financial interests exist.