
Abstract
The atrial natriuretic peptide (ANP) hormone is secreted by cardiac atrial myocytes and acts to regulate blood pressure homeostasis in humans. Previous research indicates ANP treatment significantly decreases the proliferation of human prostate cancer cells, pancreatic adenocarcinoma, and breast cancer cells. Minimal studies have been conducted with regard to ANP regulating tumor suppressor genes and steroid hormone receptors in breast cancer cells. Our study analyzed the effects of ANP in combination with 17β-estradiol (E2) and antiestrogen treatments on p53 and ERα levels in T-47D breast cancer cells. Preliminary studies through Western blot analysis showed that ANP treatment decreases p53 and ERα expression levels in a concentration-dependent (10–100 nM) manner. Treatment with ANP alone, at a 100 nM concentration, causes a decrease of p53 and ERα expression compared with Cs (control stripped), but with E2 and antiestrogen combinations, expression of both protein levels decreased compared with treatments without ANP. Combined treatment with E2, an estrogen antagonist, and ANP decreased cellular proliferation compared with treatments without ANP, except in the case of raloxifene (RAL). Our studies indicate that ANP has potential as a therapeutic breast cancer treatment and should inspire further studies on the molecular mechanism of ANP in T-47D breast cancer cells.
Introduction
Breast cancer is one of three cancers commonly diagnosed in females in the United States. 1 Breast cancer progress through varying developmental stages can result in invasive breast cancer; one of the later stages of development is ductal carcinoma in situ (DCIS). DCIS is an intraductal neoplastic proliferation of breast epithelium cells and does not invade the breast stroma through the basement membrane layer.2,3 It has been suggested that this stage of breast cancer, known as DCIS, depends upon the hormone estrogen for proliferation, thus making it a hormone-dependent form of breast cancer. The T-47D human breast cancer cell line implemented in this study was isolated from infiltrating ductal carcinoma of the breast and is hormone dependent for estrogen receptor (ER) and progesterone receptor.2,3
Varying hormones influence the stages of development, expression, and regulation of breast cancer cells. Protein products, primarily oncogenic proteins and tumor suppressor proteins, require precise balance in expression to maintain appropriate cell growth. Mutations of oncogenic and tumor suppressor proteins during cell cycle stages can lead to irregular cellular proliferation. Previous studies from our laboratory have shown that steroid hormones regulate the expression of tumor suppressor proteins and ERs in breast cancer cells.4–9 The proliferative effects of estrogen on breast tissue are mediated through ERs, which may induce transcription of estrogen responsive genes.10,11
Hormonal treatment of DCIS functions most effectively when targeting ER binding sites. 12 T-47D breast cancer cells are ERα dependent, which provides them with heightened susceptibility to therapeutic mechanisms due to their ability to elicit receptor-mediated effects.
17β-estradiol (E2) binds ERα within cancerous cells, promoting cellular proliferation through upregulation of ERα. Antiestrogenic hormones, such as ICI 182,780 (a pure ER antagonist), and the selective estrogen receptor modulators (SERMs) RAL and tamoxifen (TAM), bind ERα to block receptor-mediated effects of E2 and inhibit cell proliferation. Antiestrogens similar to those used in this study are common hormone therapy options for ER-positive breast cancer.12,13
The T-47D cell line exhibits a mutation in p53 expression due to a missense mutation at codon 194, which alters leucine to phenylalanine. 14 T-47D cells possess high levels of p53 protein when administered with 5–10% fetal bovine serum (FBS)-containing medium. 8 The protein p53 is responsible for the promotion of DNA repair and apoptosis in normal cells. Mutations in p53 have been linked to variable levels of cellular proliferation in most cancers. 6 The binding of E2 to ERα results in the upregulation of p53 as shown in previous studies. 15 Given the presence of a p53 mutation in the T-47D cell line, tumor cells are no longer suppressed through this protein, resulting in uncontrolled growth of cancerous cells.
Atrial natriuretic peptide (ANP) is one of four peptide hormones cleaved from ANP prohormone, and is most commonly known to be involved in blood pressure regulation. 16 Previous research has shown ANP to inhibit cell proliferation in prostate, breast, colon, and pancreatic adenocarcinoma cancer cells at varying concentrations.17,18 One breast cancer study performed on athymic mice displayed 33% elimination of human breast adenocarcinoma when ANP was administered. 19 Some studies, using ANP concentrations from 1 to 100 nM, have witnessed a decline in viable cells of prostate and pancreatic cancers. However, when these same ANP concentrations were administered to nonmalignant human lung, kidney, or prostate cells, no decline in cellular viability was perceived. 18 This suggests that the cardiac hormone, ANP, delivers selective cell death to cancerous cells and spares normal, healthy cells from these anticancer effects.
Studies indicate the anticancer effects of ANP, through metabolic targets, may be reproducible in other cancers. This could be accomplished through the inhibition of protein kinases, such as those present in T-47D breast cancer cells, which control growth promoting signals.20,21 The possible mechanism of ANP as an anticancer agent may be driven by the intracellular messenger system, cyclic guanosine monophosphate (GMP), formed by the activation of enzyme guanylate cyclase. The natriuretic peptide receptor-A (NPR-A) contains a catalytic portion of guanylate cyclase. Research has shown that cyclic GMP has a strong anticancer effect on the pancreatic cancer cells. But studies have not established that NPR-A presence or activation in cancer cells is necessary for the anticancer activity of ANP. Some data in the literature have hypothesized that the antiproliferative effects of ANP in cancer cells may be due to inhibition of the Wnt pathway through a complex crosstalk mechanism.6,22 This mechanism is an ANP metabolic pathway that promotes its anticancer effects in vivo. The presence of ANP's inhibitory effects on these biological mechanisms leads to a decrease in breast cancer cell growth.
In research focused on ANP's influence in breast adenocarcinoma cells, ANP inhibited DNA synthesis specifically during the cancerous cell's G2-M phase, resulting in inhibition of cellular proliferation. 23 The actions of ANP on tumor suppressor proteins and hormone receptors in breast cancer cells require further study to conclusively determine its effects on cancerous cell growth. The focus of this study is to investigate ANP's effects on ERα and p53 levels in T-47D cells and to work toward determining whether these effects are enhanced in the presence of antihormonal treatments.
Materials and Methods
Materials
TAM, LY 139481 (RAL), and ICI 182,780 were purchased from Tocris Bioscience (Ellisville, MO). Anti-p53 (p53/80) monoclonal antibodies were purchased from BD Biosciences (San Jose, CA). Estradiol (E2), ANP, insulin, and activated charcoal were purchased from Sigma (St. Louis, MO). RPMI-1640, antibiotic antimycotic solution, l-glutamine, and 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES) were purchased from Hyclone (Logan, UT). Anti-ER and NPR-A monoclonal antibodies, RIPA lysis buffer, goat antimouse secondary antibody, and protease inhibitors were purchased from Santa Cruz laboratories (Santa Cruz, CA).
Cell culture and treatment with ligands
T-47D cells were obtained from American Type Culture Collection (Manassas, VA). Cells were routinely cultured at 37°C and 5% CO2 in RPMI-1640 medium with phenol red and supplemented with the following: 2 mM l-glutamine, 25 mM HEPES, antibiotic antimycotic solution (100 units/mL penicillin, 0.1 mg/mL streptomycin, and 0.25 mg/mL amphotericin B), 0.14 IU/mL insulin, and 5% heat-inactivated FBS or single-stripped fetal bovine serum (SSFBS). Cells were originally subcultivated into 5% FBS medium and allowed to grow for 48 h. Cells were then switched into SSFBS medium for 6 days before 2-day interval treatments with ligands. Control cells were cultured only in the presence of serum-containing medium for the entire duration of the study. Upon treatment, six-well plates were treated with 2 μL of each ligand for a 24-h duration. Stock solutions of the ligands (ANP, E2, and antiestrogens) were prepared in ethanol at 1000-fold higher concentration, and aliquots were reduced upon direct addition of ligand (10 μL) to 10 mL of the SSFBS culture medium. ANP concentrations ranging from 10 to 100 nM were utilized for concentration dependency studies, whereas for hormone studies, a treatment with a combination of 100 nM ANP with hormones and antihormones was used.
Extraction of cells
Cellular protein extraction occurred in 500 μL of extraction buffer (RIPA lysis buffer, 0.4 mM phenylmethylsulfonyl fluoride (protease inhibitor), 3 μL/mL protease inhibitor cocktail, 2 mM Na orthovanadate). Cells were removed from culture plates using disposable cell scrapers, and a high-speed supernatant (HSS) of the extract was prepared through centrifugation at 15,000 g for 15 min at 4°C. HSS was frozen and stored at −80°C until further use.
Quantification of proteins
Sample protein concentrations were determined using a standard protein assay (Bio-Rad, Hercules, CA) based on the Bradford assay method. The linear regression method was used to calculate the protein concentration of the individual samples, which were normalized before loading.
SDS-PAGE and Western blot analysis
The proteins in HSS were heat denatured and 30 or 50 μg total protein per lane (for p53, respectively) was resolved on Bio-Rad TGX mini gels under denaturing conditions. Proteins were transferred to Immobilon polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA) at 100 V for 1 h in a Tris-glycine buffer system containing 0.025% SDS and 25% methanol, and using a Bio-Rad transblot cell. Membranes were blocked in Tris-buffered saline (TBS) supplemented with 0.1% tween-20 (TBS-T) plus 10% nonfat dry milk (NFDM) for 1 h. Membranes were then probed for 1 to 4 h in TBS-T plus 5% NFDM containing a 1:1000 dilution of anti-p53 or anti-ER or anti-NPR-A monoclonal antibodies. The membrane was then washed for 30 min with three changes of TBS-T, reblocked for 30 min with TBS-T plus 10% NFDM, and incubated in the same solution plus a 1:1000 dilution of horse radish peroxidase-conjugated goat antimouse IgG secondary antibody. After a final wash, p53 specific bands were observed by the enhanced chemiluminesence (ECL) method through Amersham instructions and blot images were obtained and quantified by the ChemiDoc XRS+ system (Bio-Rad, Inc.). After immunoblotting, PVDF membranes were stained with coomassie blue to ensure complete protein transfer and correct normalization against total protein levels. Protein levels were normalized by probing through antiactin monoclonal antibody, according to manufacturer's protocol for actin bands. Image Studio Lite program version 3.1 (LI-COR Biosciences, Lincoln, NE) was used to quantify the protein band densities for each Western blot.
Immunofluorescence and confocal microscopy
T-47D cells were fixed on cover slips with 1% formalin in phosphate-buffered saline (PBS), permeabilized with ice-cold acetone and methanol, and washed three times with PBS. Staining procedures were performed in a humidified chamber at 25°C. Cells were incubated in 10% goat serum (Sigma) to suppress nonspecific binding of IgG, followed by 3 h incubation with anti-ERα monoclonal antibody. Cells were then incubated for 2 h with antimouse IgG conjugated with Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA). Cover slips were washed in PBS and incubated for 2 min in 1 μg/mL 4′, 6-diamidino-2-phenylindole (DAPI) dissolved in PBS. Cells were washed in PBS, mounted with Fluoromunt-G (Electron Microscopy Sciences, Hatfield, PA), and stored in the dark at 4°C. Distribution of three-dimensional fluorescent structures was analyzed using a Nikon Digital Eclipse CI plus confocal microscope. NIS elements software (Nikon Instruments, Melville, NY) was used for noise reduction and three-dimensional image reconstruction.
Cellular proliferation analysis
Cell proliferation studies were conducted (30,000/well) in triplicate into 12-well plates. Cells were plated for 2 days in medium containing 5% FBS and then withdrawn from endogenous stimuli by culturing for 6 days in SSFBS in the presence of ligands. Fresh ligand-containing medium was replaced in 2-day intervals. After a 7-day incubation with combinations of E2, ANP, and antiestrogens, cells were washed, trypsinized, and suspended in Isoton II diluent (Beckman Coulter, Brea, CA). Number of cells was determined by Coulter Counter model Z2 (Coulter Electronics, Inc., Hialeah, FL).
Statistical analyses
The results are expressed as mean ± SEM. Statistical significance was determined by Kruskal–Wallis test followed by post hoc analysis using Mann–Whitney U test. The p-values were adjusted for multiple testing corrections using the False Discovery Rate. Differences are considered significant at p < 0.05 with asterisks representative of *p < 0.05, **p < 0.01, and ***p < 0.001 in all figures. Statistical analyses were facilitated by SPSS for Windows version 11.5 (SPSS, Inc., Chicago, IL).
Results
Concentration-dependent effects of ANP on p53 and ERα
To determine the concentration-dependent effects of ANP on p53 and ERα, T 47D cells were cultured with ANP for 24 h at concentrations of 10, 50, and 100 nM. Proteins were extracted, quantified through the Bradford method, and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. Protein bands were observed using Bio-Rad Chemi Doc XR. The concentration-dependent effect of ANP was determined and compared with the control cells grown in medium containing 5% dextran-coated charcoal (DCC)-stripped FBS without the addition of ANP and Cs cells grown in medium containing 5% FBS without ANP (Cw).
ERα expression levels of control and ANP-treated T-47D cells, at ANP concentrations ranging from 10 to 100 nM, are represented in Figure 1. Compared with the controls, ANP initially increased ERα expression at 10 nM concentration. However, as ANP concentrations increased to 50 nM, ERα expression increased when compared with Cw and decreased when compared with control (Cs). By increasing ANP levels to 100 nM, ERα expression was decreased in comparison with the control (Cs), as is shown in Figure 1. These data display decreasing ERα levels in a concentration-dependent manner from 10 to 100 nM ANP.

Concentration-dependent effects of ANP on ERα protein expression levels in T-47D cells. T-47D cells were treated for 24 h with 10–100 nM ANP and subjected to SDS-PAGE and Western blot analysis. The relative intensities of each band were compared with vehicle control. Representative blots from three independent experiments are shown. ANP, atrial natriuretic peptide; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Figure 2 displays p53 expression level results when T-47D cells were under controlled conditions and treated with 10 to 100 nM ANP concentrations. ANP exerted a concentration-dependent effect on p53 expression when samples were compared with the controls, showing significantly decreased p53 levels as ANP concentrations increased to 10, 50, and 100 nM. The greatest decrease in p53 expression was witnessed at 100 nM ANP, causing this concentration to be chosen as optimal for further testing of p53 hormonal/antihormonal effects.
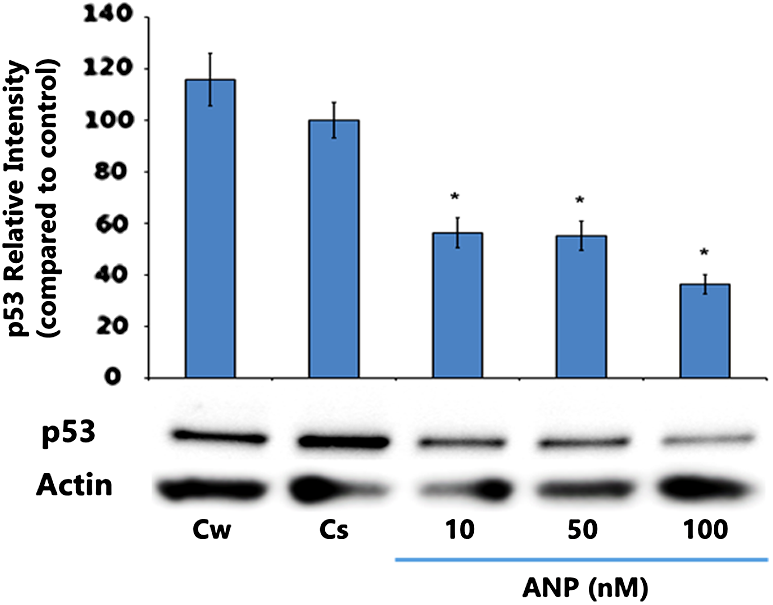
Concentration-dependent effects of ANP on p53 protein expression levels in T-47D cells. T-47D cells were treated for 24 h with 10–100 nM ANP and subjected to SDS-PAGE and Western blot analysis. The relative intensities of each band were compared with vehicle control with statistical significance at p < 0.05. Representative blots from three independent experiments are shown.
Hormonal and antihormonal effects on p53 and ERα
T-47D cells were cultured, as previously described, and treated for 24 h with combinations of ANP (100 nM), E2 (1 nM), and antihormonal treatments (ICI [1 μM], TAM [1 μM], and RAL [1 μM]). Treatment combinations utilized in this study include E2, E2+TAM, E2+RAL, E2+ICI, ANP, E2+ANP, E2+ANP+TAM, E2+ANP+RAL, and E2+ANP+ICI. Protein was extracted, quantified through the Bradford method, and subjected to SDS-PAGE and Western blot analysis. This process was utilized to study the effects of ANP with hormone and antihormone on the steroid receptor and tumor suppressor protein.
Figure 3 shows Western blot results for ERα expression levels following the varying treatment combinations. Compared with the control (Cs), treatment with E2 alone presents significant downregulation of ERα, as expected. Treatment with ANP alone or with E2+ANP displayed downregulation of ERα expression when compared with the control. Combined E2+TAM treatment shows significant downregulation, whereas E2+RAL shows little to no downregulation. When cells were treated with E2+ICI, there was a significant downregulation of ERα compared with the control, as expected, due to ICI being a pure ER antagonist. Combination treatments with E2, ANP, and either TAM or RAL resulted in decreased ERα expression compared with the control. Furthermore, treatment with E2+ANP+ICI displays significant downregulation of ERα expression compared with the control.

Effects of ER agonists and antagonists in combination with ANP on the levels of ERα protein expression. T-47D cells were treated with 100 nM ANP, 1 nM E2, 1 μM ICI, 1 μM TAM, and 1 μM RAL, either alone or in combination, for 24 h. SDS-PAGE and Western blot analysis of ERα were conducted. The relative intensities of each band were compared with the control. Statistical significance was determined at p < 0.05 with one asterisk denoting p < 0.05 and two asterisks for p < 0.01. Representative blots from three independent experiments are shown. E2, 17β-estradiol; ER, estrogen receptor; TAM, tamoxifen.
Figure 4 represents expression levels of p53 through Western blot. Compared with the control, the largest p53 expression increase is viewed in the presence of E2. This significant upregulation of p53 was expected through our previous studies, which showed that E2 treatment, at physiological concentrations (0.1–1 nM), increases p53 expression.13,15 When the antiestrogens ICI or RAL were added to the treatment with E2, they were observed to block the estrogenic effects of p53 expression through the decreased expression compared with E2 treatment alone. However, TAM treatment in combination with E2 did not portray blocking abilities as efficient as the other antiestrogens, as exhibited by p53 expression levels similar to E2. E2+ANP treatment displayed decreased expression compared with E2 treatment alone, as did treatment with E2+ANP+TAM compared with the treatment without ANP. These decreased p53 expression levels, compared with similar treatments without ANP, suggest that ANP can block E2 effects, similar to that displayed with antiestrogens. The combination of E2, ANP, and antiestrogen treatments (TAM, RAL, or ICI) all show decreased p53 expression in comparison with the combination treatments of E2 and antiestrogens, thus illustrating that these antiestrogens exhibit antagonistic activity with respect to ER. This downregulation further suggests that ANP, in combination with antiestrogens, out-competes E2 with respect to p53 expression.
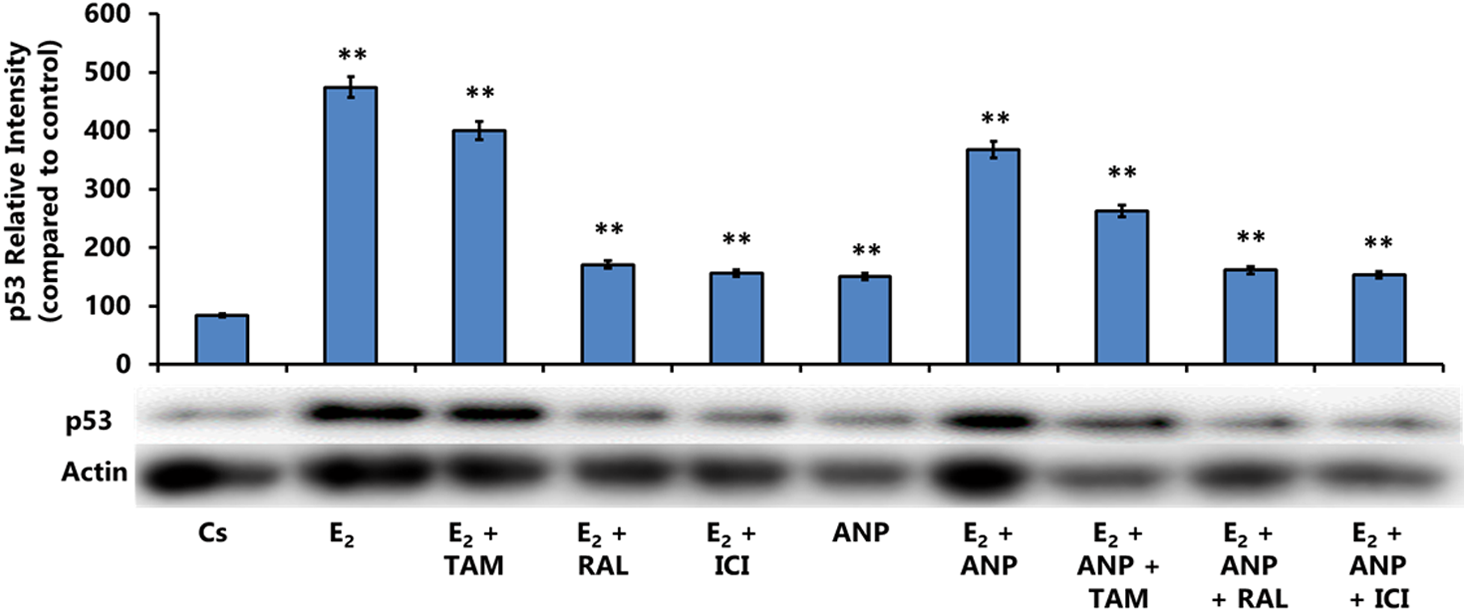
Effects of ER agonists and antagonists in combination with ANP on the levels of p53 protein expression. T-47D cells were treated with 100 nM ANP, 1 nM E2, 1 μM ICI, 1 μM TAM, and 1 μM RAL, either alone or in combination, for 24 h. SDS-PAGE and Western blot analysis of p53 were conducted. The relative intensities of each band were compared with the control, with two asterisks denoting p < 0.01 statistical significance. Representative blots from three independent experiments are shown.
Concentration-dependent effects of ANP on NPR-A protein expression levels in T-47D cells
T-47D cells were cultured for 24 h with ligands of ANP concentrations 10–100 nM to examine the concentration-dependent effects of ANP on NPR-A expression. Proteins were extracted for quantification utilizing the Bradford method, and SDS-PAGE was conducted to analyze the proteins through Western blot techniques. Bands were observed through Bio-Rad Chemi Doc XR, and ANP concentration treatments were compared with control cells either grown in medium containing 5% DCC-stripped FBS (Cs) or medium containing 5% FBS (Cw).
Figure 5 displays T-47D cells examined for NPR-A expression levels at ANP concentrations ranging from 10 to 100 nM. Compared with the Cs, ANP exhibited significant downregulation of NPR-A expression across all concentrations, with the greatest decrease witnessed at 10 nM ANP.
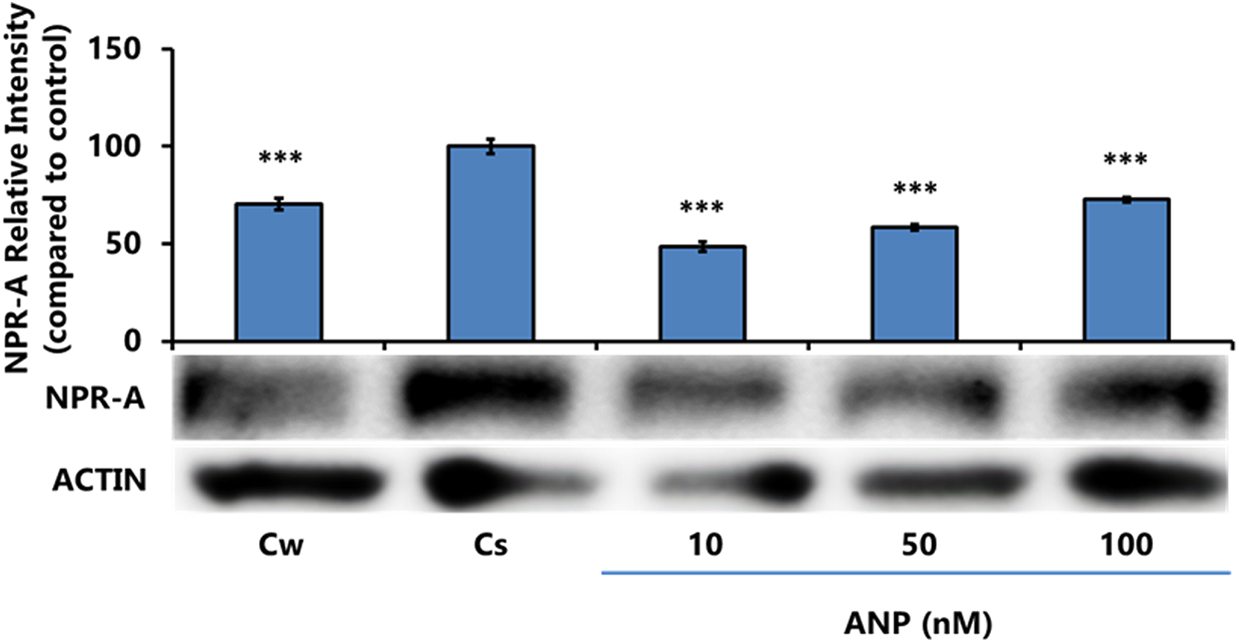
Concentration-dependent effects of ANP on NPR-A protein expression levels in T-47D cells. ANP at concentrations of 10–100 nM was treated for 24 h in T-47D cells. These cells were then subjected to SDS-PAGE and Western blot techniques to determine NPR-A protein expression. Relative densities for each sample were compared with the control, with statistical significance determined at p ≤ 0.05 with three asterisks denoting p < 0.001. NPR-A, natriuretic peptide receptor-A.
Effects of ER agonists and antagonists in combination with ANP on the levels of NPR-A protein expression
ANP (100 nM), E2 (1 nM) treatment, and antihormonal treatments (ICI [1 μM], TAM [1 μM], and RAL [1 μM]) were utilized for 24 h in T-47D cells. Combination treatments were as follows: E2, E2+TAM, E2+RAL, E2+ICI, ANP, E2+ANP, E2+ANP+TAM, E2+ANP+RAL, and E2+ANP+ICI. Extracted proteins were quantified via the Bradford method and the effects of various ANP treatment conditions were examined by SDS-PAGE techniques.
Figure 6 portrays the results of NPR-A expression levels following varying hormonal treatments. When E2 was treated with ANP, significant downregulation occurred similar to that with E2. Likewise, when ANP was added to the combination E2 and ICI treatment, significant downregulation occurred as compared with the control to levels near E2 with ICI. Treatments of E2, in combination with TAM or RAL with or without ANP, all displayed similar significance in downregulation compared with the control.

Effects of ER agonists and antagonists in combination with ANP on the levels of NPR-A protein expression. Treatment was implemented on T-47D cells for 24 h either alone or in conjunction with 100 nM ANP, 1 nM E2, 1 μM ICI, 1 μM TAM, and 1 μM RAL. Proteins extracted from these cells were then analyzed through SDS-PAGE and Western blot techniques to assess NPR-A expression levels. Band intensities were compared with the control and statistical significance was determined at p < 0.05 with two asterisks denoting p < 0.01.
Effects of ANP, E2, and antiestrogens on T-47D cell proliferation
T-47D cells were grown in triplicate in 12-well plates with 30,000 cells per well in medium (FBS) containing growth factors for 2 days. DCC-stripped FBS containing medium supplements with ligands were replaced at 2-day intervals for 6 days. Number of cells and viability were determined using the Coulter Counter model Z2 and results are shown in Figure 7.

Effects of ER agonists and antagonists in combination with ANP on T-47D cell viability. T-47D cells were treated for 6 days, either alone or in combination, with 100 nM ANP, 1 nM E2, 1 μM ICI, 1 μM TAM, and 1 μM RAL, and cell viability was determined by propidium iodide staining and image cytometry. Statistical significance was determined at p < 0.05, with one asterisk denoted as p < 0.05 and two asterisks denoted as p < 0.01.
With combination E2+ANP treatment, compared with the control, a significant cell proliferation increase was present. With addition of antiestrogens, ICI, RAL, or TAM, to E2, cellular proliferation of breast cells decreased compared with E2 alone. E2+ICI displays inhibitory effects, regarding cellular proliferation, to levels below the control. This cellular proliferation decrease is expected with ICI with ER-mediated upregulation of p53 expression during cellular proliferation. 9 E2+RAL portrays a slightly less inhibitory effect on cellular proliferation; however, E2+TAM is the least effective treatment combination in inhibiting cell growth. These data correlate with previous results, determining that the antiestrogen, ICI, displays a better ability to compete against E2 for ER binding than TAM. 9 Combination treatments using E2+ANP+TAM resulted in decreased cellular proliferation compared with treatments using only E2 and the antiestrogen, showing that ANP overrides the effect of E2+TAM on T-47D cells to decrease cell growth.
Effects of ANP, E2, and antiestrogen treatments on the immunolocalization of ERα
The cytolocalization of ERα was determined using confocal microscopy. T-47D cell nuclei were stained utilizing DAPI (seen in blue) for observation and Cy3 (seen in red) to stain ERα protein localization within the cells, as shown in Figure 8. DAPI-stained images, displaying cell nuclei location, were merged with images stained to observe ERα. From the images, it can be determined that ERα is located within the nuclei of T-47D cells under all treatment conditions. Furthermore, E2, ANP, and E2+ANP conditions show increased ERα expression, determined from the intensified immunofluorescent stain visible in these three treatments. This supports our Western blot data, demonstrating increased ERα expression under the same conditions.

Effects of ANP on the immunolocalization of ERα in T-47D cells. Image analysis was performed by confocal microscopy involving immunofluorescence to determine the cytolocalization of cellular ERα. Treatment, either alone or in combination, with 100 nM ANP, 1 nM E2, 1 μM ICI was implemented. The images of the cells stained for the nuclei and stained for ERα were merged to further demonstrate that ERα is localized exclusively in the nuclei.
Discussion
The data from this study indicate that ANP promotes significant decreased expression of p53 with ANP levels administered at the concentrations of 10, 50, and 100 nM. ANP also promotes decreased cellular proliferation as compared with E2 treatment alone and in combination ANP and antiestrogens.
These data suggest that ANP is ICI sensitive, which may support ANP being an ER-mediated compound. These data further exhibit the antiestrogenic effects of ANP, providing it with the ability to inhibit E2 effects. Further studies are necessary to understand ANP's role and its influence on tumor suppressor proteins and ER-mediated pathway.
Previous studies from our laboratory have shown that T-47D cells treated with E2 in medium containing charcoal-treated serum display increased cellular proliferation, increased expression of p53, and downregulation of ERα.6,15 The goal of this study was to demonstrate ANP's effects on T-47D breast cancer cells and the molecular mechanism of ANP on ER and p53 expression.
The previous results are consistent with our study; E2 treatment induced an increase in cellular proliferation and p53 expression level. However, when these same treatments were combined with ANP, the opposite effects were observed. Optimal ANP concentration was determined to be 100 nM, which induced a significant decline in p53 protein quantity compared with the control. Treatment with E2+ANP decreased p53 expression levels, which initially increased with E2 treatment alone. ERα, which downregulated in the presence of E2 alone, was similarly downregulated when treated with E2+ANP. However, cellular proliferation was decreased by E2+ANP+TAM compared with E2. These data correlate with previous studies showing the antiproliferative effects of ANP on cancerous cells. 24 Our study suggests that ANP inhibits the effects of E2 and may exhibit antiestrogenic abilities.
Similarly, when E2+TAM or ICI was administered, p53 expression decreased when compared with E2 alone. With the addition of ANP to these treatments, p53 expression displayed further downregulation. 9 These findings may imply ANP's antagonistic activity with respect to ER. When E2+RAL treatment was implemented, p53 expression significantly increased compared with the control, but the addition of RAL to E2 treatment decreased expression. However, when ANP was added to the treatment, p53 levels remained similar to those without ANP, demonstrating the competitive nature between ANP and RAL for estrogen-binding sites.
We examined the effects on ANP on its own receptor, NPR-A, in breast cancer cells. The expression level increased in higher concentration, indicating that ANP is upregulating NPR-A expression. Studies have shown that estrogen and the SERMs (TAM and RAL) increased ANP levels, promoting cardiovascular protection in rats. These effects may be mediated through NPR-A receptors or crosstalk between ERα and NPR-A receptors.25,26 Treatment with ANP alone or with E2 increased the expression of NPR-A in breast cancer cells and these effects were reduced with antiestrogen in combination with E2 and ANP. Further studies are warranted to examine the molecular mechanism of NPR-A effects by hormone and antihormone in breast cancer cells.
With respect to ERα expression levels, combination of ANP and E2 treatment shows a downregulation of ERα compared with E2 or ANP treatment alone. This mechanism may be mediated through another crosstalk receptor signaling pathway and requires further investigation. When E2+ANP+ICI treatment is administered, significant downregulation occurs compared with the control and E2+ANP treatments. Similarly, the addition of ANP to the E2+TAM treatment causes ERα expression downregulation. These effects imply that ANP is ICI sensitive and show that ICI competes better than TAM for E2 binding sites.
With the E2+ANP treatment, cell proliferation increased with respect to the control and E2 treatments alone. In addition, E2+ANP+RAL treatment had a more significant increase than E2+ANP treatment alone. Supporting the ICI sensitivity of ANP, when ICI was added to E2+ANP treatment, cellular proliferation decreased. Similarly, cell growth declined when E2+ANP+TAM treatment was administered. These findings suggest that ANP's antiproliferative abilities may be induced by its inhibition of various protein kinase growth-promoting signals.20,21 These results may illustrate ANP's therapeutic potential as an anticancer compound. 22
Footnotes
Acknowledgment
Funding was provided by Center of Biomedical Research and Prevention Research Center at Oakland University, Rochester, Michigan.
Author Disclosure Statement
No competing financial interests exist.