
Abstract
Compared to routine diagnostics, screening for pathogens in outbreak situations, with or without intentional release, poses demands on the detection technology to not only indicate the presence of already known causative agents but also novel and unexpected pathogens. The metagenomic approach to detecting viral pathogens, using unbiased high-throughput sequencing (HTS), is a well-established methodology with a broad detection range and wide applicability on different sample matrices. To prepare a sample for HTS, the common presequencing steps include homogenization, enrichment, separation (eg, magnetic separation), and amplification. In this initial study, we explored the benefits and drawbacks of preprocessing by sequence-independent, single-primer amplification (SISPA) of nucleic acids by applying the methodology to artificial samples. More specifically, a synthetic metagenome was divided into 2 samples, 1 unamplified and 1 diluted, and amplified by SISPA. Subsequently, both samples were sequenced using the Ion Torrent Personal Genome Machine (PGM), and the resulting datasets were analyzed by using bioinformatics, short read mapping, de novo assembly, BLAST-based taxonomic classification, and visualization. The results indicate that even though SISPA introduces a strong amplification bias, which makes it unsuitable for whole-genome sequencing, it is still useful for detecting and identifying viruses.
The development and implementation of high-throughput sequencing (HTS) technologies during the past 5 years have opened up new possibilities for investigating complex clinical and microbial samples using the emerging methodology metagenomics. Metagenomics is defined as the study of a collection of genomes from a mixed community of organisms and usually refers to the study of microbial communities. 4 Viral metagenomics focus on the viral fraction of a metagenomic sample. Originally relying on tedious cloning and Sanger sequencing for characterization, 5 the metagenomic field advanced greatly with the introduction of HTS. 6 The new ability to directly assess the complete genetic diversity in a sample also allowed more specific HTS-based metagenomic approaches to be developed for rapid detection of unexpected new pathogens and characterization of unculturable microorganisms.7-9 These approaches are usually comprised of several common presequencing steps including sample homogenization, target enrichment, and amplification.10-12 In this study, we focus on a specific step of the methodology for viral metagenomics: the whole-genome amplification.
The material obtained from a clinical sample after enrichment by reducing the background of the host (eg, by nuclease treatment or ultracentrifugation) is often in such small quantity that amplification is required before sequencing. This can be achieved without the need to know the genome composition of the sample a priori by using a nonspecific amplification technique such as multiple displacement amplification (MDA).13-15 However, MDA is time consuming and has on several occasions been demonstrated to introduce biases into the amplified material.16-18 Sequence-independent, single-primer amplification (SISPA) was used early on as an alternative full-genome amplification method for unspecific detection of viruses. 19 As such, SISPA was considered an alternative when methodologies for genome presequencing amplification in metagenomics were developed.20,21
Synthetic metagenomes are a predefined combination of microorganism in an in vitro constructed sample that can be used to test the various methodologies applied for sample preparation, sequencing, and data processing. This is done to validate and show proof of concept for metagenomic approaches and changes to established methodologies. 22 This is advantageous since most obtained metagenomes are inherently complex due to the amount of microbes, sample matrices interference, and bias introduced by analysis. It is therefore technically impossible, or at least extremely challenging, to accurately depict the composition of a metagenome. Thus, the focus has been on partial microbiomes and, more recently, synthetic metagenomes for the subsequent analysis of changes in laboratory procedures and sequence methodology.
The Ion Torrent Personal Genome Machine (PGM) is a benchtop HTS platform that works by scalable semiconductor sequencing. The scalability stems from the use of different sized ion sequencing chips, which can generate from 10 Mb up to 1 Gb of sequence data per run, with read lengths in the range of 100-400 bp. 23 The total output can hence be chosen according to user needs and the scale of the experiment. Applications for which the PGM is commonly used include sequencing of small genomes, sets of genes (amplicon sequencing), ChIP-seq, and selected microbiome or virome studies. 24 Compared to other HTS platforms, the PGM represents an affordable and rapid benchtop system with flexible capacity, 25 which makes it suitable for use in an outbreak situation, as recently demonstrated by Mellmann et al. 26 We therefore selected the Ion Torrent PGM as our platform for HTS in this study.
The comparisons of metagenomic datasets can be used to find likely etiological factors for specific conditions; this is most commonly achieved by comparing healthy and diseased cases. It also offers a means for evaluating methodological changes.27,28 Although bioinformatically possible, the standard approach is associated with a rather long process of analysis involving quality control of the dataset, filtering of reads, assembly of raw reads, homology search, and taxonomical mapping. 29 For assessment of methodological changes, it is more convenient to perform a mapping comparison using known genomes. With a synthetic metagenome, this can be performed quickly and will give accurate results, which can be depicted as changes in coverage (reads per base) or recovery of whole-genome sequences. 22
The present work was performed as part of the methodology and technology evaluation section of the AniBioThreat project, which dealt with detection of unknown viruses and investigating new methodologies for increased preparedness against infectious diseases in the EU. A small synthetic viral metagenome was constructed to investigate whether the use of SISPA, a commonly used method for preamplification in viral discovery,30-32 introduces artifacts that can affect the outcome of pathogen detection and retrieval of complete genomes.
Materials and Methods
Synthetic Metagenome
In total, 4 viruses with known genomic sequences were chosen for the construction of the synthetic metagenome. These 4 viruses were selected because they are important pathogens and because they are well-characterized and properly described in the literature. Care was taken to include both RNA and DNA viruses. The 4 viruses chosen were human adenovirus type 2 (Ad2), African swine fever virus (ASFV), avian paramyxoviruses type 1 (APMV-1), and avian influenza A (AIV). Both APMV-1 and AIV were kindly provided by S. Zohari after culturing in the BSL-3 laboratory at the National Veterinary Institute (SVA), Uppsala, Sweden. The genomic contents of these agents were extracted by using a combination of TRIzol and RNeasy columns (Qiagen) as previously described 33 and converted into cDNA by using Superscript First-Strand Synthesis Kit (Invitrogen). ASFV genomic DNA was kindly provided by N. LeBlanc at SVA, and Ad2 genomic DNA was generously provided by C. Svensson, Uppsala University, Uppsala, Sweden. All viral nucleic acid samples were quantified using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies) and pooled to create a representative synthetic viral metagenome with 1 virus in excess and the other 3 at lower concentrations in the background (see Table 1). To enable the effects of SISPA to be assessed, by comparing unamplified and amplified samples, a fraction of the metagenomic sample was diluted 1,000 times and amplified using SISPA as previously described. 21
Composition of the Synthetic Metagenome
Sequencing
Ion Torrent Sequencing was performed at the Uppsala Genome Center, SciLifeLabs, Uppsala, Sweden. In brief, libraries were built using the Ion Xpress plus fragment library kit (LifeTechnologies), followed by size selection using the E-Gel CloneWell system (LifeTEchnologies). The quality of generated libraries was assessed using the Agilent Bioanalyzer (Agilent Technologies) with Agilent High Sensitivity DNA Kit (Agilent Technologies), combined with Ion library quantification kit qPCR (LifeTechnologies). Approved libraries were subjected to emulsion PCR using the Ion PGM 200 Xpress Template Kit (LifeTechnologies). Final samples were each loaded onto Ion 314 chips and individually sequenced using an Ion Torrent PGM. The results were delivered in raw SFF format through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX), Uppsala University, Sweden.
Bioinformatics
The sequence data were transferred into the high-capacity computing system of the SLU Global Bioinformatics Center, where quality controls of the datasets were performed according to the PrinSeq methodology. 34 For assessment and assignment of the sequence reads, the Burrows-Wheeler Aligner (BWA)35,36 was used to align the reads toward the viral reference genomes. The resulting alignment files in BAM format were processed using the TABLET alignment viewer 37 and BamView 38 for estimation and visualization of sequence coverage.
To enable assessment of SISPA as a preamplification step for resequencing of viral genomes, the datasets were also de novo assembled using the MIRA assembler. 39 The CodonCode Aligner software (CodonCode Corporation) was used to align the assembled contigs along the reference genomes to allow the generation of longer consensus sequences from overlapping contigs. Pairwise comparisons of consensus sequences with reference genomes were performed by using the needle program of the EMBOSS package. 40
The taxonomic distribution of sequence reads was based on the results from BLASTn searches against a local copy of the NCBI's nucleotide database using NCBI's standalone BLAST package.41,42 Final taxonomic assignment and visualization were performed by using the MEGAN 4 software 43 with the following lowest common ancestor (LCA) settings: Min support 5, Min Score 50, Top Percent 10, and Min complexity 0.44. The BLAST results were manually parsed by using a custom Biopython script. 44
Results
Sequencing Results and Detection of Viral Sequences
Performing the sequencing on separate Ion 314 chips yielded approximately 36 and 63 Mbp of sequence data, with an average read length of approximately 115 bp, for the unamplified and SISPA-amplified samples, respectively. Mapping of the reads against the viral reference genomes demonstrated that we had sufficient data for detection of all viruses in both samples (see Table 2).
Number of Reads Mapped by BWA to Each Reference Genome and Estimated Average Coverage
Differences in Coverage Indicate Amplification Bias
To visualize the differences between the unamplified and SISPA-amplified samples, the coverage for each position in the Ad2 genome (the only virus with an average coverage larger than 2) was plotted (see Figure 1). This revealed that although the SISPA-amplified sample yielded almost 2 times higher average coverage (1,460 compared with 844), the distribution of the sequencing reads was strongly biased, leaving parts of the genome almost empty while other parts had a coverage of several thousand. In contrast, the unamplified sample displayed a rather even distribution of sequencing reads along the complete genome.
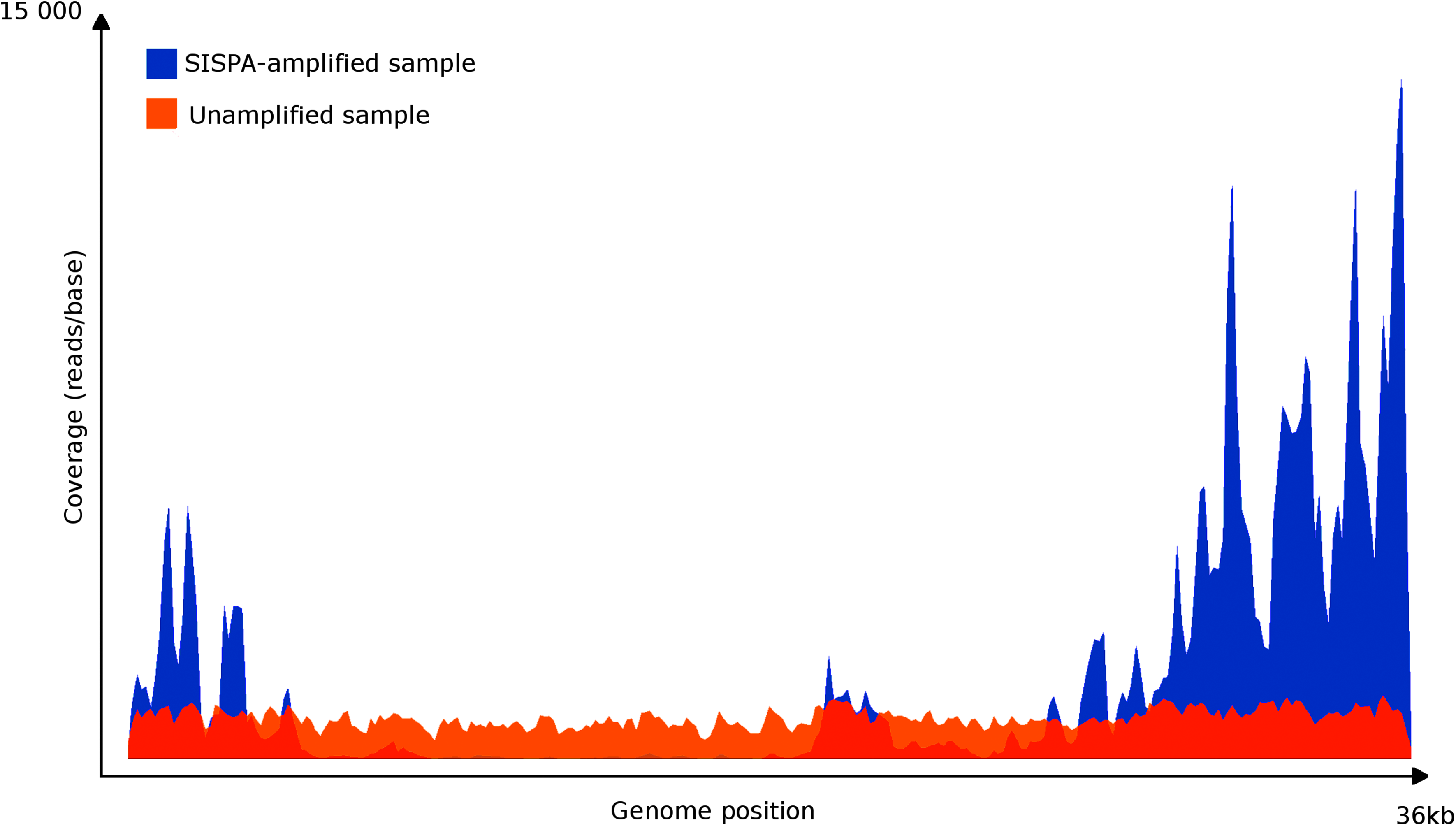
Comparison of coverage by mapping the reads to the Ad2 reference genome using Bamview. Amplified and unamplified samples are mapped together overlapping. Shown is the discrepancy between the amplified approach and the unamplified approach.
Retrieval of Whole-Genome Sequences
Sequencing reads could be assembled into contigs for all viruses present in the synthetic metagenome using MIRA with the de novo settings. However, comprehensive whole-genome coverage was available only for Ad2. A consensus sequence was produced for both the unamplified and the SISPA-amplified sample by aligning the assembled contigs with the reference Ad2 genome (GenBank AC_000007). While the nucleotide similarity with the reference genome, as shown by the Needle program, was 94.9% for the consensus sequence of the SISPA-amplified sample, the unamplified displayed a nearly identical resemblance (99.9%). The major difference was that the consensus sequence of the SISPA-amplified sample contained several gaps with a total size of approximately 1.8 kb. In contrast, the consensus sequence of the unamplified sample was only 35 bp shorter than the Ad2 reference genome, which was largely due to missing base pairs in regions with homopolymers.
Taxonomic Classification
The results from a metagenomic study are most commonly reported by classifying short reads and/or assembled contigs into taxonomic groups. All obtained sequencing reads were therefore mapped toward the NCBI nucleotide sequence database using BLASTn with default parameters. The resulting alignments were used for the assignment of reads into taxonomic groups using the software MEGAN 4. The number of reads assigned to each of the viral groups present in the synthetic metagenome are illustrated in Figure 2 and Table 3, as a direct comparison between the unamplified and SISPA-amplified samples. Interestingly, no reads were assigned to the taxonomic group of influenza A, and the numbers of reads assigned by MEGAN to the other viral groups are in general lower than what was obtained with direct mapping against viral reference genomes using BWA (compare with Table 2). However, parsing of the BLAST results, using a custom Biopython-script, readily picked out several homology hits to influenza A (data not shown). An adjustment of MEGAN's LCA settings revealed the influenza reads in the unamplified sample (data not shown). The needed adjustments were, however, outside of the normally recommended settings, which led to the introduction of artifacts from accepting reads of low quality. Another interesting observation is the presence of reads classified as Porcine Parvovirus 4 in the SISPA-amplified sample but not in the unamplified sample. Since this virus has been investigated in our laboratory, it is most likely due to an unintentional introduction during the SISPA amplification procedure and should be regarded as a contamination.
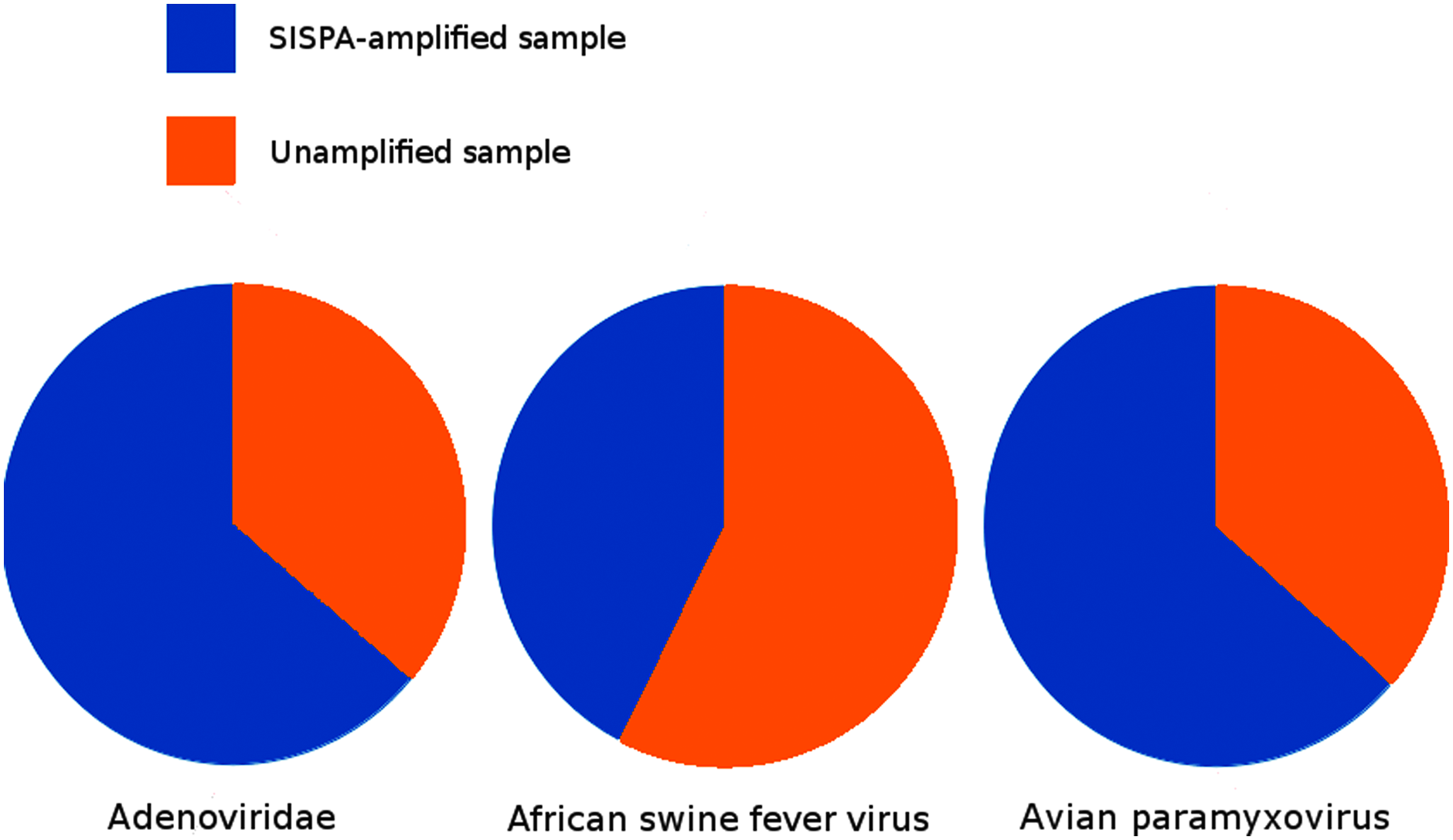
The number of sequencing reads in taxonomic groups classified by MEGAN using LCA parameters Min support 5, Min Score 50, Top Percent 10, and Min complexity 0.44. As noted in Table 3. Results for both AFSV and APMV-1 are low compared to the results in Table 2, which should be taken into account studying the figure.
MEGAN Classified Reads Using LCA Parameters Min Support 5, Min Score 50, Top Percent 10, and Min Complexity 0.44
Discussion
The results indicate that SISPA is a powerful tool for amplification and detection, but less suitable for the retrieval of complete viral genomes due to a strong amplification bias. This is in line with previous findings regarding the effect of PCR-based methods for whole-genome amplification. 45 The estimation of average coverage, or sequencing depth, is normally based on the assumption that all the mapped reads are evenly distributed along the genome. This might not always be the case, which makes it advisable to assess the variation in read distribution across the reference genomes as a standard control. The use of a synthetic metagenome is analogous to the process controls that are commonly being used in routine diagnostics—for example, providing a spike to a sample with a known organism early in sample preparation to use as process control. 46 With or without the SISPA-introduced bias, the components of the synthetic metagenome were discernible in quantities sufficient for detection of all viruses using either the BWA or BLAST approach but not with the MEGAN tool.
MEGAN is a useful and widely applied tool for the exploration of metagenomes, as evidenced by several publications.47,48 However, it appears that even though sequences of 100 bp are considered to be long enough to identify a species with MEGAN, perhaps better results would have been achieved with longer lengths, since shorter read lengths have been correlated with under-prediction. 43
As indicated by Table 2, the SISPA-amplified sample not only showed bias in amplification (as in coverage over the genome), it also shifts the result toward the dominant genome in the composition. This indicates another bias introduced by the amplification, where the dominant genome seems to be favored for amplification compared to the underrepresented. Combined, these 2 biases can produce serious problems in detection of viruses in relatively low quantities compared to the dominant genome in the sample. These results are comparable to those of previous investigations into low-input amplification of viral genomes as well as whole-genome amplification by SISPA.45,49 If this holds true in a larger sample set, it would strongly discourage the use of SISPA for metagenomics where the target agent is suspected to be in the background—for example, analysis of complex diseases, or for semi-quantitative analyses. The amount of reads classified in the 3 areas of unassigned reads (not assigned, no hits, low complexity) in MEGAN also greatly increase with amplification (see Table 3). Moreover, MEGAN completely failed to identify the influenza A reads and only processed them properly with heavily modified LCA parameters and only in the unamplified sample. A possible explanation can be that the segmentation of the influenza genome gives the classifier problems; even though enough reads exist to properly map in all segments, there are not enough reads to reach the minimum requirement for classification with MEGAN's algorithm. In the case of using metagenomics as a tool for detection, this possibility should be considered; MEGAN is not suited for detection of critical reads as much as it is a visualization tool for datasets. The MEGAN analysis also illuminates the old problem with PCR contamination in molecular genetics laboratories. Porcine parvovirus was not part of the constructed synthetic metagenome and is most likely the effect of contamination from amplicons during the SISPA-amplification.
In contrast, the use of short read mapping for detection of sample content proved to be extremely efficient. This opens up opportunities to use automated systems in larger screening efforts to align sequence reads directly against a list of selected agents with known genomes, such as the select agents list of the Centers for Disease Control and Prevention (CDC). This approach would enable a quick multiplex detection of various pathogens similar to the pioneering work being done with HTS profiling of antibiotic resistant bacteria. 50
The subfield of microbial forensics made its breakthrough in 2001 in the United States following the Amerithrax attack using letters containing anthrax spores. Based on the previous genomic knowledge of the organism (Bacillus anthracis), profiling the agent used for the attack provided valuable forensic data for the investigators. 51 The use of HTS technologies, both for providing genomic information and aiding in detection, is a subject discussed in both microbial forensics as well as in biotracing (eg, the 2011 EHEC outbreak in Germany and in the wake of the Amerithrax attack).52-54
The HTS methods hold merit for use in microbial forensics because of several inherent properties. The whole-genome shotgun approach provides the methodology with the least amount of bias for characterization of viral and microbial communities. The sensitivity of detection by metagenomic approaches rival that of PCR but with a broad detection range. 55 The result of HTS is the sequence information, thereby enabling not only detection but also classification of the microorganism by genotyping. As a consequence, it is possible to detect both synthetic biological constructs as well as naturally occurring emerging organisms.
Microbial forensics relies on producing accurate classification of microorganisms in forensic samples. Combined with metadata from the investigation as well as previous knowledge about the detected microorganisms, this enables traceability of the sample. It also enables tracing of microorganisms used for a crime. Forensic samples, on the other hand, suffer from low input material and possible contaminations. SISPA in its turn enables highly unspecific amplification of genomic material in a sample. Combined with a broad but specific detection method such as metagenomics, a broad picture of the microbial community of a forensic sample can be produced, even with low input material as a starting point.
Conclusion
Metagenomics is a powerful tool for detecting nucleic acid in a sample without previously known information about the target. The limitations previously described hold little credence as technology moves forward into ever more affordable sequencing. With the wide range of possible methodologies developed over the past 5 years for HTS, the potential applications in virology are enormous. From a preparedness perspective, we are looking at a technique that is as specific as the other molecular methods but still holds the possibility of broad range detection and unprecedented multiplexing capabilities.
Coupled with selective bioinformatics, as well as the standard broad metagenomic approach, there is also the possibility to quickly assess a sample for known agents (eg, select agents list). This will also provide in-depth information about the viral and microbial communities, thereby detecting possible emerging viruses as well as disruptions in the normal microflora. Both methods could be wrapped in the same fully automated software suite to speed up the bioinformatics analysis process.
In an article by Stanhope, the design for metagenomics experiments is comprehensively deduced from a modeling perspective. 56 This proves to be of relevance for most virologists looking into detection of viruses in a metagenomic sample. In a sample with a dominant genome and a small viral genome, such as a segmented RNA virus genome, the possibility to recover enough reads for detection is lower than in a balanced sample with equal size genomes. Here the possible metadata can help in designing the experiment (such as the clinical description as well as host, sampling method, and sample preparation), but care must be taken to not underestimate and thereby get a false-negative detection.
Keeping in mind the inherent bias of SISPA previously discussed here, it still enables minute quantities of starting material to rapidly be amplified for use with most HTS technologies. However, with the introduction of low-input library preparation kits for the common HTS platforms, the need for preamplification before preparation of sequencing libraries might be declining. The findings indicate that SISPA (1) introduces bias to the distribution of sequence reads and thereby coverage; (2) shifts the composition of a sample in favor of overrepresented genomes; and (3) increases the amount of unclassified sequences in a metagenome.
Metagenomics relies heavily on low input material methods, and as such SISPA has been one of the more commonly used methods for amplification. With increased development of alternative approaches as well as diversification of the use of metagenomics, the use of SISPA with its inherent bias will not likely persist as a method of choice for amplification. SISPA is, however, rapid and easy to perform and most laboratories can employ it for amplification, making it a valid technique for enhanced detection when whole-genome retrieval and exact quantification of microbial community compositions are of less importance. In light of this study, further research is warranted to assess the detection limit of SISPA by also introducing background noise (ie, host material) to the artificial sample. It would also be valuable to conduct a larger study comparing SISPA and other rapid presequencing amplification methodologies, such as optimized linker amplified shotgun library (LASL), in order find a less biased alternative for quantitative metagenomics.
Footnotes
Acknowledgments
This work was supported by the Award of Excellence (Excellensbidrag) provided to SB by the Swedish University of Agricultural Sciences (SLU). The authors would also like to acknowledge support of Uppsala Genome Centre and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at Uppsala Genome Center has been funded by RFI/VR “SNISS” Swedish National Infrastructure for large Scale Sequencing and Science for Life Laboratory, Uppsala. Writing of this article has been supported by the framework of the EU project AniBioThreat (Grant Agreement: Home/2009/ISEC/AG/191) with financial support from the Prevention of and Fight against Crime Programme of the European Union, European Commission—Directorate General Home Affairs. This publication reflects the views only of the authors, and the European Commission cannot be held responsible for any use that may be made of the information contained therein.