
Abstract
Diagnostic DNA analysis using polymerase chain reaction (PCR) has become a valuable tool for rapid detection of biothreat agents. However, analysis is often challenging because of the limited size, quality, and purity of the biological target. Pre-PCR processing is an integrated concept in which the issues of analytical limit of detection and simplicity for automation are addressed in all steps leading up to PCR amplification—that is, sampling, sample treatment, and the chemical composition of PCR. The sampling method should maximize target uptake and minimize uptake of extraneous substances that could impair the analysis—so-called PCR inhibitors. In sample treatment, there is a trade-off between yield and purity, as extensive purification leads to DNA loss. A cornerstone of pre-PCR processing is to apply DNA polymerase-buffer systems that are tolerant to specific sample impurities, thereby lowering the need for expensive purification steps and maximizing DNA recovery. Improved awareness among Laboratory Response Networks (LRNs) regarding pre-PCR processing is important, as ineffective sample processing leads to increased cost and possibly false-negative or ambiguous results, hindering the decision-making process in a bioterrorism crisis. This article covers the nature and mechanisms of PCR-inhibitory substances relevant for agroterrorism and bioterrorism preparedness, methods for quality control of PCR reactions, and applications of pre-PCR processing to optimize and simplify the analysis of various biothreat agents. Knowledge about pre-PCR processing will improve diagnostic capabilities of LRNs involved in the response to bioterrorism incidents.
Rapid and sensitive nucleic acid analysis methods are of vital importance in the response to a possible bioterrorism attack. The polymerase chain reaction (PCR) has become the backbone of contemporary diagnostic DNA analysis through its high target specificity, low limit of detection, and rapid analysis. In PCR, a specific DNA fragment is copied multiple times, ideally enabling detection and quantification of a bioterrorism agent even when only a few target cells are present. Several quantitative PCR (qPCR) assays have been developed for biopreparedness purposes, targeting different agents such as Bacillus anthracis, Francisella tularensis, and the smallpox virus.14-16 The use of multiplexing—that is, simultaneously analyzing 2 or more genetic markers in 1 reaction—has increased our ability to target several agents and even fingerprint specific species.14,17 Recently, universal assays targeting several agents have been proposed.18,19 The strength of this approach is that no prior assumptions on the type of agent need to be made, lowering bias as well as the need for several parallel analyses. The methods employ new detection and identification principles, as opposed to the “classic” principles using specific probes in qPCR and separation by fragment size in capillary electrophoresis. In one, the dissociation pattern of the DNA markers generated with universal primers was studied through high-resolution melting. 18 This enabled the differentiation of 16S rRNA gene fragments from different organisms, giving a specific identity for each of 100 relevant bioterrorism agents. In the other, electrospray ionization mass spectrometry was applied for detection and approximate quantification of PCR products from several possible bioterrorism agents. 19 There, specific primer pairs were used, and identification was based on the differing molecular weights of the amplicons. Next-generation sequencing is a powerful new technique with potential for highly discriminatory analysis of biothreat agents, and it has been shown to enable differentiation between individual strains of Bacillus anthracis and Yersinia pestis. 20 The continuous development of specific PCR-based assays is an important preparedness measure against bioterrorism.
However, irrespective of the assay or detection principle used, all PCR-based analysis systems share some common challenges. In a bioterrorism crisis, decision makers need accurate and timely information, and the source is often impure biological samples with low levels of target. A rapid analysis process, with a high level of automation, will elevate the throughput. A great analytical challenge is the extraneous substances that pollute biological samples from the environment, impairing amplification and reducing the analytical limit of detection. 21 Additionally, the concentration of target nucleic acids (ie, cells, spores, and viruses) is often low, so large samples need to be taken for subsequent concentration of nucleic acids.
Following sampling of surfaces, air, or water, a heterogeneous and dilute sample must be converted into a concentrated, amplifiable nucleic acid extract. Pre-PCR processing21,22 is an integrated concept in which the issues of analytical limit of detection and simplicity for automation are addressed in all steps leading up to PCR amplification (ie, sampling, sample treatment, and the chemical composition of PCR) (Figure 1). The sampling method should maximize target uptake and minimize uptake of PCR-inhibitory substances. In sample treatment, there is a trade-off between yield and purity, as extensive purification leads to DNA loss. 23 Additionally, the buffers and reagents used in DNA extraction and purification should be compatible with the PCR in order to support amplification.24,25 A cornerstone of pre-PCR processing is to apply DNA polymerase-buffer systems that are tolerant to specific sample impurities, thereby increasing the analytical success rate and lowering the need for costly sample treatment procedures where DNA is lost.26,27 The overall aim of pre-PCR processing is thus to streamline the analysis process and maximize the amount of amplifiable DNA in relation to the robustness of the applied PCR assay. The aim is not to generate the purest nucleic acid extract possible.
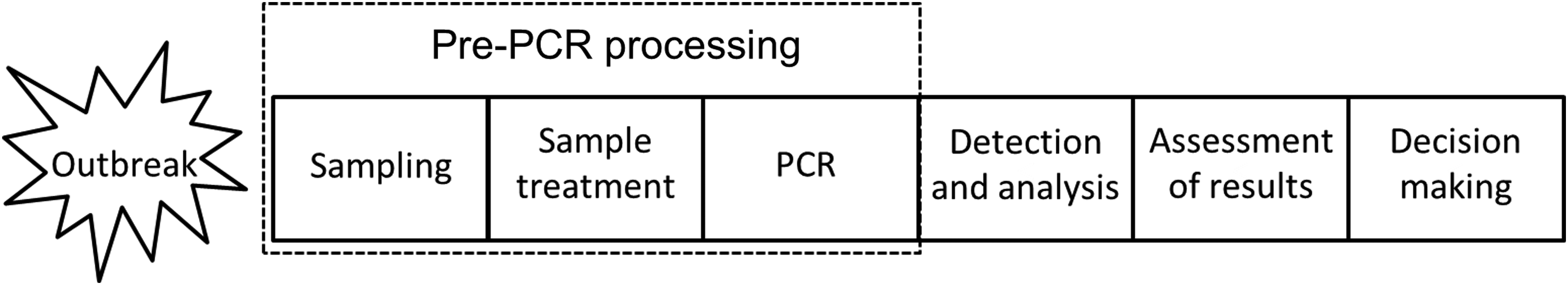
Overview of the DNA Analysis Process in Response to a Disease Outbreak or Bioterrorism Attack. The aim of pre-PCR processing is to provide a high analytical success rate in rapid, high-throughput analysis. A cornerstone of pre-PCR processing is to apply inhibitor-tolerant DNA polymerase-buffer systems in PCR, thereby lowering the need for costly sample treatment procedures where DNA is lost.
The issue of PCR inhibition has so far been most frequently discussed in the fields of microbial analysis of foods and feeds21,28,29 and in forensic DNA analysis30-32 due to the variable and complex nature of the targeted biological samples. In these fields, just as in agroterrorism and bioterrorism preparedness, relevant samples can be retrieved from any given background material, resulting in impure DNA extracts. Knowledge about PCR inhibitor effects and pre-PCR processing is important for existing LRNs in order to improve their diagnostic capabilities. Most diagnostic laboratories use commercially available DNA purification kits and PCR master mixes with proprietary content. This has benefits regarding standardization and quality control of reagents but has also turned PCR-based analysis into a black box: Troubleshooting is difficult when several parameters are unknown, and it is complicated to make appropriate improvements to the analysis systems.
This article is focused on the effects of PCR-inhibitory substances relevant in bioterrorism preparedness, methods for quality control of PCR reactions, and applications of pre-PCR processing to optimize the workflow and analytical success rate for various bioterrorism agents. The sections on PCR inhibition mechanisms, monitoring PCR inhibition, and robust DNA polymerase-buffer systems benefit from information from other scientific fields in which diagnostic PCR is employed: archaeology, clinical diagnostics, food and feed analysis, and forensics. The section on sampling and sample treatment is dedicated to challenges facing biopreparedness DNA analysis. Our aim is to provide laboratories involved in biopreparedness with knowledge on the limitations of diagnostic PCR, thereby enabling efficient sample processing, sound assessment of results, and competent troubleshooting.
PCR Inhibitory Substances and Their Mechanisms
Some substances have been identified as PCR inhibitors and completely or partly characterized regarding their molecular PCR-inhibitory mechanism(s) (Table 1; for a more extensive review, see Hedman and Rådström 22 ). Presence of PCR inhibitors results in impaired reaction kinetics, reduced limit of detection, and lowered reproducibility. In a bioterrorism incident, PCR-inhibitory substances that are not dealt with can have a devastating effect, hindering detection even though the specific agent is present in the sample. Any substance interfering with one or more of the molecular reactions involved in PCR (ie, denaturation, primer annealing, binding of polymerase to primer-DNA complex and primer extension) can have an inhibitory effect on amplification 21 (Figure 2). Categorizing PCR inhibitors is therefore a difficult task. As the exact mechanisms of most inhibitors are as yet unknown, the common classification is based on the affected targets, leading to 3 main groups of inhibitors: (1) DNA polymerase inhibitors, (2) nucleotide/nucleic acid inhibitors, and (3) fluorescence inhibitors (in qPCR). DNA polymerase inhibitors can either affect the enzyme directly (eg, by binding to its active site) or indirectly (eg, by chelating essential Mg2+ ions). Nucleotide or nucleic acid inhibitors can block amplification by binding to primers, single-stranded DNA (ssDNA), and/or double-stranded DNA (dsDNA) or by simply degrading nucleic acids. Fluorescence inhibitors interfere with the detection of amplicons in qPCR—for example, by forming precipitates blocking fluorescence, interacting with the fluorophore, or disturbing the probe's functionality or polymerase's 5′-3′ exonuclease activity (Table 1).
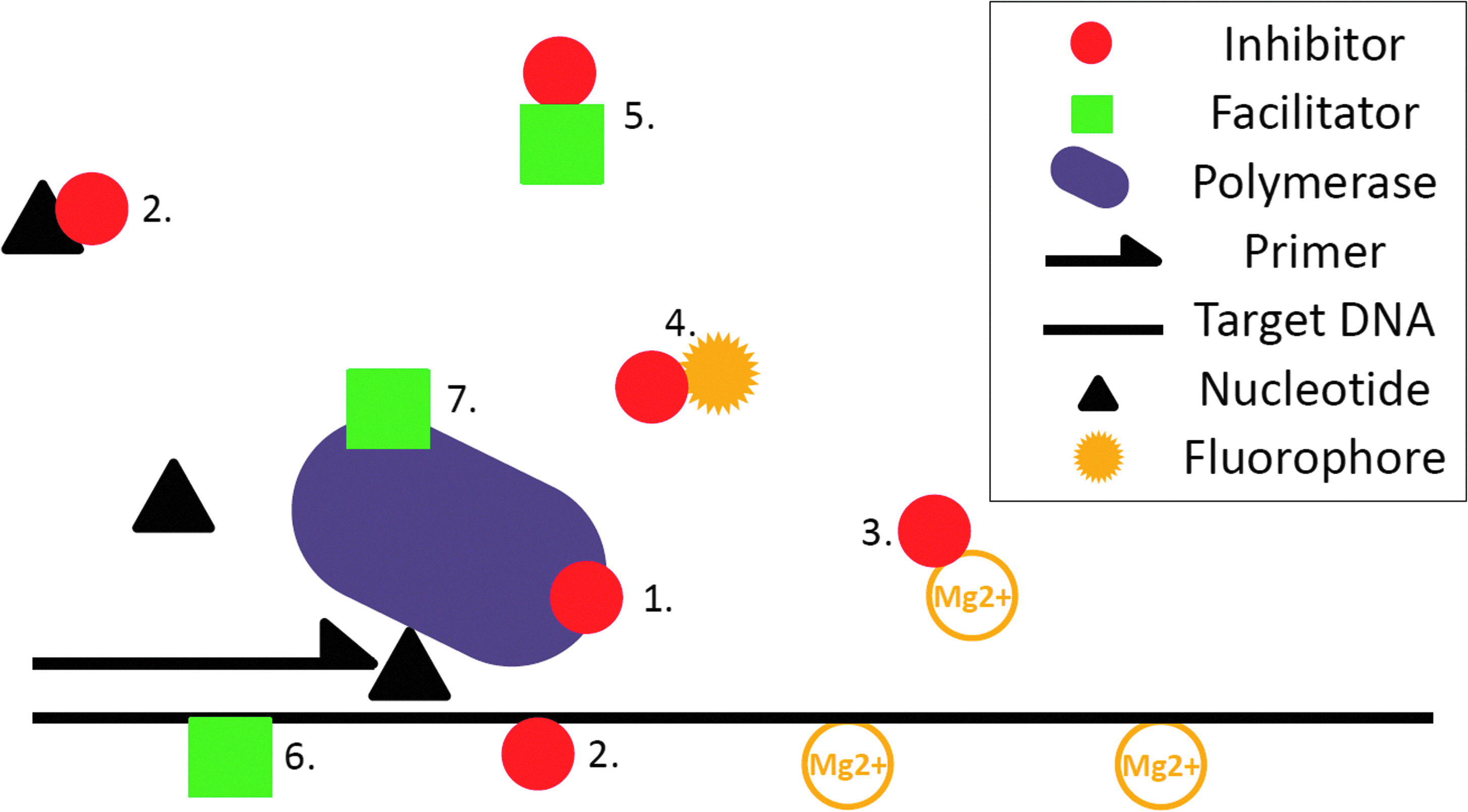
The Complexity of PCR Inhibition. A single PCR-inhibitory compound can affect the reaction in several ways, and different inhibitors can have different effects. Inhibitors may (1) bind to or degrade the DNA polymerase; (2) bind to nucleotides, primers, or the DNA template; (3) chelate cations; and/or (4) prevent the release of fluorescence from detection dyes. PCR facilitators may counteract inhibition by (5) serving as a target for inhibitors or bind to inhibitors; (6) stabilizing single-stranded DNA; and/or (7) improving the activity of the DNA polymerase.
Overview of PCR-Inhibitory Compounds and Ions
Presumed mechanism(s). Inhibitors affecting the ion composition will affect the DNA polymerase (cofactor) and annealing properties of primers and probe system through nucleotides/nucleic acids and/or the qPCR fluorescence detection.
Humic, fulvic, and tannic acids, as well as other polyphenolic compounds from the degradation of organic material, are potent PCR inhibitors that are present in, for instance, soil and bone.33,56-58 Humic acids may affect the DNA polymerase in 2 ways: by binding to its active site, thereby lowering enzymatic activity, and/or by binding to the enzyme elsewhere, introducing conformational changes that impair the binding of the polymerase to the primer-DNA complex. 34 However, humic acids are heterogeneous substances—in solution no 2 molecules are exactly alike—and purified humic acid purchased from a company may have different inhibitory properties compared to humic acids from real environmental samples. 35 This heterogeneity may be the reason why different studies, using different preparations of humic acids and different PCR assays, have come to different explanations for their PCR-inhibitory effects. Opel et al. 32 argue that humic acid is a strict nucleic acid inhibitor, not affecting the DNA polymmerase, which binds sequence-specifically to DNA, thereby lowering the amount of available template and also affecting the melting temperature of the amplicons. Sutlovic et al. 34 also note that binding of humic acids to DNA could make it less accessible for amplification, but they view the impact as a combined DNA polymerase/nucleic acid effect, as shown by the ability to relieve inhibition by increasing the amount of DNA polymerase.
Suitable concentrations of mono- and polyvalent metal ions are important for successful PCR amplification, ensuring complete denaturation, efficient and specific primer annealing, and high DNA polymerase activity. Ca2+, K+, and Mg2+ are quite prevalent in cells and are thus released in DNA extraction. Mg2+ is a co-factor for the DNA polymerase and increases the affinity between the primers and DNA by lowering the electrostatic repulsion between the phosphate backbones of the strands. Increasing the amount of Mg2+ thereby generates a higher amplicon yield but decreases specificity. Ca2+ ions compete with Mg2+ for the binding sites on the polymerase, thereby inhibiting PCR by directly affecting polymerase activity.32,36 K+ ions are often used in the PCR buffer to ensure suitable ion content. Changing the amount of K+ ions or introducing other ions (eg, Na+) may disturb amplification 24 by affecting both DNA polymerase activity and the primer annealing conditions.
Nucleic acid inhibitors are molecules that affect the melting temperature of primers and DNA (eg, ions), bind to DNA, and/or alter the DNA conformation in such a way that primer annealing or extension is negatively affected. Nontarget DNA may inhibit amplification by sterically preventing the primers from annealing, or by providing nonspecific binding sites for the primers. 37 Immunoglobulin G in blood plasma forms a complex with ssDNA, thereby affecting primer annealing and the level of free primers. 38
The fluorescing dsDNA binding dye SYBR Green I, used for qPCR amplicon detection, may partly inhibit PCR when used at the recommended concentration. 39 SYBR Green I fluoresces with high intensity when bound to dsDNA compared to the unbound dye, and increased fluorescence thereby signifies an amplicon increase. However, the exceptional DNA affinity of SYBR Green I can have an inhibitory effect by stabilizing dsDNA, thereby hindering denaturation. Dyes with lower affinity, such as SYTO-13 and SYTO-82, showed no PCR-inhibitory properties. 39 SYBR Green I and various other DNA-binding dyes also bind ssDNA with different affinities, depending in part on the ion content in the buffer. 40 This may lead to primer shortage and/or hinder primer annealing if too high dye amounts are used.
The detection system in qPCR is influenced by a specific set of inhibitors—that is, those that interfere with the fluorophores, probes, or fluorescence measurements. These inhibitors may not affect the actual amplification of DNA but can still produce false-negative results or underestimated DNA concentrations. Changes in the ion content of the reaction could affect the success of probe hybridization, causing a lowered fluorescence signal. Molecules may interfere with the DNA polymerases' 5′-3′ exonuclease activity, thereby inhibiting TaqMan probe hydrolysis. 41 Humic acids quench the fluorescence of SYBR Green I, probably by binding to the fluorophore or by collisional quenching. 42 Surface-bound SYBR Green I fluoresces most, and humic substances may also prevent surface binding. Hematin in blood and indigo dye may interfere with the binding of SYBR Green I to dsDNA or have a quenching effect. 32 Materials that are strong in color may also interfere with fluorescence detection by simply obscuring the signal.
Monitoring PCR Inhibition in Biopreparedness Analysis
An internal amplification control (IAC) (ie, an “alien” DNA fragment of known concentration) may be included in biopreparedness qPCR assays to monitor the quality of the reactions and to avoid false-negative results.59,60 The presence of PCR inhibitors or other problems related to amplification can result in either complete failure to amplify the IAC or an elevated IAC quantification cycle (Cq) value.
For absolute quantification, dilution series of pure DNA with known concentrations are analyzed to obtain a standard curve. The standard curve is then used to calculate the DNA concentrations of the samples. For correct DNA quantification, it is important that the reaction kinetics of the sample be the same as for the DNA used to obtain the standard curve. Concentrations could be seriously underestimated if the amplification efficiency of the sample is lower than for the DNA standard (eg, due to PCR inhibition). 61 Tichopad et al. 62 proposed a bivariate kinetic outlier detection (KOD) method to evaluate the kinetics of individual reactions by comparing them with ideal standard reactions, in which first and second derivative maxima of a fitted sigmoidal model are calculated.62,63 The 2 values are then combined to give 1 χ2 distributed value. High values imply that the reaction kinetics are significantly different from the kinetics of pure reactions. The method can be used to identify kinetic outliers in quantification—that is, samples that may not be properly quantified using the standard curve—or to estimate the level of inhibition in a reaction. The KOD method has been shown to be more reliable in identifying reactions affected by PCR inhibitors than various methods for calculating amplification efficiency from individual amplification curves. 62 For a review of KOD methods, see Bar et al. 64
Inhibitory compounds that have a direct effect on DNA, lowering the amount of available template, may impair amplification and elevate Cq values without affecting amplification efficiency or being flagged as kinetic outliers. 32 Thus, an IAC is beneficial in the bioterrorism agent qPCR assay to ensure that all inhibitory effects are detected. When designing an appropriate IAC, several criteria must be considered.
The IAC primer sites may be equal to those of the target, 65 or they may be different, creating a need for separate primer pairs. 66 In the first case, there is competition for the primers, affecting the amplification efficiency of both the template and the IAC. Incorporating a second primer pair may increase the risk of primer-dimer formation and nonspecific primer annealing.
If the IAC concentration is too high, the amplification of the target may be negatively affected. The appropriate number of IAC molecules depends on the assay, but generally low numbers are preferable—for example, from around 20 to 100 copies per reaction.65,67,68 Since the amplification of target DNA competes with IAC amplification, even when different primer pairs are used, the Cq value for the IAC may be elevated when levels of target DNA are high, generating a false inhibitory effect. 69
Amplification of the IAC should ideally be somewhat more easily disturbed by impurities than the target. The size of the IAC is important in this respect, since shorter fragments are generally more readily amplified in the presence of inhibitors than longer ones. 70 An IAC amplicon slightly longer than the target is therefore recommended. 65
IACs can also be applied in multiplex biomarker assays for conventional PCR with capillary electrophoresis detection. Zahra et al. developed 2 IAC fragments, 90 and 410 base-pairs, for a forensic DNA typing system with target amplicons ranging from 100 to 350 base-pairs. 71 The longer IAC fragment proved to be more sensitive to inhibition from heme and humic acid compared to the shorter one, making the ratio between their respective fluorescence peak heights a measure of the inhibitory effect. Other than adding IACs to multiplex conventional PCR assays, each marker's electropherogram peak height can be used as a measure of analytical quality.72,73 The intensity of the allelic peaks, corresponding to the number of generated amplicons, and the balance between peaks give information on the impact of PCR inhibition. Intensity and balance can be presented as separate measures 73 or combined to 1 measure using principal components analysis.26,72
Inhibitor-Tolerant DNA Polymerase-Buffer Systems
DNA polymerases from different sources differ in their tolerance to various PCR inhibitors, 24 making the choice of DNA polymerase a vital part of setting up a PCR assay for detection and quantification of a certain bioterrorism agent in a specific environment. Additionally, the composition of the PCR buffer (ie, the pH, ion content, and presence of PCR facilitators) affects amplification efficiency and PCR inhibitor-tolerance.25,74,75 These facts are often overlooked, and most end-user laboratories and researchers simply use the DNA polymerase-buffer system that is provided with the commercial kit or master mix they purchase, possibly affecting the limit of detection negatively. This section highlights the differences in PCR inhibitor-tolerance between different DNA polymerases, and the possibilities to increase analytical robustness by optimizing the content of the PCR buffer.
Inhibitor-Tolerance of Thermostable DNA Polymerases
High-quality thermostable DNA polymerases are a vital component of PCR. 76 Any compound that interferes with their enzymatic activity will reduce the formation of specific PCR products.24,26,51 The DNA polymerase obtained from the thermophilic bacterium Thermus aquaticus (Taq) and its commercial derivatives are most widely used because of their high thermostability and good processability, 77 but also because of availability and “old habit.” Polymerases from a range of other organisms are also commercially available (eg, Thermus thermophilus [Tth], Thermus flavus [Tfl], and Pyrococcus furiosus [Pfu]), differing in catalytic properties (eg, having 3′-5′ and/or 5′-3′ exonuclease activity), extension rates, and fidelity.77,78
Hot-start DNA polymerases provide higher target amplicon yields than natural variants79,80 and can be more tolerant to impurities than other polymerases; 35 this is partly explained by the continuous release of active polymerase throughout PCR cycling. Hot-start DNA polymerases are inactive at room temperature, either by binding of a heat-sensitive antibody to the active site of the polymerase 81 or by covalently binding a blocking molecule to lysine residues within and outside the active site, 82 and they are activated by the heat in the initial denaturation step.
Increasing the amount of DNA polymerase is a means to titrate out polymerase inhibitors and has been shown to relieve inhibition caused by humic acid. 33 However, the success of this approach is limited and depends on the amounts and types of inhibitors present. When analyzing crime scene samples of various types, doubling the amount of AmpliTaq Gold improved amplification for moist snuff, but no or minor improvements were seen for chewing gum and cigarette butts, 27 indicating that inhibitors in the latter had a stronger negative effect on the DNA polymerase activity.
Several DNA polymerases originating from Taq are more susceptible to inhibitors found in blood, bone, foods, feces, and soil than Tth-based polymerases.24,52,53,83 Moreover, notable inhibitor-tolerance differences have been found between different Taq-based polymerases.26,27,83,84 The commonly used polymerase AmpliTaq Gold performs poorly in the presence of inhibitors from various substances, including blood, soil, and bone.24,52,83,84 For over a decade, AmpliTaq Gold was the standard DNA polymerase in forensic DNA analysis. However, when analyzing impure crime scene samples, such as cigarette butts and swabs from cans and bottles, 3 alternative DNA polymerases (Bio-X-Act Short, ExTaq Hot Start, and PicoMaxx High Fidelity) performed significantly better than AmpliTaq Gold, generating a higher number of complete DNA profiles. 26
During recent years, new DNA polymerase variants have been developed with the objective of improving PCR inhibitor-tolerance. Using a protein engineering approach, random mutations have been introduced into natural Taq and its N-terminally truncated version KlenTaq.84,85 Subsequent screening for inhibitor-resistant properties led to the development of DNA polymerases with improved tolerance to blood and soil components, one bearing the commercial name OmniTaq. However, although OmniTaq is tolerant to various inhibitory compounds, it does not necessarily provide any improvements compared to natural Taq polymerase in terms of limit of detection and general amplification efficiency. 26
Molecular breeding and compartmentalized self-replication have been used to engineer chimeric, inhibitor-tolerant DNA polymerases with elements from various Thermus polymerases (eg, Taq, Tth, T. oshimai, and T. brockianus). 35 One of these engineered DNA polymerases, called 2D9, contained 81 mutations compared to natural Taq and showed considerable tolerance to inhibitors present in soil and bone. Since inhibitors such as these are likely to form colloids, it was suggested that 2D9 may interact less with colloids. However, 2D9 was not successful in the analysis of whole blood, indicating that it may not be suitable for broad-range PCR-inhibitory bioterrorism samples.
Blending DNA polymerases is one way of achieving desirable properties in a PCR assay—for example, to improve the fidelity of Taq polymerase assays by adding a small amount of a polymerase with proofreading capacity (3′-5′ exonuclease activity), such as Pfu polymerase.86,87 Combining 2 inhibitor-tolerant DNA polymerases with complementary properties has been shown to provide a DNA polymerase-buffer system with broader-range inhibitor-resistant properties. 27 Apart from complementarity, the DNA polymerases in the blend (ExTaq Hot Start and PicoMaxx High Fidelity) exhibit synergy: Not only do the DNA polymerases function together; they amplify each other's performance in impure reaction conditions.
Relieving Inhibition by Modifying the PCR Buffer
For most Taq polymerases, a Tris buffer with pH 8.3 (measured at room temperature) is recommended. Elevating the pH of the Tris buffer to 9.0 or more has been shown to relieve inhibition caused by leukocytes. 88 Replacing the Tris buffer with a zwitterionic buffer such as tricine may improve inhibitor-tolerance, as shown for direct amplification of whole blood. 89
Increasing the amount of Mg2+ ions can counteract inhibition from chelating agents, DNA intercalating dyes, and Ca2+ ions.36,90 Apart from optimizing the core reagents, PCR facilitators can be added to the buffer. Various facilitators have been used to increase the specificity and fidelity of PCR, and several substances have the capacity to relieve PCR inhibition (Figure 2, Table 2).
PCR Facilitators Used to Alleviate Inhibition
Classification according to AbuAl-Soud.157
Presumed mechanism(s).
T4 bacteriophage gene 32 product.
Bovine serum albumin (BSA) is the most commonly used PCR facilitator, and it is added to the buffers supplied with several commercial DNA polymerases. BSA is a blood-tissue transport protein that binds fatty acids (lipids) and other organic molecules. Its excellent binding capacity makes it suitable for relieving various types of PCR inhibition. BSA has been suggested to bind inhibitory compounds such as heme, phenols, and melanin,25,56,94 thereby protecting the polymerase. BSA may also act as a competitive target for proteases. 95 A range of different BSA concentrations have been used to counteract inhibition, and the optimal concentration depends on the assay and the nature of the sample (Table 2). BSA has been shown to outperform other facilitators such as dimethyl sulphoxide (DMSO), polyethylene glycol (PEG), and Tween 20 for removing inhibitory effects from blood, feces, and meat samples. 25
Different types of PCR facilitators may provide complementary or synergistic effects. Two recently developed facilitator blends have been shown to increase the tolerance to blood and soil inhibitors.40,96 Both blends apply the same osmoprotectant, the disaccharide trehalose, as a key component. Trehalose was complemented with the detergent NP-40 and L-carnitine, or propanediol. However, positive interactions between PCR facilitators depend on their nature and cannot always be expected. 25 On the contrary, combining facilitators may induce inhibition. 119 Overloading of facilitators will also inevitably lead to inhibition.46,97
Biothreat Agent Sampling, Sample Treatment, and PCR
Sampling
Prior to the 2001 terrorist actions with envelopes containing Bacillus anthracis spores in the US postal system, there were no standardized policies for sampling procedures for bioterrorism preparedness. 98 Following these events, substantial work has been focused on developing and evaluating sampling methods for spores on various surfaces, for PCR-based analysis99,100 as well as for classical microbial analysis.98-102 Recently, standards for “Sample Collection and Swab Sample Collection of Visible Powders Suspected of Being Biothreat Agents from Nonporous Surfaces” and “Operational Guidelines for Initial Response to a Suspected Biothreat Agent” have been developed.103,104
Cotton swabs are commonly used as a part of the PCR analysis chain in all fields applying nucleic acid analysis. A pre-PCR processing protocol with cotton swabs has been developed for Yersinia enterocolotica. 105 However, cotton swabs and swabs of other materials are limited by the size of the surface they can cover. When screening for possible bioterrorism agents, large surface areas must be sampled to avoid false-negative results, meaning that ordinary swabs are not ideal. A biological sampling kit (BiSKit) applying a thick foam material enables sampling of a surface area of 1 m2 (wood laminate and metal) for subsequent PCR analysis. 100 BiSKit foam performed better than swabs made of cotton and foam for both surface types, explained in part by the smaller area covered by the swabs (around 100 cm2). Gauze can also be used for sampling of large surfaces, as shown for carcass sampling. 106 Tape lifting is gaining popularity in forensic DNA analysis and has been successfully used for sampling of human cell material from diverse items such as clothes, skin, handguns, and shoe insoles.107-111 A piece of adhesive tape is pressed against the object a number of times, covering a larger area than a swab. The cells adhere to the tape, which is subsequently placed in a tube for DNA extraction. Tape lifting could possibly be applied in bioterrorism preparedness for sampling of inert surfaces.
The aim of surface sampling is to efficiently release and recover cells, spores, and viruses. However, impurities will also be released and collected in the process. Depending on the sampling method, more or fewer impurities will be absorbed, affecting the amplifiability of the generated extracts. In a study comparing surface decontamination strategies, a swipe sponge and a foam swab gave higher levels of PCR inhibition compared to a Heavy Wipe cloth. 112 Sampling efficiency can be affected by the sampling method but also by interactions between the surfaces and the microbes. For virulent strains of Bacillus anthracis and Yersinia pestis, the sampling efficiency was lower for hydrophobic surfaces (vinyl and plastic) compared to hydrophilic surfaces (glass and stainless steel). 113 It was suggested that this was due to specific cell surface receptors or capsular material, as nonvirulent strains did not show any differences between the surface types.
In direct sampling, as opposed to the indirect swabbing procedures, a piece of the material carrying the target cells is directly submitted to sample treatment. Examples are soil, water, and fabrics. Generally, direct sampling provides a high DNA yield but often leads to problematic levels of PCR inhibitors.57,114
Sampling of large air volumes can be performed by filtration 115 or by applying instruments that concentrate the air content into a small amount of liquid, such as the SKC Biosampler and the SpinCon system.115,116 If infectiousness of viruses needs to be studied, liquid sampling is preferable. 115 Inevitably, microorganisms naturally present in the environment, and dust and pollutants will also end up in the concentrated air samples. This can have a detrimental effect on analytical limit of detection. In a study employing liquid air samples spiked with possible bioterrorism agents such as Coxiella burnetii and Yersinia pestis, the environmental background elevated the limit of detection up to 1 log unit depending on the assay. 116 However, some of the assays tested were not affected by the background, although only a simple bead beating procedure was applied.
Sample Treatment
Sample treatment generally includes (1) eluting cells/spores from the sampling material, (2) cell lysis, and (3) DNA purification. In these steps there is a trade-off between yield and purity. Physical separation of cells from complex background material prior to lysis (eg, by differential centrifugation methods 117 ) can improve purity. However, cell separation methods are time-consuming, and differential centrifugation generally gives poor recovery rates (below 50%). 118
The method used for releasing cells from the sampling material has a great impact on the analytical limit of detection. For both F. tularensis and B. anthracis cells, centrifuging the sampling material immersed in PBS buffer using the Swab Extraction Tube System (SETS), consisting of an inner tube with a small hole and an outer collection tube, gave a 1 log unit lower limit of detection compared to sonication and vortex procedures.119,120 The better performance of SETS was verified using nylon flocked swabs, rayon swabs, polyester swabs, and cotton swabs. It was hypothesized that centrifugation provided the most efficient transfer of liquid from the sampling material to the extraction tube.
Cell lysis can be achieved using various approaches: chemical (eg, applying detergents), enzymatic (eg, applying proteinases), physical (eg, heating), or mechanical (eg, bead-beating). Combining 2 or more of these (eg, heating and proteinase K treatment) can elevate the lysis efficiency. Following lysis, PCR-inhibitory substances co-extracted with DNA need to be handled. Preferably, this should be done by customizing the DNA polymerase-buffer system as described above. Otherwise, extensive DNA purification is often needed,114,121,122 leading to DNA loss.23,123 The degree of loss depends on both sample type and purification method. Recovery rates from about 10% to 85% have been reported when comparing different purification principles and methods for a certain sample type.23,124 It is necessary to choose wisely when applying commercial DNA extraction/purification kits for biological samples of limited amount and purity. In a comparison among 5 extraction kits for detection of F. tularensis spiked in soil samples, the limit of detection differed up to 3 log units between the best and the worst kit. 125 As most kits have undisclosed contents to some extent, it is difficult to know whether a specific purification kit would work for a specific sample type without testing it.
Nonlinear electrophoresis applying synchronous coefficient of drag alteration (SCODA) is a promising new tool for DNA purification for complex environmental samples, as shown for plants and soils.126,127 There, DNA is concentrated and separated from impurities by changing the direction and amplitude of the voltage over an agarose gel. SCODA was shown to remove humic acids more efficiently than other available purification techniques and kits, including direct current electrophoresis, chromatography, and PowerSoil DNA isolation kit. 127 The reported recovery rates span from 38% to 73%, with differences between different soil types.126,127 Considering the extra work, cost, and DNA loss, SCODA, as well as other purification methods, should be applied only when the downstream analysis demands it—that is, when the best possible DNA polymerase-buffer system fails due to impurities.
Automation and Point-of-Care Analysis
Automation of nucleic acid purification with homogeneous extract as starting material is quite straightforward. There are several commercially available systems, most based either on DNA binding magnetic beads or spin columns.128-130 For cell samples on filter paper, such as FTA, it is easy to set up an automated analysis chain where PCR is performed on paper discs and DNA extraction/purification is not needed. 131 Automating cell elution and lysis for large, heterogeneous samples with low amounts of target cells, as in forensic DNA analysis and bioterrorism preparedness, is more difficult and currently not possible using any validated robotic systems. In order to optimize limit of detection and speed of analysis, it is advisable to automate the post-lysis procedures, or nucleic acid purification (if needed after optimizing the inhibitor-tolerance of the DNA polymerase-buffer system), PCR/qPCR setup, and post-PCR analysis setup. Cell elution and lysis can instead be simplified and improved by reducing manual handling and the number of tube transfers. Quick 1-tube DNA extraction procedures, such as a method based on EA1 proteinase,132,133 will probably form an important base for efficient sample processing in the future. Dedicated research is needed to make these types of quick sample treatment methods more efficient, both regarding throughput and analytical limit of detection.
The development of efficient analysis procedures in bioterrorism preparedness should preferably go in 2 directions. First, central laboratories with special facilities for handling bioterrorism agents in a safe manner are needed to build up cost-effective, automated, high-throughput analysis chains for various microbial targets. There the bulk of the samples are analyzed—for example, if several farms must be investigated to confirm the presence or absence of B. anthracis in the wake of an outbreak. Second, mobile point-of-care analysis systems are needed to provide quick results on the site of an outbreak. Lab-on-a-chip (or, so far, rather lab-on-a-truck) solutions can be applied. Miniaturized PCR-based analysis systems for detection of pathogens have been developed, using real-time fluorescence measurements 134 and integrated capillary electrophoresis separation. 135 However, the bottleneck for lab-on-a-chip–based analysis is turning a large heterogeneous sample into a highly concentrated nucleic acid extract that fits into nanoliter-scale analysis. Development of sampling and sample treatment methods suitable for lab-on-a-chip applications is therefore a major challenge in developing truly usable biopreparedness point-of-care analysis systems.
Conclusions
Diagnostic PCR plays an important part in the detection of biothreat agents because of its high specificity and low limit of detection. Pre-PCR processing is the concept of viewing sampling, sample treatment, and the chemical composition of PCR as an integrated chain and, preferably, optimizing all these steps in one combined study. A cornerstone in pre-PCR processing is to apply inhibitor-tolerant DNA polymerase-buffer systems, thereby reducing the need for extensive DNA purification where DNA is lost.
It is vital to improve the awareness about pre-PCR processing issues among LRNs involved in agroterrorism and bioterrorism preparedness, in order to enable streamlined analysis processes, efficient troubleshooting, trustworthy quality control, and an optimal analytical success rate. Implementation of pre-PCR processing validated protocols will improve the diagnostic capability of LRNs. This is crucial since diagnostic PCR is applied to detect low levels of biothreat agents in various PCR inhibitory samples. Applying pre-PCR processing principles in the optimization of biopreparedness DNA analysis chains ensures reliable data for decision makers involved in a bioterrorism incident.
Footnotes
Acknowledgments
The writing of this article was supported by grants from the Swedish Civil Contingencies Agency (Anslag 2:4 Krisberedskap) and was performed in the framework of the EU project AniBioThreat (Grant Agreement: Home/2009/ISEC/AG/191) with financial support from the Prevention of and Fight against Crime Programme of the European Union, European Commission—Directorate General Home Affairs. This publication reflects the views only of the authors, and the European Commission cannot be held responsible for any use that may be made of the information contained therein.