
Abstract
In the field of diagnostic microbiology, rapid molecular methods are critically important for detecting pathogens. With rapid and accurate detection, preventive measures can be put in place early, thereby preventing loss of life and further spread of a disease. From a preparedness perspective, early detection and response are important in order to minimize the consequences. During the past 2 decades, advances in next-generation sequencing (NGS) technology have changed the playing field of molecular methods. Today, it is within reach to completely sequence the total microbiological content of a clinical sample, creating a metagenome, in a single week of laboratory work. As new technologies emerge, their dissemination and capacity building must be facilitated, and criteria for use, as well as guidelines on how to report results, must be established. This article focuses on the use of metagenomics, from sample collection to data analysis and to some extent NGS, for the detection of pathogens, the integration of the technique in outbreak response systems, and the risk-based evaluation of sample processing in routine diagnostics labs. The article covers recent advances in the field, current debate, gaps in research, and future directions. Examples of metagenomic detection, as well as possible applications of the methods, are described in various biopreparedness outbreak scenarios.
In biopreparedness there are several subsections, all equally important, and they can be divided in 2 groups: capacity building and capacity evaluation. 3 Capacity is built according to intelligence and risk assessment; training is then required to evaluate effectiveness and provide further input for capacity building. Capacity build-up and research fulfill important roles as sources of intelligence, tools to battle threats, and a methodology for implementation, evaluation, and risk assessment. The AniBioThreat European Union project aims at increasing the European Union's (EU) capacity to counter animal biological threats by building biopreparedness. 4
Animal Biological Threats
Animal biological threats can be of 2 types: threats directed toward animals and threats aimed at the human population by distribution through animals. 5 With direct threats, the production chain is targeted, and such attacks can lead to economic loss as well as having a socioeconomic impact and an effect on morale, in addition to the logistical challenges of handling the outbreak itself. In the second type, animals are a means to an end, the human target. The OIE estimates that up to 80% of the pathogenic agents suggested for use in bioterrorism have zoonotic origins and as such can be harbored by both animal and human hosts. 6 Given the often close proximity of farm production plants to populated areas, as well as the high density of animals in production facilities, large-scale outbreaks, whether artificial or natural, can have serious economic and health consequences if not controled.
Detection Methods
Rapid detection and proper risk assessment are crucial tools in countering biological threats. It is important to quickly detect and confirm the specific infection for effective outbreak control. Studies from Canada estimated that a 1-week delay in implementing control measures for a severe acute respiratory syndrome resulted in a 2.6-fold increase in the mean epidemic size and an extension of 4 weeks of the mean epidemic duration. 7 The traditional culture-based methods, as well as the immunologic and nucleic acid–based methods, suffer from a number of drawbacks when dealing with novel or unknown organisms. There are no validated culture-based methods for all organisms, and sometimes methods have poor specificity. Immunological and nucleic acid–based methods have a high specificity, making them unable to (or to very little extent) detect organisms outside of their target range. In contrast, metagenomics, a method in which the combined genomes of all organisms present in a sample are analyzed, represents a broad range, high-throughput methodology for detection that requires little or no prior knowledge of the target. This can be of crucial importance when encountering a novel or unexpected target, as it removes the initial lag caused by identification issues.
Metagenomics
Metagenomics, the genetics subfield studying the combined genomes of a sample and their interaction, is a quickly growing field. Not only does it offer a rapid high-throughput method for mapping interactions in a microbial community, but it also circumvents the need for culturing, which enables the study of the unculturable majority of microorganisms in a microbial community. 8 Next-generation sequencing (NGS) has been suggested as a future application that can improve the response to a bioterrorism incident in the food chain. 9 Metagenomics can, for instance, be useful in enabling earlier discovery of novel emerging viruses and bioterrorism incidents.10-12 However, using metagenomics approaches in bioterrorism response requires interoperability between biodefense databases. 13 Metagenomics also represents a new logistics problem for laboratories implementing the technology: data storage. The falling price of sequencing not only allows for large-scale sequencing investigations but also enables production of enormous amounts of data. Given that the price of storage is steadily increasing and that the computational overhead for analysis is rising with dataset complexity, this is one of the largest obstacles when implementing large-scale metagenomics for screening and diagnostics. 14
Target-Specific Approach
The traditional approach for DNA sequencing was introduced in the 1970s by Sanger, and this method is capable of retrieving up to 1 kb of sequence data at a time. 15 Even though Sanger sequencing has been used for several metagenomics projects, the high demand for low-cost sequencing has caused a shift to use of NGS platforms.16,17 Because of the high genetic diversity of most microbial communities, it is currently difficult to obtain enough sequence depth to sample any gene with sufficient coverage to get meaningful information on the diversity or population characteristics. To overcome this difficulty, target-specific metagenomics, which aims to obtain sequence reads from specific genes of interest based on prior knowledge of sequence information, can be a useful tool. 18
The first step in a metagenomic investigation is the amplification of single targets using polymerase chain reaction (PCR); the products are then sequenced either by Sanger sequencing or NGS methods.19,20 Target-specific metagenomics generates low amounts of data compared with whole-genome sequencing (WGS), thus allowing analysis of pathogen diversity in communities in a shorter time. This method is therefore useful for screening of pathogens in samples before WGS.
Whole-Genome Approach
The whole-genome approach was pioneered to detect viruses using broad range PCR methods in the late 1990s and early 2000s.21,22 With the introduction of affordable sequencing, and in the wake of the technological developments during the Human Genome Project, massive paralleled Sanger sequencing became a reality and with it the ability to sequence large amounts of clonal libraries at once. 23 Following the first few pioneering experiments, several important discoveries were made within 5 years, proving that our understanding of viral diversity in host species (in this case, human) was so far largely unexplored.24-27 Given the introduction of NGS platforms in 2005, the field quickly expanded to encompass this new technological breakthrough. Several successful studies and proof of concept articles were produced in a relatively short span, and in 2012, a total of 198 articles using deep sequencing for detection of viruses were published. Whole-genome based approaches also were used for the bioterrorism investigation of the anthrax letters in the Amerithrax case. 28
The whole-genome methodology involves the isolation of genetic material from the sample, enrichment toward the target, enrichment using nonspecific amplification strategies, and finally deep sequencing of the sample. Strategies for sample preparation (extraction of genomic material and enrichment of target-specific material) and amplification of material vary greatly between groups. 29 Some favor the unmodified version that was first pioneered (with both enrichment toward target organism as well as amplification), while others suggests that this might introduce undue bias into the datasets. 30 Both methodological approaches have merit and are proven principles described in several articles in the literature.
Technical Review of Metagenomics
Two things are of outmost importance for metagenomic applications: to preserve the metagenome as intact as possible, and to provide as much contextual information as possible. For possible forensic or epidemiologic use, the contextual metadata provided with the sample might be just as important as the sample itself.
Sampling
Metagenomics presents a unique challenge to first responders and field personnel performing the sampling when it comes to deciding what to sample. Because metagenomics has a proven efficiency for detection in solids, liquids, and environmental samples, the need for awareness of one's surroundings for correct sampling is essential. There are many different sample types to be considered, including clinical, water, vegetables, feed, and environmental samples. These sample types have various metadata that should be evaluated for complete analysis of the results.
For clinical samples, the OIE terrestrial manual is the gold standard for methodologies considering animal health and animal welfare, including disease control and surveillance sampling.31,32 The main samples to be taken from a live animal are blood, feces, skin, genital tract swabs and semen, eye and nasal discharge, and milk. For postmortem investigations, the normal guidelines for necropsy should be applicable. 33 When sending samples, a risk assessment according to OIE guidelines, as well as the guidelines for the select agents list, should be made concerning possible risks associated with entry, exposure, and the consequences thereof. 34
Sample Treatment
Depending on the sample type, different pretreatments can be used. Each sample type requires a specific protocol, and various types of methods for extraction of nucleic acids are available.35-37 If extraction of nucleic acids of microorganisms is associated with a host (eg, plants or animal parts), selective lysis are required to ensure minimal contamination of nucleic acids from the host in the subsequent analysis. 38
For liquid samples, selective filtration is an opportunity, whereas for solid samples separation and isolation of cells is an alternative strategy.16,39,40 Liquid samples can be represented by both water and various fluids from animals. The ability of the metagenomics approach to detect not only known but also unexpected and unknown microorganisms has been extensively evaluated and proven to work for liquid samples.24,41-43 The general procedure is to concentrate the sample and then take a fraction of the sample for sequencing. Solid material can be from environmental samples (eg, swabs from surfaces), food, feed, or whole organs or partial animal parts. Extraction from soil and sediments (eg, environmental samples) is often more difficult than pure cultures because of the presence of inhibitors. The inhibitor can originate from soil compounds that might inhibit the extraction or compounds that co-purifiy with the nucleic acids, such as humic acids, which can interfere with downstream applications.44-46 For these types of samples more thorough sample treatments are needed. 47 The sample preparation has been shown to influence results from metagenomics analysis—for example, when introducing a freezing step to store the samples before analysis 48 and when starting with a culture enrichment step. 49
Since 9/11 and the Amerithrax case, microbial forensics has been developing at great speed. When a forensic investigation of an outbreak is performed, it would be prudent to perform the examination in the way that human medical forensics are conducted, if results are to be included in a possible forensic investigation at a later point. The literature lists several procedures to be taken for preserving and stabilizing samples used for forensic microbiology. 50 Three main areas can be of interest: extraction techniques and applications, preserving and stabilizing samples, and assessing and controling sample contamination. All of these are extensively covered in other literature and will not be discussed here in more detail. 32 Figure 1 provides a schematic view of sampling and sample preparation procedures.
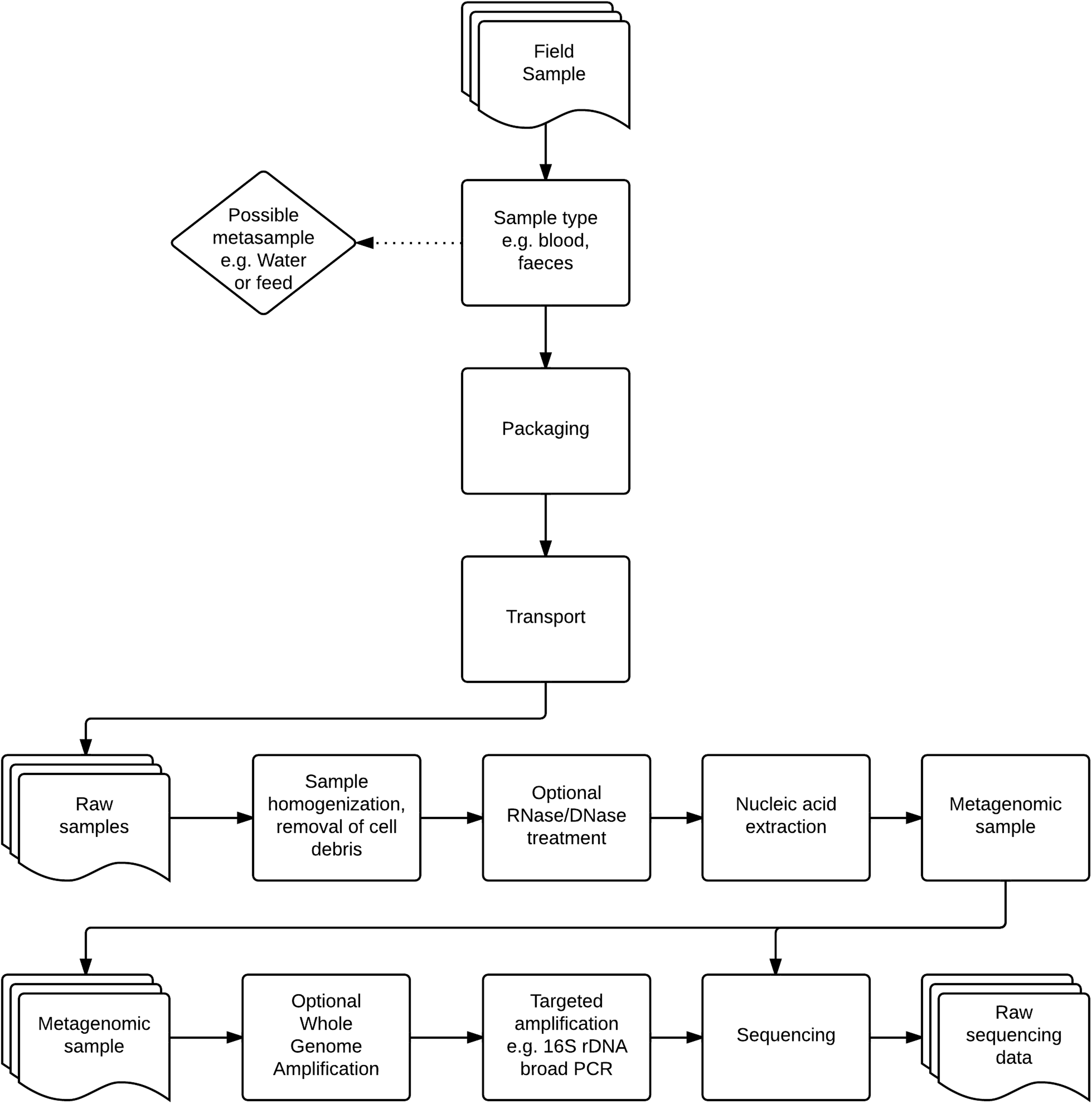
Samples are collected in the field. Following classification and collection of possible metasamples (samples related to the incident site such as feed or water for the animals), risk assessment is performed on site, and samples are packaged in accordance with OIE recommendations and international rules for transport of biological specimens. As samples arrive at the diagnostic lab, they are homogenized, cell debris is removed, and, depending on agents of interest, an optional DNase/RNase step is performed to degrade parts of the metagenome. Nucleic acid extraction is performed on the samples, and they are stored. Samples can now pass through 2 optional steps: whole genome amplification or targeted amplification (eg, 16S rDNA amplification). Sequencing is performed, and raw sequencing data are produced.
Sequencing Technologies
For a considerable amount of time, the long read length paradigm held true for metagenomic studies; thus, technology was chosen based on how long a read it could produce. This resulted in a number of high-profile novel virus articles, as well as microbial 16S rDNA sequencing to characterize microbial communities. As read length was the main criterion for choosing technology, the Massively Parallel Pyrosequencing, 454 Roche, was the primary choice in metagenomic studies incorporating NGS.39,51-53
The introduction of the Four-Color Reversible Chain Termination (Illumina, HiSeq) technology allowed unbiased broad-range sequencing of the microbial community. In this approach, relatively short reads are sorted and classed into subgroups and thereafter analyzed. Even though this method has proven useful, the shear amount of sequence reads, as well as time-consuming analysis and lack of specificity, are factors limiting an extensive use of this technique.54-57
In 2011 LifeTechnology released the Personal Genome Machine (PGM). The PGM employs a technique called sequential electrical detection, in which sequencing of the template strand is performed inside a microchip, directly transferring the released ions into the semiconductor, thereby reading the sequence by the current produced.58,59
Choosing Sequencing Technologies
The choice of sequencing technology for metagenomic investigations is based on 4 main factors: length of sequences, depth of sequencing/throughput of technology, speed of sequencing, and automation availability.60-62 Length of sequences still plays a major role in allowing easy alignment toward genomes of interest, spanning possible repetitive regions, and providing sequence islands in the target genome for further sequencing. 63 Depth and throughput of sequencing depends on (1) how selective you have to be with your sample (selection of parts of the microbiome or presequencing sample preparation), and (2) how many samples you can multiplex (design of assay). The speed of sequencing is of utmost importance from a biopreparedness perspective; technologies taking up to 2 weeks to finish a run are of limited use in an outbreak situation. Finally, automation and the possibility of making the metagenomic approach interoperable in the current automated diagnostic pipeline are factors to consider.
Table 1 presents a compilation of data as given by the sequencer manufacturers adjusted for experimental results, including possible applications, costs for running and acquiring the instrument, and speed for a single run. In 2012, Loman et al provided an excellent initial overview of the newly introduced benchtop sequencers. 60 The IonProton was introduced after the experiment, as were the increased read-length chemistry for MiSeq and IonTorrent. Suggesting a single technique is therefore complicated. At the time of the writing of this article, MiSeq held the lead in throughput for the 250bp chemistry and has a compelling sample preparation, whereas the IonTorrent provides longer read lengths and faster sequencing turnover. The capabilities of the benchtop sequencers do, however, regardless of platform, provide medium-sized laboratories with a good capability to provide rapid metagenomic analysis of clinical samples on a medium scale. The technology will also likely be developed considerably in the near future, making it simpler and having even more capacity.
List of Sequencing Technologies, Their Properties, and Possible Applications
Note. The list includes the benchtop version if possible (454 Junior, Illumina MiSeq, and Ion Torrent/Proton Torrent), speed of sequencing as well as possible mean read length, and total data output together with procurement costs and running costs.
Bioinformatics and Processing of Data
An integral part of NGS technologies is the postsequence processing of data. After the data have been produced by the chosen sequencing technology, the output is often an abundance of sequence reads with little or no meaning to the untrained eye. Postsequencing processing is applied to raw sequence data to perform quality control (QC), filter reads for contamination, map reads toward relevant genomes, assemble genomes (or partial genomes), and perform homology searches and taxonomic/phylogenetic studies.29,64,65 This is shown in Figure 2 as a flowchart of methodologies and implementation of methodologies for analysis.
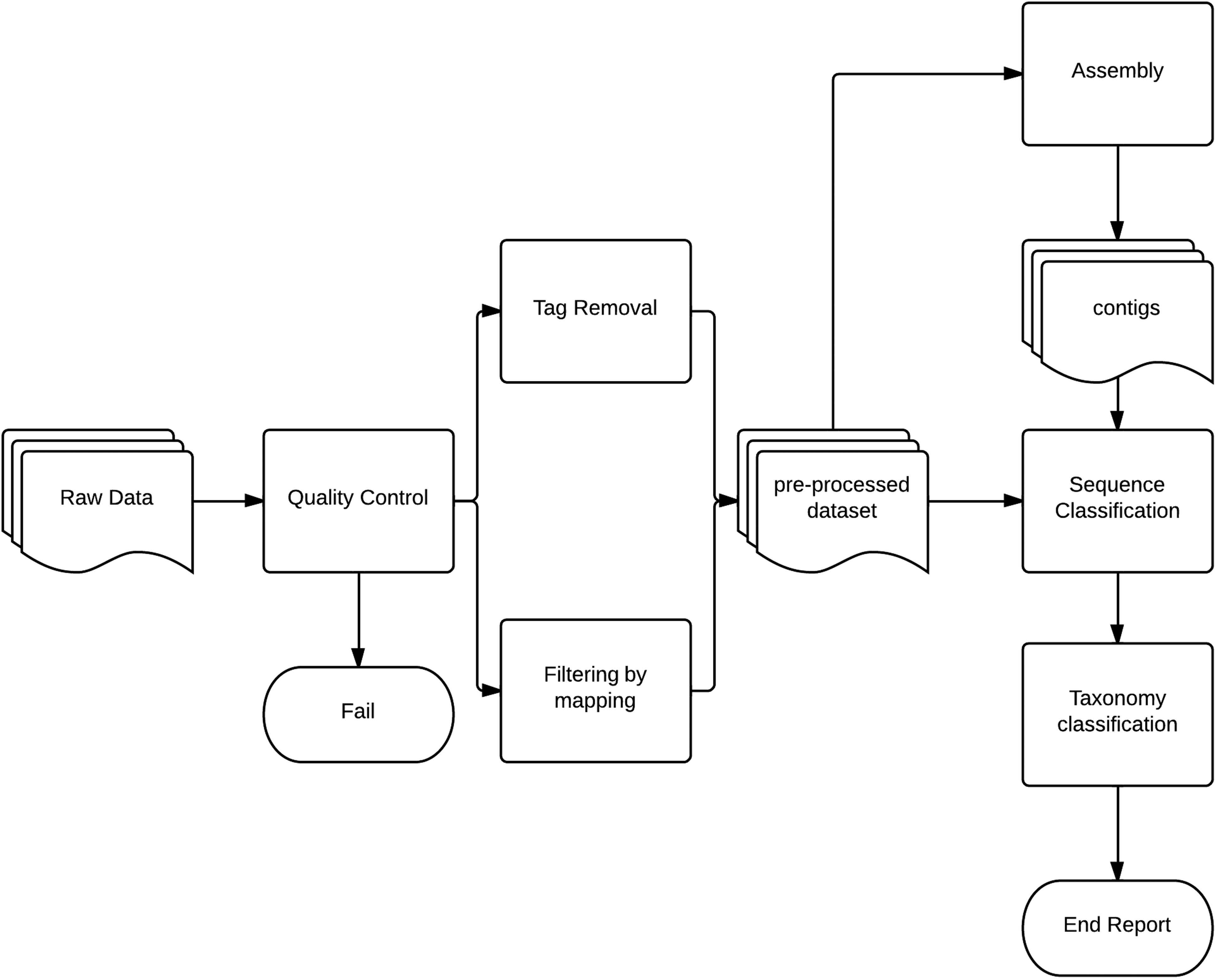
Raw data from sequencing is checked for quality irregularities. Tags from sequencing technology and/or preamplification step are removed, and reads are optionally filtered toward host genome or known contaminating genome (eg, process controls, etc). The preprocessed dataset is split into 2 paths, 1 performing assembly of the reads and 1 mapping the reads directly. Products from assembly are then subjected to the same sort of homology search as the single reads. Finally, the datasets are combined and evaluated for taxonomic classification.
Quality of datasets for metagenomic analysis consists of several parts. First, base calling is generally performed by the vendor implementation, crafted into the technology itself, filtering away reads of dubious quality and providing the user with an output from the sequencer. 66 After the initial filtering, the resulting dataset is usually composed in a data format containing all the relevant information in 1 file. Most vendor pipelines also include the possibility for directly mapping the data toward a genome, giving the user the possibility to map sequences toward several microbial organisms or to map sequences toward the host genome for removal. Mapping of reads toward a genome is functional when a known genome is present; this can either be a host genome (to more efficiently filter the reads and get an estimate of host contamination) or added process controls.67,68 This step is normally not used extensively for metagenomic analysis since the strength lies not on what you know is in the sample but in the broad range of detection. Users can then apply several different quality checks on the dataset to determine its validity for the experiment.
During processing the identification and removal of systematic artifacts is also of great importance, as bias introduced this way can skew the analysis and provide a shifted final picture of the metagenome. 69 By combining several tools, the user can remove most artificial duplicate reads, clean away tag sequences, assess guanine-cytosine content, trim the sequences, and provide initial filtering by quality, ambiguous base, and sequence complexity. 70 After initial filtering, as much as 80% to 95% of the initial read counts can be removed, depending on the presequencing processing.55,71 This provides the user with a considerably easier dataset to handle. Combined with classification of sequence reads, assembly of short reads can render additional information about the organisms present in the metagenome. Because of the complex nature of metagenomic datasets, assembly can introduce various in silico artifacts into the dataset. 72 These artifacts can be of a different nature, but usually include chimerical contigs and ultradeep contigs. Assemblers are continuously developing new applications, and several metagenomic assemblers have been introduced in the past few years.73,74 These assemblers aim at taking the complexity of the metagenome into the assembly, thereby offsetting the possible assembly bias. Moreover, these provide individual unique sequences into the final dataset, thus preserving data for the final analysis.
The final step of a metagenomic analysis is the classification of the sequences. Classification is usually performed by homology-based methods, but both composition-based methods and phylogenetic methodologies exist. These 3 main approaches are sometimes also combined for increased speed and accuracy. (See the review by Bazinet and Cummings for extensive information about classification. 75 ) It is noteworthy that homology-based methods have high accuracy and can be fast when using a small database for the query. This provides a possible use for rapidly mapping of a metagenomic sample toward, for example, the select agent, giving fast information about the agents present in a sample. 76
When reporting metagenomic results, it is important to take into consideration the metadata. The Genomic Standards Consortium provides guidelines for reporting metagenomes. Combined with proper sample documentation according to the OIE sampling standards, these guidelines will suffice for most in-depth analyses of metagenomes for several applications, including preparedness questions or basic science. 77
Metagenomics-Based Approaches in Outbreak Investigations
There are several examples of how metagenomics-based approaches have been used in investigations aimed at finding the source and origin of a disease outbreak, including bacteria, viruses, and parasites.42,78-81 We present 2 examples to explain the usefulness of applying metagenomics in an outbreak situation when other diagnostic tools were found to be insufficient.
Regions in North Rhine-Westphalia, Germany, and in the Netherlands started in 2011 to see a decline in milk production, and several clinical signs such as fever and diarrhea in cows were identified. Conventional methods failed to establish the source of the infection, and reasons for the decreased milk production were unclear. The Friedrich-Loeffler Institute, Germany, therefore launched a metagenomic study using the 454 Genome Sequencer FLX technology. During sequence analysis, a homology toward 7 orthobunyavirus sequences was discovered. Using repeated sequencing of the original library rendering the reads, a partial viral genome was recovered. Filling the gaps with conventional Sanger sequencing, the Schmallenberg virus was described on a genome level.43,82
Another case was the enterohemorrhagic Escherichia coli (EHEC) outbreak in Germany in 2011. This case demonstrated how sequencing technologies could be used in the early stages of an outbreak to characterize the causative agent of the outbreak. 83 The outbreak strain was shown to be a hybrid, which means that the current molecular methods used for detection, relying on either detection of virulence genes that are linked to known bacterial serotypes or detection of only the virulence genes, were found to be unable to detect and correctly characterize the outbreak strain. The use of WGS enabled identification of sequences that were specific to the outbreak strain. 78
Discussion
The evolution of high-throughput sequencing (HTS) technologies toward inexpensive equipment and sequencing runs is slowly making an impact in clinical laboratories. As with all technology introductions, laboratories are now faced with the enormous task of standardizing methodologies and procedures and incorporating results from the new technology into the communication and command structure. In outbreak situations with unknown agents, the criteria for detection are, however, somewhat less stringent than in routine diagnostics. Therefore, NGS-based technologies and methodologies can be a powerful tool in preparedness for detecting unexpected microorganisms. 84
Diagnostic Use of Metagenomics
The major obstacles to diagnostic use of metagenomics are the timeframe of the analysis and the lack of experienced personnel to perform the data analysis. The first point is slowly resolving itself as the benchtop sequencers today are moving into a 2-day preparation and sequence timeframe. This is comparable to culture-based and serological methods. The second point is more critical, as the lack of trained informatics personnel is at the moment a serious problem. There is to our knowledge no available and thoroughly tested standard NGS methodology for use in an outbreak. Consequently, there is a need to develop standard operating procedures (SOPs) for field-based sampling by veterinarians or first responders, as well as to develop sample preparation protocols in clinical laboratories. Core facilities, for example, in the EU, should engage in interoperability exercises to ensure common understanding of the methodologies, to exchange expertise, and to identify research needs.
In an outbreak situation, where the origin of the causative agent is not known, the extraction of nucleic acids in a representative manner is the major hurdle for success. Extraction of nucleic acids directly from the sample might introduce bias of the purified DNA or RNA, which in the end could produce misleading results. The methods used for lysis of the cells affect the composition of the total nucleic acid present, since harsh lysis methods that are needed for extraction of DNA from some bacteria (eg, Gram-positive bacteria) might degrade DNA or RNA from others. 85
Current research has been focused on the detection of novel viruses, a process performed differently depending on, for example, group, sample accessibility, experimental planning, and technology used for NGS. In virology, metagenomics is a proven methodology for detecting unexpected or novel viruses in clinical samples. 84 When it comes to the bacterial field, metagenomics has mainly been used for investigation of large bacterial communities, such as the human gut, or for environmental samples.16,86 However, the use for diagnostic purposes is emerging.
Additionally, the discussion about sequence-based microbiology must be taken further. The same caveat as with PCR-based technologies exists; finding DNA or RNA of an organism does not prove the presence of a living or intact form of that organism. Several factors can produce false-positive results, and with the need for some assembly in several NGS technologies the risk for technological artifacts increases. For well over 100 years, Koch's postulates have been seen as a gold standard for detection of pathological entities. Late in the last century, these postulates came under question with the broad introduction of nucleic acid–based methods into diagnostic microbiology. With the subsequent introduction of NGS methods, there is a clear need for microbiologists to redefine the criteria for how to define causative agents, as reviewed extensively by others.84,87
Theoretical Applications in Diagnostic Preparedness
Metagenomics is today one of the few molecular methods for detection of novel microorganisms, as well as a tool with extreme throughput capabilities. It is our belief that in metagenomics there is a methodology for the unbiased detection of the microflora of a sample, thus offering a diagnostic capability for tracing introduced organisms at an unprecedented level. In other words, metagenomics can detect several pathogenic organisms in 1 sample, including genetically modified versions of known pathogens. Several different disciplines in the preparedness structure could use such a method applied to microbial forensics, smuggling, outbreak investigations, and R&D needs.
Microbial Forensics
Microbial forensics aims at detecting microorganisms from a forensic perspective—that is, detection of microorganisms provides forensic clues to investigators. 88 In a preparedness structure, the forensic field can be seen as a part of the epidemiologic work of tracking the origin of introduction. There is also the direct forensic approach in the possible deliberate spread of microorganisms where metagenomics can provide both traceability and detection.
Customs and Smuggling
It is feasible to theorize that high-resolution metagenomics will provide profiling indicating the geographic origin of individual animals. As such, one could hypothesize that it is possible to use metagenomic profiles of the gut microbiota to determine the origin of animals. Previously, we discussed the broad range of detection, the ability to handle a large amount of samples, and the multiplexing capability. This fits well into the customs perspective: large amounts of material to be tested, unknown origin, unknown agents, and possible grave consequences if organisms slip the grid. Given the movement of illegal animals in the EU and globally, large-scale sequencing for broad screening might be implemented in the near future. This would also include the legal and illegal trade of food commodities.
Determining the Causative Agent of an Outbreak
In microbiology, and virology in particular, metagenomics is an important technique to strengthen the capability to handle previously uncharacterized microbial agents and to provide diagnostic laboratories with previously unattainable capabilities for handling outbreaks.
Research Needs
In general, the time from sample to proof of presence of a potential pathogen needs to be speeded up considerably. All steps in the analytical chain (Figure 2) can be improved and simplified, as well as automated. At the moment focus should be put on the bioinformatics, to develop easy-to-use programs that ideally can warn of the presence of potential pathogenic microorganisms in a given sample. A follow-up on this would be to, in the same set, suggest primers for fast real-time PCR detection of the detected organisms, coupled to an oligonucleotide synthesizer that synthetizes new oligonucleotides that can be used for further strengthening of the proof of the presence of the given pathogen.
In virology there is a clear need for validation of methodology toward a larger sample set of domestic animals. Producing a gold standard for metagenomes, artificial or naturally occurring, would also prove beneficial. As with most molecular techniques involving virology, isolation of the complete viral genome from a clinical sample should be standardized and implemented in diagnostic laboratory procedures. As discussed, the need for establishing whole-genome amplification methods, selective toward microorganisms, and incorporating them into the workflow might be of great use, but the increasingly powerful sequencing equipment might evolve to provide solutions that make this need redundant.
Discussion about the cut-off and reporting of results must also be taken into consideration for implementation in a preparedness setting. Possibly harmful information such as the presence of a pathogen on the restricted list or novel organisms must have standardized procedures for validation.
For large-scale viral screening, the metagenomic approach can be combined with bioinformatics solutions to select for only viral genomes, greatly speeding up the process of aligning sequence results toward possible agents. This combined with a backlog procedure for analyzing the whole metagenome, preferably automated, could greatly improve the rate of detection of novel and emerging viruses.
Further research is also needed for bacteria. The major need is for optimized and standardized methods for sampling and extraction of nucleic acids. Standardized methods should also include ways to quantify possible bacterial contamination to investigate if what is seen is out of the ordinary.
Another area that needs more focus is the link between genotype and phenotype. The uncertainty in predicting the phenotype solely on genomic or metagenomic data implies that the culture-based techniques never will be completely replaced. 89 Furthermore, for certain toxin-producing bacteria (eg, Clostridium botulinum) it is not necessarily the bacterium itself that is the causative agent, but the toxins. The presence of active toxins cannot be identified by genome-sequencing techniques. For this reason, other techniques are needed such as the Endopep-MS, in which the presence and activity of toxins is investigated. 90
Conclusions
This article has identified potential applications of metagenomics technologies for biopreparedness applications. For instance, metagenomic approaches for molecular detection of microbial agents are important in a biopreparedness perspective and hold great merit for laboratory response capabilities. Not only does it improve on the current molecular methods in throughput and flexibility, it also presents the only currently unbiased high-throughput methodology for broad screening of samples. Metagenomes as a research field also provides new insights into biomarkers produced by the microorganisms connected to animals, enabling new tracing of food and feed as well as possible forensic applications.
Footnotes
Acknowledgments
Writing of this publication has been supported by the framework of the EU project AniBioThreat (Grant Agreement: Home/2009/ISEC/AG/191) with financial support from the Prevention of and Fight against Crime Programme of the European Union, European Commission—Directorate General Home Affairs. This publication reflects views only of the authors, and the European Commission cannot be held responsible for any use that may be made of the information contained therein.