
Abstract
Microbial forensics is an important part of a strengthened capability to respond to biocrime and bioterrorism incidents to aid in the complex task of distinguishing between natural outbreaks and deliberate acts. The goal of a microbial forensic investigation is to identify and criminally prosecute those responsible for a biological attack, and it involves a detailed analysis of the weapon—that is, the pathogen. The recent development of next-generation sequencing (NGS) technologies has greatly increased the resolution that can be achieved in microbial forensic analyses. It is now possible to identify, quickly and in an unbiased manner, previously undetectable genome differences between closely related isolates. This development is particularly relevant for the most deadly bacterial diseases that are caused by bacterial lineages with extremely low levels of genetic diversity. Whole-genome analysis of pathogens is envisaged to be increasingly essential for this purpose. In a microbial forensic context, whole-genome sequence analysis is the ultimate method for strain comparisons as it is informative during identification, characterization, and attribution—all 3 major stages of the investigation—and at all levels of microbial strain identity resolution (ie, it resolves the full spectrum from family to isolate). Given these capabilities, one bottleneck in microbial forensics investigations is the availability of high-quality reference databases of bacterial whole-genome sequences. To be of high quality, databases need to be curated and accurate in terms of sequences, metadata, and genetic diversity coverage. The development of whole-genome sequence databases will be instrumental in successfully tracing pathogens in the future.
In order to produce results with a quality that is sufficient for use in criminal proceedings (especially given the serious consequences of an inaccurate identification or analysis), strong operational capabilities and high levels of scientific understanding are required. On the operational side, the capabilities in sampling, sample preservation, and sample preparation need to be refined, and methods and strategies need to be appropriate and validated.7,8 Throughout the entire analytical chain, from sampling to the interpretation of results, forensic awareness and chain of custody procedures need to be implemented. 9
The theoretical foundations of microbial forensics have mainly been a merging of theories from the field of human genetic forensics and viral and bacterial phylogenetics.10,11 In this article, we focus the discussion on forensics applied to bacterial agents. For a recent comprehensive review on forensic analyses of viruses, see, for example, Wilson et al. 11
There are major differences between humans and bacteria in terms of the mechanisms of reproduction and the processes that create genetic variation. The 2 therefore have very different population structures, which in turn leads to differences in how genetic similarities and differences between individuals should be interpreted.
In humans, the genetic variation between individuals primarily arises from the pairing of and recombination between homologous chromosomes during meiosis. This variation is exploited in human forensics to determine whether a DNA sample from a crime scene can be connected to a suspect (individual matching). The probability of a coincidental match (ie, a case in which 2 samples coincidentally have identical profiles) in DNA fingerprinting of Caucasian Americans is 1.74×10−15. 12
Bacteria, on the other hand, reproduce asexually through cell division, and so every daughter cell would be genetically indistinguishable from its ancestor were it not for a variety of mechanisms that introduce genetic variation in bacterial populations. 13 These mechanisms make it possible for a cell to differ from its ancestor and can also cause unrelated organisms to contain shared sequences. Therefore, when comparing individual strains of a microbial species, a complete or near-complete match between individual strains does not necessarily reflect identity. Similarly, minor genetic differences between strains do not necessarily exclude the possibility that they might originate from the same source. Microbial species, and even groups of organisms within a microbial species, differ in their capacity for genetic variation. Some species are very prone to shuffling parts of their genome or integrating genetic material from other organisms into their own genomes, while other species or lineages within species do so rarely. In the latter type of organisms, commonly called clonal, the genetic content hardly changes over generations. Many of the most pathogenic bacterial species have clonal, or even monomorphic, population structures.13-15
For decades, microbiologists have used a wide range of methods to compare different organisms to determine how they relate to one another. 16 Collections of reference strains have been created to which new isolates can be compared in order to analyze population structures and the relationships between the species' individual strains. While microorganisms once were characterized based on their phenotypes (the organism's observable characteristics), technological developments have made genetic analyses increasingly more important. Most bacterial genetic comparisons that have been reported to date rely on genetic marker systems, in which each marker reflects a specific part of the microbial genome. The assumption when using systems of this type is that individual isolates that share marker profiles are related. However, such analyses can sometimes falsely suggest associations between strains or fail to identify relationships that are in fact there. They may also be incapable of resolving closely related strains, such as those of clonal or monomorphic organisms.13,14 The low variability of certain highly pathogenic clonal bacterial pathogens has until recently hampered comparisons between individual isolates because minor differences have been difficult or impossible to detect.17-19
Sequencing and analysis of the entire genome of an organism (whole-genome sequencing and analysis) can reveal subtle genetic differences that would not be detected by older methodologies. As such, this technology provides a range of useful new options for differentiating between individual strains of monomorphic highly pathogenic agents. Until recently, the process of whole-genome sequencing and analysis was painstaking, costly, and time consuming. However, the recent development of the so-called next-generation sequencing (NGS) technologies has revolutionized biology by greatly reducing the time and money required. 20 As a result, the number of laboratories and commercial companies that perform whole-genome sequencing is growing rapidly.
These recent developments have made it possible to whole-genome sequence the entire genome, not only of a few type strains (well documented strains, chosen to represent its species or subspecies) but of numerous individual isolates from a given species. It is expected that ongoing progress in this area will enable the construction of reference strain databases with whole-genome sequences, which will permit more accurate and reliable analyses of genetic relationships within bacterial species. This will create new opportunities in both molecular epidemiology and microbial forensics, facilitating investigations at every step.
Stages in Microbial Forensic and Epidemiologic Investigations
Microbial forensic investigations and epidemiologic investigations are performed for different reasons. An epidemiologic investigation aims to find the source of an outbreak (infectious sources) and to clarify its routes of transmission in order to hinder its further spread and reduce the risk of future outbreaks through effective preventive measures. In contrast, microbial forensics investigations are undertaken to determine whether a crime has been committed, to find the perpetrator, and to gather evidence of sufficiently high quality to be used in a criminal trial. A microbial forensics investigation will therefore be considered only in very special cases—for example, when crimes involving illicit handling of biological agents are suspected. Such offenses may be intentional, as in bioterrorism or biocrime, or unintentional, as when caused by carelessness or negligence.
Microbial forensic investigations have been described as consisting of 3 interrelated stages: identification of the biological agent(s) responsible for an event (identification); characterization of the event as either intentional or unintentional (characterization); and, if the event is deemed illegitimate, attribution of use to a specific perpetrator (attribution). 21 Attribution relates to identifying the person(s) responsible for the attack. 22 The 3 stages can be regarded as an analytical stairway with 3 steps, where the required quantity of whole-genome sequence reference data and level of understanding of the pathogen's population genetics increase as the stairway is ascended (Figure 1).
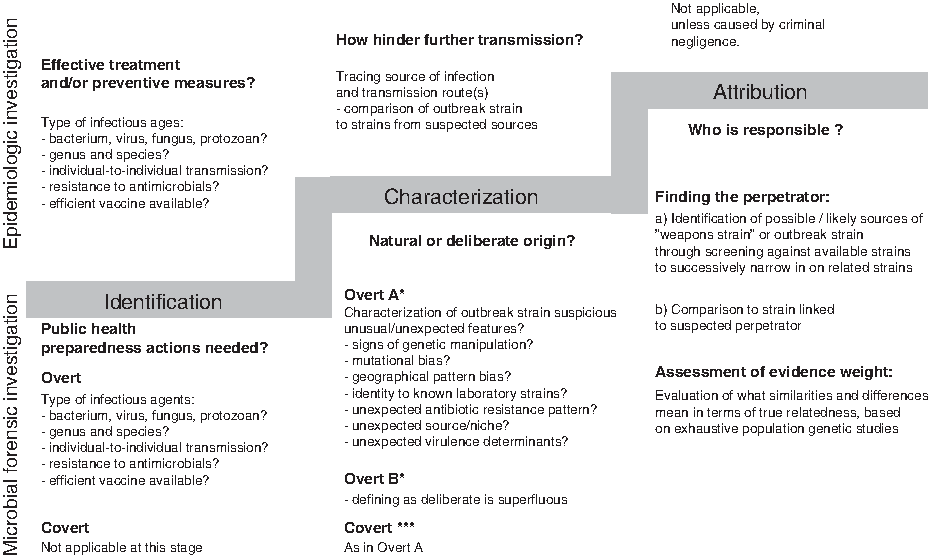
The 3 stages of microbial forensic and epidemiologic investigations:
Many of the questions asked during the first 2 stages of a microbial forensics investigation are identical to those encountered in an epidemiologic investigation, and the same methods and technologies are generally used to answer them (Figure 1). However, the third step is unique to microbial forensics. At this stage, in addition to more “traditional” forensic analyses of recovered materials from the crime scene (eg, analysis of human DNA, fingerprints, and fibers), 23 detailed analyses are conducted of the attack strain. Epidemiologic and microbial forensic investigations can be conducted in parallel, and over time they may diverge and again converge. It is also possible that an epidemiologic investigation may produce findings that suggest a deliberate or unintentional release of a pathogen and thus lead to the initiation of a microbial forensic investigation.
Deliberate distribution of pathogenic microorganisms can be done overtly, in which case the perpetrators will announce that they have taken action, or covertly, in which case they will avoid calling attention to the pathogen's release. 24 The working procedures followed during a microbial forensics investigation, and the extent of the first 2 parts of the investigation, will be influenced by whether the attack is overt or covert and especially by whether investigators are in possession of an attack strain (as was the case during the investigation of the anthrax letters in the United States, for example).
Identification
A fully covert attack will not be detected until signs of disease appear in exposed animals or humans. Identification of the pathogen responsible will thus be performed as part of the animal or public health response rather than as part of the microbial forensic investigation. In an overt attack scenario, however, where the perpetrator announces the attack and attack strain samples are available from the outset, identification of the organism will also be a part of the forensic investigation. This will also be the case when authorities act on intelligence information and thereby come into possession of an attack strain before an attack has been carried out.
In an overt attack, where threat samples can be analyzed before exposed individuals develop signs of disease, the rapid and timely identification of the species (or subspecies) of the attack organism will be of paramount importance when deciding on appropriate animal or public health responses. Knowledge of the pathogen's identity will provide important information on its capacity to cause disease and whether it is transmissible between individuals, factors that greatly influence the extent and dynamics of an outbreak. Other information that would be important at this stage includes what antibiotics could be used to treat or prevent the disease and whether there is a vaccine capable of preventing the disease. Such information can be gained through a combination of several more traditional, and often time-consuming, analyses. Whole-genome sequence analysis, on the other hand, has been shown to be a powerful method for providing all information relevant for bacterial strain identity resolution (ie, information capable of resolving strains at every level, from family to isolate) 25 as well as timely information for prediction of, for example, pathogenic potential and resistance to treatment, 26 and thus allows different investigative questions to be addressed simultaneously.
Characterization
The second step of the microbial forensic investigation analytical workflow (characterization) involves determining whether the outbreak is intentional or unintentional in origin (Figure 1). If a perpetrator has announced intent to release a pathogen before signs of disease have appeared, this step will be superfluous. This would also be the case if authorities had made findings indicating that a release had occurred (eg, equipment for airborne release) before signs of disease become evident. In other scenarios, such as those where an outbreak has unusual properties and is therefore suspected to have resulted from a deliberate attack, or where an individual or group claims responsibility after signs of disease become apparent, it would be necessary to determine the cause of the outbreak. Rapid identification of an attack as deliberate is important as it increases the possibility of obtaining evidence (“traditional” as well as microbial) against the perpetrator(s) before it gets destroyed or obscured by the passage of time. Likewise, in the event that an outbreak initially was suspected to be deliberately caused but later proven to be of natural origin, providing the public correct information as quickly as possible would be important to avoid unnecessary anxiety.
During an outbreak, anomalies in various epidemiologic characteristics, such as antibiotic resistance patterns, geographical occurrence of the disease, transmission routes, and general outbreak intensities and dynamics, have been proposed as potential indicators of deliberate release of a biological agent.27-30 Similarly, anomalies in the organism itself can serve this purpose—and especially if the outbreak is caused by a clonal organism where genetic variation (by norm) is more limited. Such anomalies might include unusual or unexpected virulence determinants (features that give the organism its capacity to cause disease), unusual antibiotic resistance patterns or genetic background to resistance, or a genetic variant that is not known to occur in the relevant geographical area. An array of traditional functional assays and PCR analyses could reveal such features, but only those that were actively searched for. Therefore, unexpected or previously unknown features would not be detected. In contrast, whole-genome sequence analysis has the power to reveal or predict unexpected or previously unknown features of the organism, such as antibiotic resistance31,32 or unusual virulence determinants. Whole-genome sequencing is already being adopted in modern molecular epidemiology investigations. The German E. coli outbreak in 2011 was the first occasion in which the causative organism was whole-genome sequenced during an ongoing outbreak.33-35 The sequence analyses revealed the causative agent to be a previously unknown pathotype, partly conferred by an unusual virulence determinant. 36 Subsequent sequencing of additional strains, collected from preexisting reference strain collections, showed that all the virulence determinants of the pathotype actually had occurred before in E. coli isolates, although not in identical combinations.37,38 This case, nevertheless, clearly demonstrates the ability of whole-genome sequence analysis to detect unexpected genetic features, and thus to reveal anomalies that could be indicators of intentional release of an organism. The prerequisite for correct interpretation of results is, however (and not only in epidemiologic but also in microbial forensic investigations), that sequence databases are comprehensive and correctly mirror the known genetic diversity of the organism in question.
Recent studies have shown that replicating bacteria enrich mutations that are beneficial in the specific environment in which they grow.39-42 Fine scale analyses of mutational patterns could therefore be used to distinguish laboratory-induced from naturally occurring genomic variation.43,44 The occurrence of a laboratory-induced mutational pattern in an outbreak strain would evoke suspicions of deliberate (or accidental) release. The detailed analyses required to detect mutations throughout the entire genome can be performed only through whole-genome sequencing. Further, whole-genome sequencing could also help to reveal signs of previous genetic manipulations of an agent,17,45 such as genetic elements introduced to facilitate genetic engineering, which would be another potential sign that the organism had been deliberately (or accidentally) released.
Taken together, the technological revolution in whole-genome sequencing will enable more reliable conclusions to be drawn from isolates obtained during the course of an outbreak and thus facilitate distinction between natural and intentional outbreaks.
Attribution
The third step in the analytical workflow (attribution) aims to find the person(s) responsible for the attack and to collect legally valid evidence that can be used to convict the perpetrator(s) in a court of law (Figure 1). This process will build on standard police methods, such as the collection of witness statements and intelligence information, as well as “traditional” forensics—for example, fingerprinting and matching of human DNA.23,46 The forensic investigation will also involve a detailed investigation of the weapon—that is, the microbial organism used to inflict harm.
At this stage, strain characterization and comparison to other strains will be performed for 2 reasons. First, it will be performed to determine if the attack strain is related to known strains or strains that can be linked to particular suspects, in order to allow the investigation to focus on a prime suspect. Second, it will be performed to collect evidence that eventually will be presented during a court trial of a suspect. Both tasks require an in-depth understanding of the population structure of the species (or lineage) to enable conclusions to be drawn on to what extent genetic similarities and differences correctly reflect relatedness. The statistical foundations of microbial forensics need to be further developed to enable robust evaluation of the value of evidence for the results of genetic comparisons, in relation to the relevant scenarios and hypotheses. 47 In forensics, including human DNA analysis, the value of evidence is typically calculated as a likelihood ratio 48 using “the logical approach ” (2 hypotheses) or “full Bayesian approach” (any number of hypotheses).47,49 Adopting these approaches for bacterial DNA analyses requires models of the genetic evolution and population structure of the species under investigation. To enable this, the existence of relevant whole-genome sequence databases is instrumental.
Most highly pathogenic bacterial threat agents are clonal or even monomorphic.13-15 Given the assumption that highly dangerous organisms would be of interest in acts of bioterrorism, it is not unlikely that microbial forensic investigations would concern agents for which it is difficult to differentiate between individual strains. Recent publications have shown the power of single nucleotide variants, detected by whole-genome sequencing, to differentiate between strains that appeared identical and therefore were not recognized as separate strains when analyzed with less discriminatory methods.50,51 The new technology also has the potential to identify relationships that are masked by many current characterization methods. 52 Furthermore, whole-genome sequences allows the extraction of the allelic states of specific markers (the marker pattern of a strain) under any possible marker-based typing system (VNTR, InDel, MLST, SNP, etc.), enabling backward compatibility with most marker-based typing system databases.
In an investigation of an illegitimate release, the highest levels of microbial analyses are warranted. Whole-genome sequencing now represents the most robust and powerful method for both discriminating between closely related strains and for reconstructing accurate phylogenies (genetic relationships). In addition, the information obtained through sequencing can be used to develop rapid and sensitive PCR identification systems53-55 that can be used to screen strain collections and thus facilitate the identification of additional strains to sequence and include in whole-genome comparisons.
Reference Strain Collections
Regardless of method used, a prerequisite for all comparisons between microorganisms is the existence of reference strain collections with accompanying information (metadata) on the strains included. To allow correct interpretations of results of comparisons, reference strain collections need to accurately represent the best possible genetic diversity of the species or lineage under investigation. They thus need to represent the best available range of geographical and temporal occurrences of the organism, as well as strains originating from different environments and hosts. Building of strain collections therefore is a continuous task. In the event of a suspected deliberate release, strains from laboratory repositories also must be considered.
As outlined above, in a microbial forensic context, whole-genome sequence analysis is the ultimate method for strain comparisons as it is useful during all stages of the investigation. Particularly during the characterization stage (Figure 1) of an investigation, time will be of critical importance. Valuable time will be lost if resources at this stage need to be invested into identifying, collecting, sequencing, and analyzing representative and relevant strains to which the (suspected) attack strain can be compared to detect unexpected anomalies in the genome sequence. Therefore, the availability of preexisting whole-genome sequence databases will be of substantial value for timely recognition of outbreaks as natural or deliberate. The time aspect would be of somewhat less importance during the attribution stage, but lengthy activities to identify, collect, and characterize strains needed to understand basic population genetics could undesirably slow down the forensic investigation. The preexistence of high-quality, whole-genome databases based on representative and relevant reference strain collections would be of great benefit for future microbial forensic investigations.
Comprehensive reference strain collections currently exist for a wide range of pathogenic organisms that affect public or animal health.56,57 Such collections are increasingly being digitized as corresponding whole-genome sequences are fed into reference sequence databases. 37 Construction of comprehensive databases for the many organisms that can be considered as relevant in a microbial forensic context will take time. Meanwhile, as existing strain collections are digitized, a useful advantage of whole-genome sequences is their backward compatibility. The allelic states of specific markers (the marker pattern of a strain), under any possible marker-based typing system, can readily be extracted from its whole-genome sequence58,59 and new improved algorithms that will facilitate this process can be envisaged. During the transition period, until more strain collections have been digitized, this possibility can be exploited to identify strains with identical or similar marker patterns that, depending on the specific situation, would be relevant for directing the investigation. Therefore, existing marker-based databases, into which extensive resources (costs and labor) have been invested, remain extremely valuable and need to be maintained for current and future needs.
The availability of comprehensive whole-genome sequences for some potential high-threat agents is more limited, a weakness that risks hampering both microbial forensics and epidemiologic investigations. As an example, Francisella isolates from Australia60,61 and Thailand 62 were misclassified at the species level due to the lack of comprehensive databases.
There also exist other challenges that need attention. Many public sequence databases have deficiencies in metadata. This weakness is, on an ongoing basis, being remedied by the scientific community through the defining of minimum standards.63,64 For databases to be acceptable in a forensic context, it will be crucial that they follow internationally accepted minimum standards for describing data and metadata. An additional major challenge will be to build databases that are not abandoned, which has been the unfortunate fate of many publicly funded projects.65,66
There is also a risk that microbial whole-genome sequence data will not be published because of commercial or scientific interests. The creation of reliable databases would therefore benefit from a means to provide full credit to data submitters.67,68 Another currently unresolved question relates to who should have access to pathogen genome databases. Some have warned that open access to genomic resources could itself pose a threat. 69 On the other hand, others have argued that this risk is overstated.70,71 In more “traditional” forensics, access to databases is limited to specifically authorized personnel in law enforcement (ie, police and forensic laboratories), and international data exchange is governed by specific regulations (eg, the Prüm Treaty in the European Union). 72 It should be acknowledged that transport of highly dangerous pathogens constitutes biosafety and biosecurity risks, which can be avoided if sequences rather than strains are exchanged between relevant organizations.
It is also vital to recognize that the creation of databases is only a first step. The sequences themselves are of little value if data cannot be analyzed, interpreted, and turned into useful information. Further, bioinformatics analysis requires a deep understanding of the population structure of the investigated agent. Bioinformatics therefore needs to be recognized as a major focus for attention and investment. 73 The large volumes of sequences generated also present significant problems in terms of data storage.74,75 Powerful and sophisticated computer systems are required to store the large amounts of raw data generated by whole-genome sequencing, to assemble the fragments it produces into whole genomes, and to compare the results obtained from different samples. In some cases, these requirements have limited the use of the new techniques. 76 However, the ongoing development of new, more highly automated software and algorithms, together with the adaptation of current sequencing methods for use with smaller genomes (such as those of bacteria), will reduce the computational power required for whole-genome sequencing and analysis of many important pathogens.
The enhanced applicability of the advancements of whole-genome sequencing and analysis in microbial forensics in an international context will require further efforts, such as the creation of comprehensive whole-genome reference strain databases of relevant biological agents, as well as further refinements in the understanding of population structures and the interpretation of results from genetic comparisons of such agents. The comprehensive nature of these and other required efforts place them beyond the reach of individual laboratories, institutions, and organizations.
Conclusions
Whole-genome sequencing provides an unsurpassed level of genetic resolution for microbial forensics. The increased availability of genetic data and scientific knowledge regarding the interpretation of microbial genetic information have increased the power of the tools available for investigating alleged uses of biological agents. However, there is a lack of high-quality whole-genome sequence databases that represent the global population structures of important threat agents, and the construction of such databases represents a major challenge for the biodefense community. In order to establish whole-genome databases of high quality, a number of criteria need to be fulfilled, including accuracy of sequences, metadata, and genetic variation coverage, and technical and security constraints need to be addressed. In addition, bioinformatics skills and statistical methods for evidence evaluation in microbial forensics need to be further developed to utilize the new possibilities.
Footnotes
Acknowledgments
The authors would like to acknowledge the EU project AniBioThreat (Grant Agreement: Home/2009/ISEC/AG/191) for inviting this article for their supplement issue. AniBioThreat is a project in the Prevention of and Fight against Crime Programme of the European Union, European Commission—Directorate General Home Affairs. This publication reflects the views only of the authors, and the European Commission cannot be held responsible for any use that may be made of the information contained therein. The writing of this article was partially supported by grants from the Swedish Ministry of Defence, Swedish Ministry of Foreign Affairs, and the Swedish Civil Contingencies Agency (project A404013).