
Abstract
This review focuses on mass spectrometric detection of protein-based toxins, which are among the most toxic substances known. Special emphasis is given to the bacterial toxins botulinum neurotoxin from Clostridium botulinum and anthrax toxins from Bacillus anthracis as well as the plant toxin ricin produced by Ricinus communis. A common feature, apart from their extreme toxicity, is that they are composed of 2 polypeptide chains, one of which is responsible for cell uptake and another that has enzymatic function with the ability to destroy basic cellular functions. These toxins pose a threat, both regarding natural spread and from a terrorism perspective. In order for public health and emergency response officials to take appropriate action in case of an outbreak, whether natural or intentional, there is a need for fast and reliable detection methods. Traditionally, large molecules like proteins have been detected using immunological techniques. Although sensitive, these methods suffer from some drawbacks, such as the risk of false-positives due to cross-reactions and detection of inactive toxin. This article describes recently developed instrumental methods based on mass spectrometry for the reliable detection of botulinum neurotoxins, anthrax toxins, and ricin. Unequivocal identification of a protein toxin can be carried out by mass spectrometry–based amino acid sequencing. Furthermore, in combination with antibody affinity preconcentration and biochemical tests with mass spectrometric detection demonstrating the toxin's enzymatic activity, very powerful analytical methods have been described. In conclusion, the advent of sensitive, easily operated mass spectrometers provides new possibilities for the detection of protein-based toxins.
Traditionally, the detection of large molecules like proteins has been carried out with immunological methods such as radio immune assays (RIAs) and enzyme-linked immunosorbent assays (ELISAs).6-11 These techniques are usually very sensitive, but they do not provide unequivocal evidence for the presence of a certain toxin, owing to the risk of cross-reactions with similar substances. 12 Furthermore, these tests do not provide information on the degree of toxin activity.
Mass spectrometry (MS) is an instrumental analytical technique that measures the mass-to-charge ratio of ions, making it possible to calculate molecular masses. In addition, by different fragmentation techniques, it is possible to use MS to measure the mass of different parts of a molecule, giving fingerprint-like results that can be used for identification purposes. MS has traditionally been used for small molecules, but during the past decades, MS-based methods for the analysis of proteins and other macromolecules have emerged.13,14 The principle of MS analysis is completely different from that of immunological methods. MS can give absolute information on the identity of a protein by measurement of its molecular mass, amino acid sequence, and posttranslational modifications. 13 Combinations of biochemical tests with MS detection are also available for activity measurements of some enzymatic toxins. This review focuses on the applications of modern mass spectrometric techniques to the determination of the highly potent protein toxins from bacteria of the Clostridium and Bacillus genus, mainly botulinum neurotoxin (BoNT) and anthrax toxins, as well as ricin from the plant Ricinus communis.
Clostridial Neurotoxins
Clostridial neurotoxins, such as tetanus toxin (TeNT) produced by Clostridium tetani and the botulinum toxins (BoNTs) produced by Clostridium botulinum, C. baratii, and C. butyrium, are high molecular weight proteins consisting of a heavy and a light chain (Figure 1). The heavy chain is responsible for internalization to cholinergic nerve terminals, and the light chain can act as a zinc-dependent protease. When activated, the light chain of these toxins attack 3 proteins involved in forming the SNARE (soluble N-ethylmaleimide sensitive factor associated protein receptors) complex, resulting in severe clinical symptoms.15-17 Tetanus (from the Greek word tetanos=to contract) is characterized by muscular rigidity and spastic paralysis due to blockage of inhibitory synapses afferent to motor neurons, while botulism (from the Latin word botulus=sausage) is characterized by flaccid paralysis caused by inhibition of the release of acetylcholine at the neuromuscular junction. 15 There are 7 serotypes (A-G) that have been identified in botulism outbreak cases in humans (BoNT/A, /B, /E, and /F), birds (BoNT/C), and cattle (BoNT/D) or isolated from soils (BoNT/G). 15 The different serotypes are about 40% similar at the amino acid level. The BoNT/C and /D serotypes can also exist as mosaic C/D and D/C forms, while the other serotypes can be divided into subtypes: /A1, /A2, and so on. 18
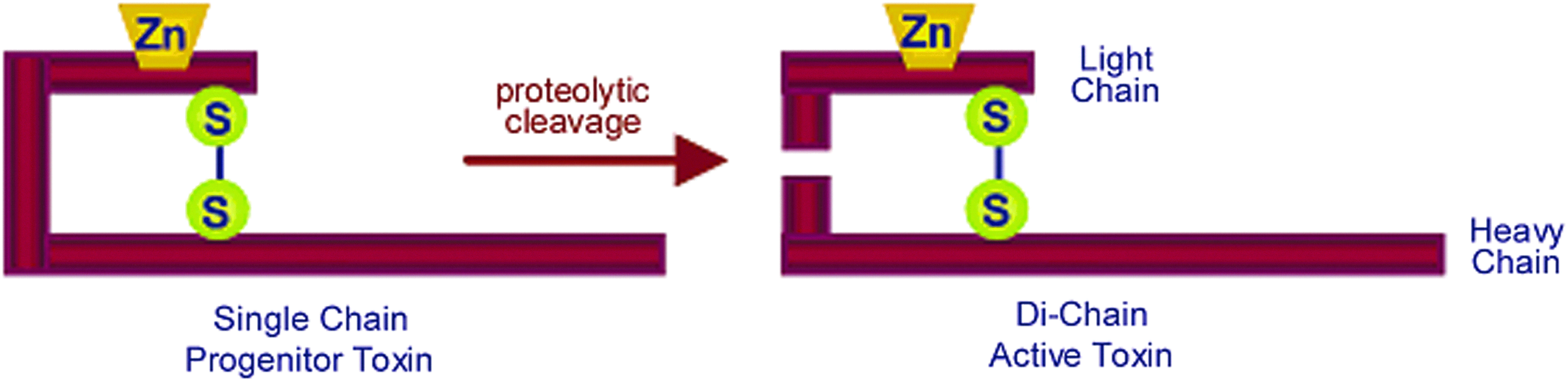
The Structure of Botulinum Neurotoxin. Endogenous proteases cleave the single chain progenitor into the active dichain form. The heavy chain is responsible for binding and internalization, while the light chain carries the zinc-dependent endoproteinase activity. Reprinted with permission from Boyer AE, Moura H, Woolfitt AR, et al. From the mouse to the mass spectrometer: detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal Chem 2005;77:3916-3924. Copyright 2005 American Chemical Society. Color images available online at www.liebertpub.com/bsp
Botulinum neurotoxins are the most potent of all bacterial toxins. The estimated lethal dose of BoNT/A for humans is believed to be about 1-2 ng/kg intravenously, 10-13 ng/kg by inhalation, or 1 μg/kg orally. 19 The extreme toxicity of these toxins can be explained by the fact that they are neurospecific enzymes—that is, one single active toxin molecule inside the synapse is all it takes to destroy the protein needed for a functional neuroexocytosis. 20 For foodborne botulism, common foods contaminated with BoNTs are low-acid vegetables, meats, or fish.21,22 Due to its extreme potency in combination with its ease of production, BoNT poses a major bioweapon threat, and the development and use of BoNT as a biological weapon has been described. 19 For botulism, antitoxins administered early can minimize nerve damage and severity of disease but will not reverse paralysis. 22 Rapid and sensitive diagnostics are very important, since botulism is a life-threatening disease and only a small amount of the toxin is needed to cause disease.
The mouse bioassay, developed by the British pharmacologist J. W. Trevan in 1927, is still considered to be the gold standard for botulinum toxin detection.22-24 In this procedure, test samples suspected to contain BoNTs are injected into mice, which are observed for up to 4 days for symptoms of botulism. The toxin serotype is determined by injecting the antitoxins (eg, anti BoNT-A, anti BoNT-B, etc), one by one, mixed with the sample. Only the mice injected with the correct antitoxin will survive. 22 With this assay only active toxin is detected, and the limit of detection is low (<10 pg/mL); but it takes several days to perform the assay, and many mice are required for each sample.18,22,23 In the past decade, several new methods have been developed, aiming to replace the mouse bioassay. Ideally, the method should detect and quantify active toxin with high specificity, sensitivity, and speed.
Immunoassays with different designs (eg, RIAs, lateral flow immunoassays, and ELISAs) have been described and previously reviewed by others.6,7,18,23,25 Advantages of ELISA methods compared to the mouse bioassay are that no mice are needed, that they are much faster (hours compared to days), and that many samples can be analyzed simultaneously on 96-well plates. Early ELISAs generally had poor sensitivity and selectivity, but newer methods with signal amplification approaches have resulted in increased sensitivity. However, these are not functional assays; thus, inactivated toxin may cause false-positive results, matrix components can affect the antibodies, and genetic variation within the different serotypes might result in false-negatives. Polymerase chain reaction (PCR) assays that target the BoNT gene have been reviewed,6,23,25 but detection of the toxin gene does not mean that the toxin itself is present in the sample.
Reliable identification of protein toxins by peptide sequencing has been achieved by van Baar for tetanus toxin, 26 and botulinum toxins type/A and /B, 27 and /C, /D, /E, and /F 28 with both electrospray (ESI) tandem mass spectrometry (MS/MS) on a quadruple-time-of-flight (Q-TOF) instrument and matrix-assisted laser desorption ionization (MALDI) TOF-MS. The sequence coverage was more than enough to identify the toxins studied. Preferably the identified peptides should be distributed across the entire protein, which is especially important to distinguish pure BoNT/C and /D from the mosaic types. 28 A few years ago, the Klaubert group developed a selective and sensitive 2-dimensional column switching nanoliquid chromatography system combined with ion-trap mass spectrometry to detect BoNT/A, /B, /E, and /F by their peptide sequence after tryptic digestion. The digestion was performed directly on the culture supernatant from different C. botulinum strains. 29 BoNTs are associated with other proteins, hemagglutinating proteins (HAs) and nontoxic nonhemagglutinin protein (NTNH), that form the BoNT complex. These can be seen as either a hindrance or benefit to MS analysis. The presence of the additional copurifying proteins can complicate identification. On the other hand, identification of specific sequences in these proteins provides additional certainty in the toxin type identification. 27 Even if these methods succeeded in detecting the toxin, they did not provide any information about its concentration or activity.
There are also assays based on the protease activity of the BoNTs. The clostridial neurotoxins are highly specific in their recognition and cleavage of VAMP-2 (vesicle-associated membrane protein 2, also called synaptobrevin), SNAP-25 (synaptosome-associated protein of 25 kDa), or syntaxin (Figure 2). Those 3 proteins are needed to form the SNARE complex, enabling fusion of synaptic vesicles with the membrane of nerve endings, resulting in the release of acetylcholine in the synaptic cleft. In mammals, TeNT, and BoNT/B, /D, /F, and /G cleave VAMP-2; BoNT/A, /C, and /E cleave SNAP-25; and BoNT/C also cleaves syntaxin. 15 In these assays synthetic peptides, containing the specific cleavage sites, are added, and the cleavage products are detected. The first assay based on the endopeptidase activities of BoNTs was described by Clifford Shone's group in 1996. 30 They used synthetic peptide substrates mimicking VAMP for BoNT/B and SNAP-25 for BoNT/A. After the endopep reaction, an ELISA was applied to detect the cleavage products. 30 Later, a step where the toxin was captured on an immunoaffinity column was included to remove potential interferences in the matrix and thus increase the sensitivity of the assay. 31 In another assay developed by Bagramyan et al, the BoNT/A proteolytic activity was determined by fluorescence detection of cleaved fluorogenic substrate peptides. 32
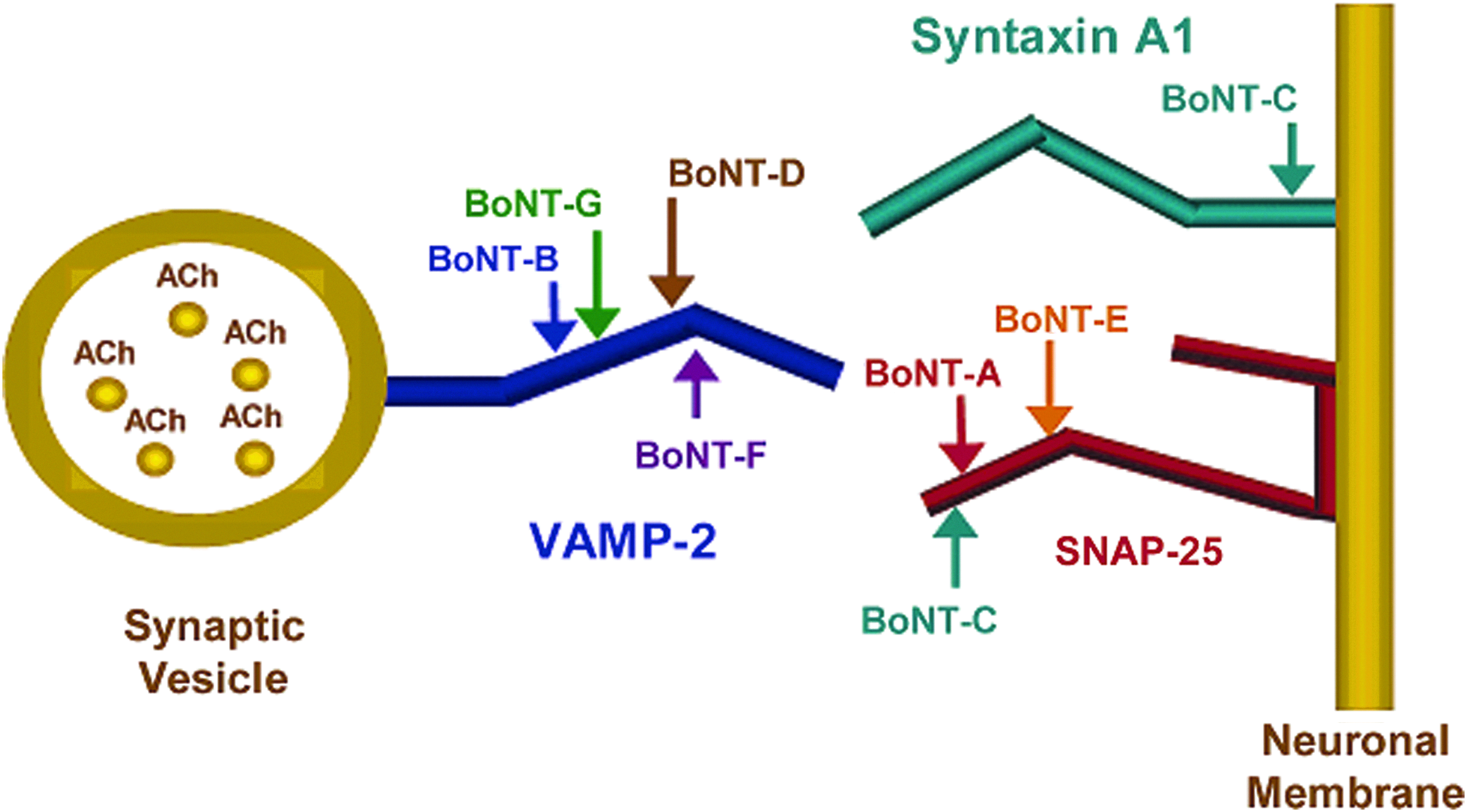
The BoNTs specifically cleave either VAMP-2 on the synaptic vesicle or syntaxin or SNAP-25 on the neuronal membrane, which are the peptides forming the SNARE complex. The complex enables the fusion of the synaptic vesicles inside the nerve cell and the neuronal plasma membrane and thus the transmission of a nerve impulse by the release of acetylcholine (ACh) to the synaptic junction. Reprinted with permission from Boyer AE, Moura H, Woolfitt AR, et al. From the mouse to the mass spectrometer: detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal Chem 2005;77:3916-3924. Copyright 2005 American Chemical Society. Color images available online at www.liebertpub.com/bsp
John Barr's group at CDC in Atlanta was the first to combine an activity-based BoNT method with MS. The assay, called Endopep-MS, was first published in 2005. 33 LC-ESI-MS/MS and MALDI-TOF-MS were used to detect the synthetic peptides mimicking the proteins SNAP-25 and VAMP-2, as well as the N- and C-terminal cleavage products formed after incubation with BoNT/A-G (Figure 3). The major advantages with the Endopep-MS assay are that it detects active toxin, is fast compared to the mouse bioassay (hours instead of days), and by mass spectrometric detection of the mass-to-charge ratio of the peptides excellent sensitivity and specificity is reached.
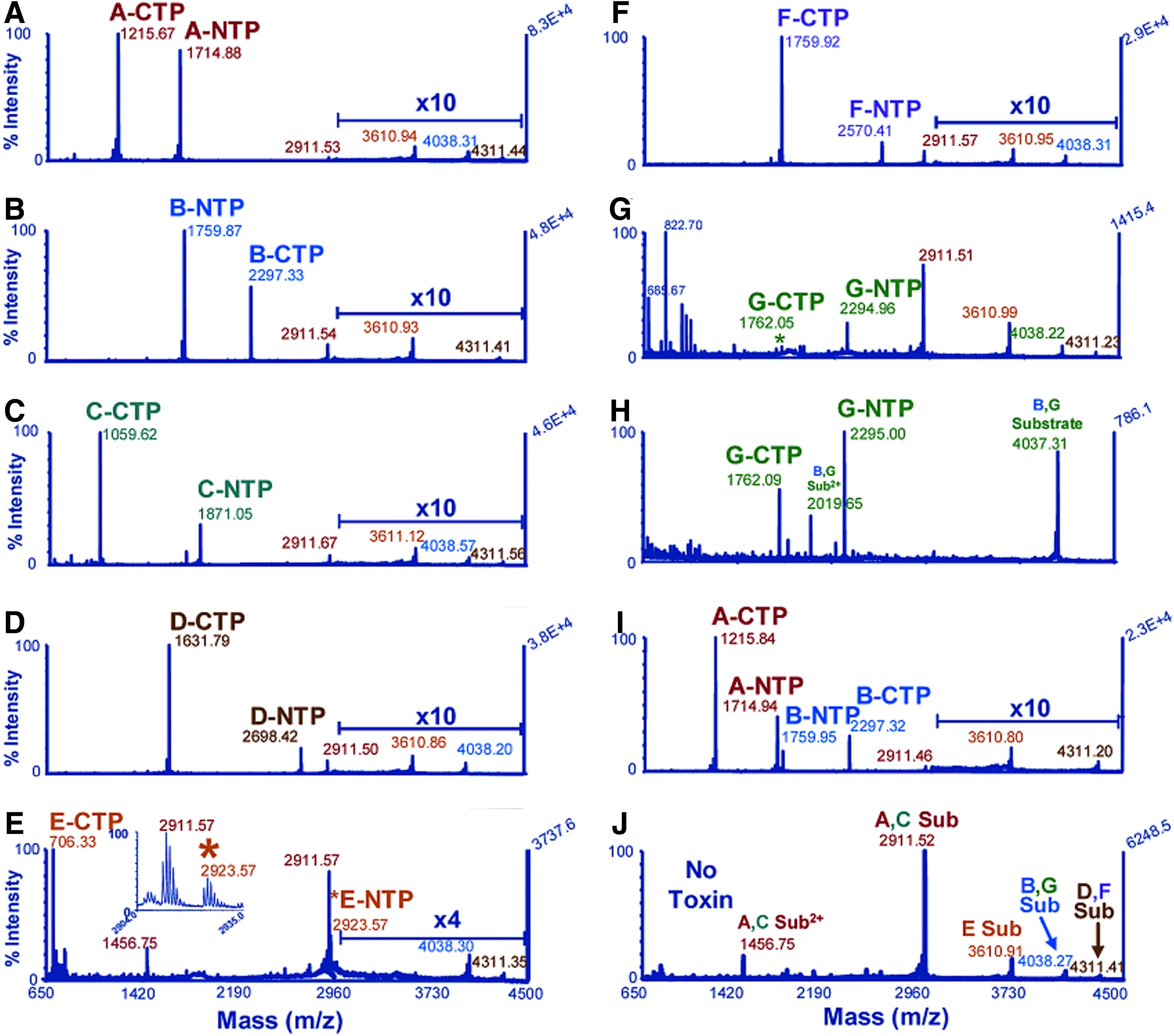
Multiplexed Detection of BoNT/A-G by MALDI-TOF-MS. Substrates (Sub), C-terminal (CTP), and N-terminal (NTP) cleavage products indicated in the mass spectra. Reprinted with permission from Boyer AE, Moura H, Woolfitt AR, et al. From the mouse to the mass spectrometer: detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal Chem 2005;77:3916-3924. Copyright 2005 American Chemical Society. Color images available online at www.liebertpub.com/bsp
Since then, the Endopep-MS method has been improved and applied on clinical samples. The addition of an antibody affinity method for purification and concentration of BoNT/A, /B, /E, and /F from serum and stool has been described.34-37 By this approach, the limits of detection (LOD) of BoNT/B, /E, and /F in serum were demonstrated to be lower than those achieved by the mouse bioassay. 34 Endogenous proteases and peptidases in stool resulted in reduced sensitivity of the assay due to nonspecific cleavage of the peptide substrate. By the addition of a salt wash step and a protease inhibitor, the LOD of BoNT/A in stool was reduced to 0.5 mouse LD50 in a 0.5 mL sample. 37 The peptide substrates for BoNT/F 35 and BoNT/A 36 have also been improved in order to achieve more effective cleavage and to reduce nonspecific degradation.
The first application of Endopep-MS to veterinary clinical samples was presented in 2011 by Hedeland et al. 38 Serum, liver, and feed samples that previously had tested positive in the mouse bioassay were confirmed to have BoNT/C activity by Endopep-MS. 38 Recently, the use of Endopep-MS for toxin detection in human serum samples, in parallel with the mouse bioassay, at 2 outbreaks of BoNT/A in France was described by Mazuet and co-workers. 39
The Endopep-MS method was further developed to include 3 levels of selectivity: capture by antibodies against the heavy chain of the toxin demonstrating the intactness of the toxin, activity determination by cleavage of synthetic peptides, and protein identification by tryptic digestion and amino acid sequencing. The entire 3-step procedure was performed in 24 hours. With this last step, the subtype of BoNT/A was determined. The additional information of subtypes can be used to connect outbreaks occurring in multiple areas or the relatedness of BoNT-contaminated products from multiple areas. 40
Anthrax
Bacillus anthracis, the bacterium responsible for the disease anthrax, is spore forming, Gram-positive, and rod-shaped.41,42 Spores can enter the host in different ways, giving rise to different forms of anthrax: dermal abrasion can result in cutaneous anthrax, ingestion in gastrointestinal anthrax, inhalation in pulmonary anthrax, and injection in injectional anthrax.42,43 Cutaneous anthrax is by far the most common form of the disease, 43 whereas the pulmonary form has the highest mortality rate. 44
There are 2 binary toxins produced by B. anthracis. These are termed lethal toxin (LeTx) and edema toxin (EdTx). Each toxin contains 2 protein types: LeTx is made up of oligomers of protective antigen (PA) and lethal factor (LF), whereas EdTx is made up of PA and edema factor (EF). 45 The process starts with PA binding to receptors at the cell surface followed by hydrolysis by proteases to a 63 kDa fragment (Figure 4).46,47 This cleavage has also been shown to occur in the blood at high concentrations in late infection. 48 The 63 kDa fragments oligomerize to form heptamers or octamers that bind LF and EF. 49 LeTx/EdTx bound to receptors is then internalized in a vesicle by endocytosis.46,47 In the case of LeTx, LF is released into the cytosol, where it acts as a zinc-dependent protease and cleaves several members of the mitogen-activated protein kinase kinase (MAPKK) family, resulting in cell death.46,47,50 When it comes to EdTx, EF stays attached to the vesicle membrane, where it works as a calmodulin and Ca2+ dependent adenylate cyclase, turning adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP), resulting in a massive increase of cAMP. 51 This interferes with intracellular signaling pathways and eventually results in edema formation. 46 A synergistic effect of the 2 toxins resulting in poor survival has also been demonstrated.52,53 Although anthrax most often afflicts herbivores, humans and other mammals are also vulnerable to infection. 42 In most cases, the disease is contracted from farm animals or animal products, 47 but, as seen in the 2001 letter attacks, anthrax can be spread as an intentional act. 54 The spores of B. anthracis can withstand rough conditions in terms of heat, radiation, and chemicals. 55 They are also small, which facilitates their transport to the alveoli in the lungs. 55 These facts make distribution of B. anthracis a likely scenario in a terrorist attack.
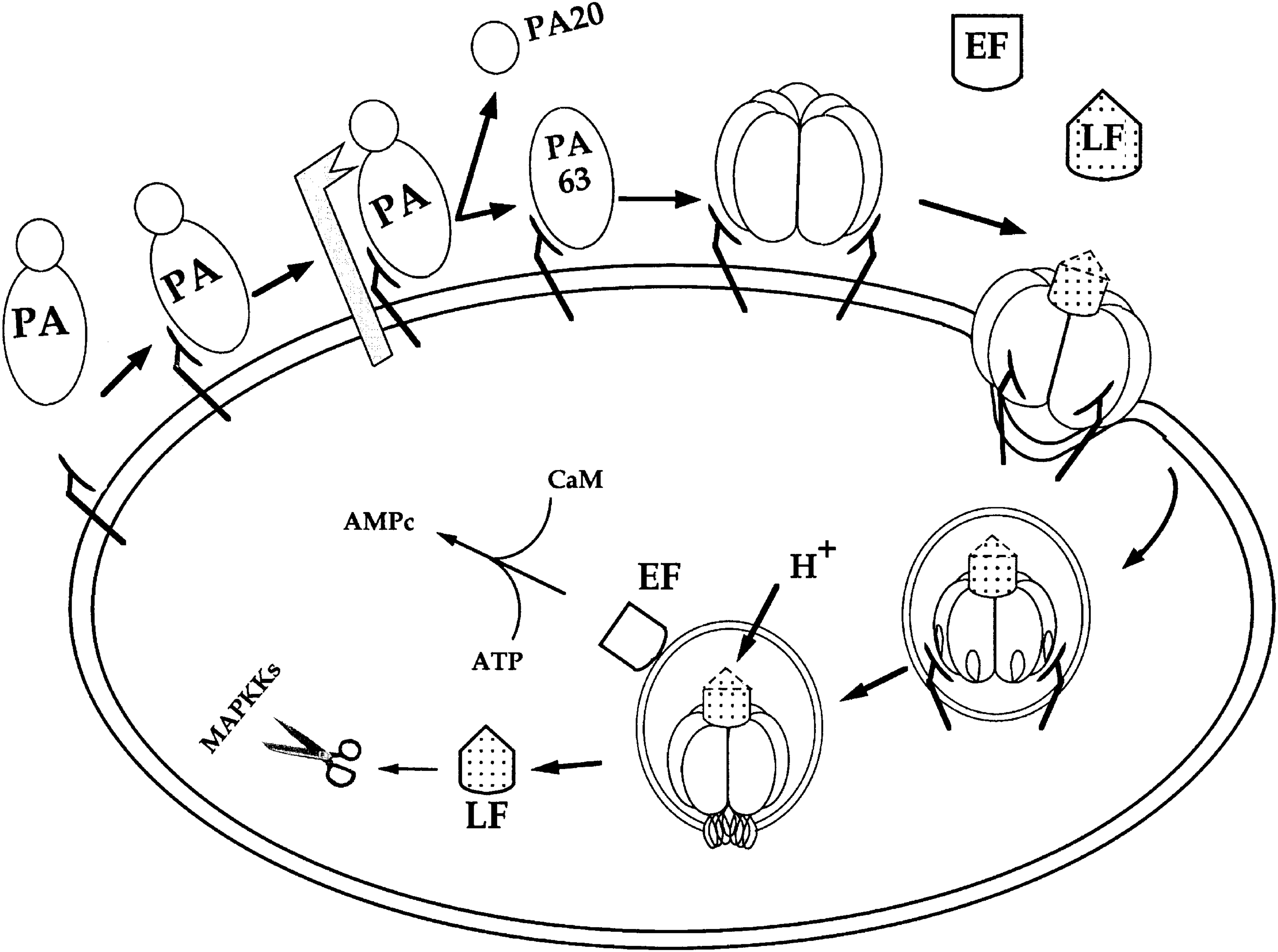
Cellular Model of Action of Anthrax Toxins. Reprinted with permission from Brossier F, Mock M. Toxins of Bacillus anthracis. Toxicon 2001;39:1747-1755. Copyright 2001 Elsevier.
The standard method for identification of B. anthracis has long been the morphological examination of growth cultures.55,56 Together with compatible clinical observations, this will often constitute a confirmed case of anthrax. 43 Today there are many different techniques available for the detection of both the B. anthracis organism and the toxin components. Examples of techniques being used for B. anthracis detection are real-time PCR, ELISA, flow cytometry, surface-enhanced raman scattering (SERS), and mass spectrometry. 57
An important factor in the virulence of B. anthracis is the production of the anthrax toxins; 55 thus, different strategies for their detection have been described. Some methods have an indirect approach, meaning they target antibodies against, for example, PA.58,59 However, the antitoxin IgG is not detectable until the body's immune system has had time to respond to the presence of the antigen (toxin). In the 2001 anthrax mail attacks, antitoxin IgG was not found until 12 days after onset of symptoms. This means that the time it takes to diagnose the disease is much longer compared to measuring toxins directly. 60
Several assays that measure 1 or more of the 3 toxin proteins have been published—for example, antibody microarrays, 61 microwave-accelerated metal-enhanced fluorescence, 62 ELISA, 8 europium nanoparticles based immunoassay, 63 or detection based on polyvalent directed peptide polymer. 64
An activity-based immunoassay for LF has been described by Bagramyan and Kalkum. 65 The method uses commercially available fluorophore-tagged peptides as substrates for LF activity measurement. Boyer et al described the first MS-based method for detection of LF in 2007. 66 This method relies on the endopeptidase activities of the toxin, a technique termed Endopep-MS and similar to the one described for BoNT earlier in this article. In the Endopep-MS method for the detection of LF, the toxin-containing sample is incubated together with magnetic beads coated with antibodies directed toward LF. Then the beads with adhered LF/LeTx are incubated in a reaction buffer containing a synthetic peptide substrate that is specifically cleaved by LF. After incubation, the cleavage products are detected by MALDI-TOF-MS. This technique has the advantage that it demonstrates not only presence of LF but also that LF is functional. To ensure specificity, antibodies for capture of LF, either in free form or as a complex with PA, are used as well as detection based on mass of the cleavage products. However, there are no descriptions in the literature of methods combining protein identification by amino acid sequencing with the Endopep-MS as is the case for BoNT and ricin.
To be able to use the method for quantification of LF, isotopically labeled versions of the cleavage products are incorporated in the analysis. 66 Proof of principle was demonstrated by the analysis of serum from rhesus macaques diseased with pulmonary anthrax. The LF concentration was at least 30 ng/mL 2 days postinfection, which was well above the LOD of 0.05 ng/mL. By further development an LOD of 0.005 ng/mL has been reached. 67 With this improvement, a real advantage of monitoring toxin has been demonstrated by showing that LF could be detected before positive status of bacteremia, positive detection of the pagA gene by PCR analysis, and positive detection of PA by antigen capture immunoassay. Detection of LF as early as 12 hours after infection in blood samples taken from rhesus macaques with pulmonary anthrax has been shown. 68
Clinical applications of Endopep-MS using MALDI-TOF-MS have been demonstrated in 2 publications. One was investigating cases of naturally acquired cutaneous anthrax in Bangladesh, 69 and the other was a case of naturally contracted pulmonary anthrax in the US. 70 The maximum measured serum levels of LF were 1.26 ng/mL and 294.3 ng/mL, respectively. The analysis of LF has also been adapted for LC-ESI-QqQ to accommodate the fact that more laboratories are equipped with those instruments. 71 Furthermore, Gallegos-Candela et al 72 have presented the possibility of differentiating between LeTx and total LF by Endopep-MS. Measurement of these levels separately is beneficial as it will aid in defining the stage of the infection.
For detection of EF, an immunoassay using ATP as a substrate for adenylate cyclase (AC) activity determination has been described. 73 However, ATP is not specific to EF since it may be converted to cAMP by host intracellular AC. Here the specificity mainly lies in the use of antibodies against EF. Further research in regards to Endopep-MS and anthrax has resulted also in the inclusion of EF AC activity detection by LC-ESI-QqQ analysis.68,74 This method was also demonstrated on serum samples from anthrax-infected rhesus macaques showing an LOD of 0.15 pg/mL. 74
The use of mass spectrometry in the analysis of anthrax toxins has been shown to provide several advantages, especially with regard to measurement of toxin activity and the risk of false-negatives compared to cultures and pagA PCR analysis, in which it was shown that a positive finding at 48 hours could revert to a negative at 72 hours.67,68 Also, growth cultures of B. anthracis from blood samples will be negative if treatment with antibiotics has commenced 54 —a situation that would not affect the MS analysis of the toxins. Quantification of LF might also prove useful in following treatment progress and in determining if additional therapeutic measures are needed. 44
Ricin
Ricin is a toxic glycoprotein of about 60 kDa from the seeds of the plant Ricinus communis. 11 It is composed of an A and a B chain linked via a disulfide bridge. 75 It is classified as a Ribosome Inactivation Protein (RIP) type II. The toxic action is initialized by cell binding and cell internalization by the B chain. 76 The chains are then separated, and the A chain, which is an RNA N-glycosidase, removes adenosine at position 4324 in 28 S ribosomal RNA. This effectively stops protein synthesis and causes cell death. The estimated LD50 for humans is quite uncertain, but numbers in the interval of 1 μg/kg body weight to 30 mg/kg body weight for an oral dose have been published. 2 However, ricin is even more toxic when injected or inhaled. The Ricinus seeds, which are called castor beans, also contain the less toxic protein ricin agglutinin (RCA) and the low molecular weight alkaloid ricinine (3-cyano-4-methoxy-N-methyl-2-pyridone). 11 The beans are used industrially for the production of castor oil, and the part of the seeds containing the toxin is left as a byproduct in huge amounts. There are many recipes in books and on the internet on how to purify ricin from castor beans. 77 The availability and ease of preparation make ricin a threat for military or terrorist use. Ricin is the only protein listed under the Chemical Weapons Convention, and it is also classified as a biological warfare agent.78,79
Traditionally, immunological methods have been used to detect ricin.11,77 ELISAs can be very sensitive but are prone to nonspecific reactions, and they may give positive responses for related proteins like RCA and even denatured ricin lacking toxic activity. 80 In vitro toxicity assays provide information on the type of toxic action but do not provide absolute evidence of which substance is present in the sample. 77 PCR-based methods detect the gene encoding the toxin but they do not provide evidence for the presence of the toxin itself. 77 The low molecular weight alkaloid ricinine has been used as a marker of ingestion of Ricinus material because it was easier to analyze than the protein ricin.81,82
In order to obtain unequivocal evidence for the presence of ricin, a sophisticated technique that can directly characterize the properties of the protein must be applied. During the past decades, mass spectrometers that can carry out this task have been developed, and in the year 2000, Despeyroux et al determined the molecular weight of ricin isoforms by MALDI-TOF-MS. 83 Increased certainty in ricin identification has been demonstrated by tryptic peptide mapping by LC-MS and MALDI-TOF-MS.84,85 However, peptide mapping is considered to give only a provisional level of identification. In order to obtain more stringent evidence for the presence of a protein, peptide sequencing is required. Fredriksson et al carried out the first identification method based on peptide sequencing for forensic identification of ricin in purified and crude extracts by LC-MS/MS using a Q-TOF instrument. 86 The method was further developed to include solvent-assisted trypsination without reducing conditions yielding peptides with intact S-S bonds. 87 This demonstrated that the A and B chains were still connected, which is a prerequisite for high toxicity. This method was also faster going from an overnight digestion to a 1-hour digestion, which shortened the response time from 24 hours to 2 hours. In applications on more complex matrices, a few methods with immuno or lectin-lactose affinity extraction combined with tryptic peptide mapping and/or sequencing have been published.88-90
The abovementioned protein-based mass spectrometric methods all give different degrees of direct evidence of the presence of ricin as they provide information on the amino acid backbone of the protein. This is an important advantage compared to purely immunological techniques, which may give false-positives by cross reaction with similar proteins and matrix components. However, the mass spectrometer does not give any direct information on the activity of the toxin, which also might be a very important factor from an investigational perspective.
The first MS-based activity test of ricin was presented in 2000 by Fabris, who detected ricin induced depurination of RNA by direct infusion ESI. 91 In 2004 an LC-MS method was published, 92 in which the release of adenine from a 14-mer RNA substrate was measured after incubation with ricin. There were no results from matrix-based samples in this study, but the authors claimed they could measure ricin in the 0.01- to 10-pmol range, which was comparable to the sensitivity of previously published LC-fluorescence-based activity methods.93,94
Becher et al 95 were the first to publish a method involving antibody capture of ricin from a complex matrix prior to an MS-based activity assay. They also selected a 14-mer RNA substrate, and they quantified the released adenine by LC-QqQ. An incubation time of 24 hours was used with the possibility of a preliminary result after 6 hours but with lower sensitivity. Antibodies against the B chain immobilized on magnetic beads were used for selective extraction of ricin. The purpose of this pretreatment step was to avoid false-positives caused by other RIPs in the sample and false-negatives incurred by RNase activity. This approach gave the advantage of proving that the toxin was intact—that is, containing both the B chain, which possesses the affinity for the antibody, and the A chain with the catalytic activity. Ricin activity was detected in tap water, bottled water, and milk spiked at 20 ng/mL. The lower limit of quantification of pure ricin in buffer solution was 0.2 ng/mL, and the lower limit of detection was 0.1 ng/mL.
A more comprehensive method that could measure 3 different properties of the toxin was presented by Kalb and Barr in 2009. 96 Their method consisted of extraction of the toxin with antibody-coated magnetic beads. The activity was then assayed by incubation of the antibody-bound toxin with a stable 12-mer DNA substrate (Figure 5). Instead of measuring the purine release, the presence of the depurinated substrate was detected by MALDI-TOF-MS in order to avoid interferences from endogenous adenine (Figure 6). Finally, the toxin was identified by tryptic digestion and LC-MS/MS-based proteomics. In this way, the method could provide information on the intactness of the toxin as the B chain had affinity for the antibodies, and the presence of an active A chain was proven by the depurination reaction. Furthermore, the following sequence analysis could differentiate between ricin and other possible related RIPs with similar immunological and enzymatic properties, such as ricin agglutinin (RCA120). After 4 hours incubation with the substrate, 500 fmol of ricin could be detected. The toxin could also be detected in amounts of 1-5 pmol after spiking more complex matrices such as milk, apple juice, human serum, and saliva. The authors claim that these levels are sufficient for detection of ricin in food. The method was further developed to include absolute quantification of ricin in beverages. 97 The concentration of the toxin was determined by measuring selected tryptic peptides using LC-MS/MS with isotope dilution. The lower limit of quantification was 10 fmol/mL (0.64 ng/mL), which was stated to be about 100,000 times lower than the lethal concentration for children in an 8 oz beverage.
Structure of the DNA Substrate for the Ricin Activity Assay Before and After Exposure to Ricin. Molecular weights of substrate and product are also included. Reprinted with permission from Kalb SR, Barr JR. Mass spectrometric detection of ricin and its activity in food and clinical samples. Anal Chem 2009;81:2037-2042. Copyright 2009 American Chemical Society.
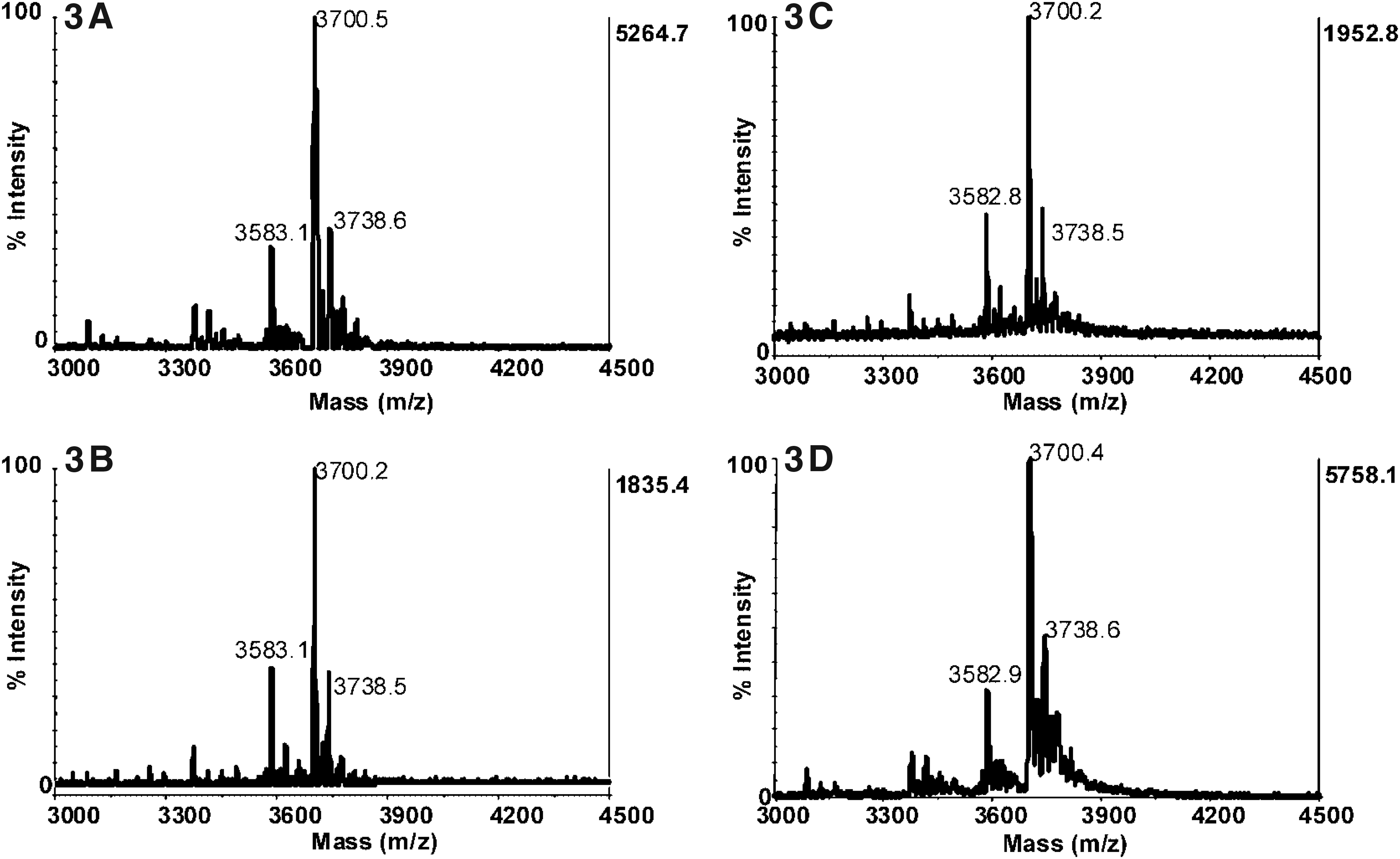
MALDI mass spectra of the DNA substrate in the presence of ricin spiked into and extracted from milk (3A), apple juice (3B), human serum (3C), and saliva (3D). The peak at m/z 3700 corresponds to the protonated intact DNA substrate, and the peak at m/z 3583 corresponds to the depurinated DNA substrate, indicating the presence of active ricin in the sample. Reprinted with permission from Kalb SR, Barr JR. Mass spectrometric detection of ricin and its activity in food and clinical samples. Anal Chem 2009;81:2037-2042. Copyright 2009 American Chemical Society.
Other ricin activity assays based on similar principles have been presented by Bevilacqua et al, 98 who used Direct Analysis in Real Time (DART)-MS for real-time monitoring of the depurination reaction, and by Antoine et al 99 who set up a general RIP activity method without the use of an immunopurification step.
In 2008, a white powder and suspected fragments of castor beans were found in a Las Vegas hotel room. 100 These materials together with swab samples were sent to CDC for analysis. A panel of different tests was performed: real-time PCR, immunoassay with time-resolved fluorescence, cytotoxicity assay, MS-based activity test, and proteomic analysis with trypsin digestion and nano-LC-MS/MS. The results demonstrated that the white powder contained active ricin. This was the first use of MS methods for the detection of ricin in real investigational samples.
Conclusions
The ability to detect protein-based toxins is of utmost importance for public health investigators and emergency responders in order to react appropriately to a terrorist attack or a natural contamination. Today, there are a number of different analytical principles available for this purpose that measure different properties of the toxins. Immunological methods that detect an antigenic property of the protein are sensitive but prone to cross-reactions, and they do not tell whether the toxin is active or not. PCR can give only indirect evidence of the presence of a certain protein as it reacts to the encoding nucleic acid. Activity assays can demonstrate a certain type of toxic reaction but not the chemical identity of the toxin. However, mass spectrometric techniques can give direct evidence of the molecular structure by measuring the molecular mass, the amino acid sequence, and posttranslational modifications. In order to establish if a disease is caused by protein toxins, and/or an organism producing them, it is crucial to know the identity of the toxin as well as whether it is intact and enzymatically active. None of the abovementioned methods alone can provide an answer to all these questions.
Recently, new very powerful methods combining the different analytical principles have been presented. These consist of an initial antibody capture step, which serves the purposes of clean-up and concentration as well as the selective recognition of a certain part of the molecule. The second step is a biochemical target reaction that demonstrates the enzymatic activity. The product of the reaction can be measured with high sensitivity and selectivity by MS. Third, the qualitative properties of the toxin can be determined by tryptic digestion and amino acid sequencing by MS. In this way, the intactness, the enzymatic activity, and the chemical identity of the toxin can be determined in a single method at toxicologically relevant levels.
Footnotes
Acknowledgments
The financial support from the framework of the EU-project AniBioThreat (Grant Agreement: Home/2009/ISEC/AG/191) the Prevention of and Fight against Crime Programme of the European Union, European Commission—Directorate General Home Affairs, and the Swedish Civil Contingencies Agency are gratefully acknowledged. This publication reflects the views only of the authors, and the European Commission cannot be held responsible for any use that may be made of the information contained herein.