
Abstract
Lentiviral vectors (LVs) are being developed for clinical use in humans for applications including gene therapy and immunotherapy. A safety concern for use of LVs in humans is the generation of replication-competent lentivirus (RCL), which may arise due to recombination between the split genomes of third-generation LVs. Although no RCL has been detected to date, design optimizations that minimize recombination events between split genome vectors would provide an added safety benefit that may further reduce the risk of RCL formation. Here we describe design elements introduced to the gag/pol plasmid with the intention of eliminating psi-gag recombination between the vector genome and gag/pol. These design changes, consisting of codon optimization of the gag/pol sequence and the deletion of the Rev-responsive element, abrogate the requirement for Rev in expression of Gag protein, thus the resulting gag/pol construct being Rev independent (RI gag/pol). We show that generating vector using the RI gag/pol construct has no effect on particle production or transduction titers. The RI and wild-type gag/pol vectors function equivalently as antigen-specific immunotherapy, potently inducing antigen-specific CD8 T cells that protect against challenge with vaccinia virus. Most importantly, the designed RI gag/pol eliminated detectable psi-gag recombination. Interestingly, we detected recombination between the vector genome and gag/pol from regions without sequence homology. Our findings imply that although unpredictable recombination events may still occur, the RI gag/pol design is sufficient to prevent psi-gag recombination.
Introduction
Lentiviral vectors (LVs) have been under development as tools for delivering nucleic acid payloads into cells, with promising outcomes for applications such as gene therapy 1 or as vaccines that can elicit immune responses to vector-encoded antigens.2–6 As compared to other viral vectors such as adenovirus or poxvirus, LVs provide several advantages (reviewed by Hu et al. 7 ) that include being able to infect nondividing cells, lack of preexisting immunity, and being able to encode all components of the vector by splitting its genome.6,8 Referred to as third-generation LVs, split genome vectors separate the lentiviral genome into a minimum of four plasmids that encode (i) the pseudotyping envelope; (ii) the gag/pol transcript, consisting of the gag gene (encoding capsid, matrix, and nucleocapsid) and the pol gene (encoding the enzymes protease, reverse transcriptase, and integrase); (iii) the vector genome which contains cis elements (including the packaging signal and long-terminal repeats [LTRs]) and encodes the antigen or gene of choice; and (iv) the Rev accessory protein. These third-generation split genome vectors minimize the chances of replication-competent lentivirus (RCL) generation, thereby providing the benefit of increased safety in comparison to earlier generation LVs.6,9,10
Of all the accessory proteins encoded by wild-type (WT) human immunodeficiency virus-1 (HIV-1), Rev is the only protein that is at minimum required for generating functional vectors derived from HIV-1. During the replication cycle of WT HIV-1, Rev is generated from spliced RNA transcripts of integrated proviral DNA in the infected cell. Rev enters the nucleus and, through interactions with the Rev-responsive element (RRE) on unspliced transcripts, facilitates the nuclear export of these transcripts constituting the full-length viral genome, which are then packaged into progeny virions.11–13 When producing third-generation LVs, two of the four vector components (the gag/pol and the vector genome transcripts) require Rev for nuclear export of their RNA transcripts. Within the vector genome, the RRE is located between splice donor and acceptor sites (Fig. 1) in order to ensure that only unspliced (full-length) transcripts will be exported into the cytoplasm and be packaged by the producer cell into vector particles. The gag/pol transcript also requires the RRE for its nuclear export, which would otherwise be inhibited due to RNA secondary structures; once in the cytosol, gag/pol is translated, yielding the vector structural and enzymatic proteins. 14

Schematic representation of genome and gag/pol constructs. A portion of the vector genome (top) is represented containing long terminal repeats (LTRs) at each end. The splice donor (SD) and splice acceptor (SA) sites flank the psi packaging signal (Ψ), the partial gag sequence and the Rev-responsive element (RRE). Components between the antigen promoter (Promoter) and the 3′ LTR are not highlighted and therefore forward slash marks (//) are used. A portion of the wild-type (WT) gag/pol (middle) is represented showing the gag (black box) and pol (gray box) genes followed by the RRE. The two components that are homologous between the WT gag/pol and the vector genome (the RRE and the first 354 bp of gag) are represented in brackets (not drawn to scale). The hypothesized region of recombination between the vector genome and the WT gag/pol are shown with connecting lines. A portion of the Rev-independent (RI) gag/pol (bottom) is represented showing the codon-optimized gag and pol genes (white boxes to distinguish from WT). The non–codon-optimized frameshift between the gag and pol genes is represented in its native codon sequence (overlapping black and gray boxes). Primers used for the psi-gag recombination assay are shown with their approximate locations on the constructs. First round polymerase chain reaction (PCR) primers are represented with closed arrows and are the pairs 709 and 710 (to detect the psi-gag recombinant) or 709 and 378 (to detect integrated vector genome). The expected amplicon for integrated vector with primer pair 709 and 378 is 1697 bp. The nested PCR primers are represented with open arrows as are the pairs 863 and 835 (to detect psi-gag recombinant with the WT gag/pol) and 863 and 864 (to detect psi-gag recombination with the RI gag/pol). The expected amplicon with either of the psi-gag recombinant primer pairs is 937 bp.
Although third-generation LVs minimize the chances of forming RCL, recombination is known to still occur between vector components despite their separation onto different plasmids. 9 Common sequences do exist between split components that may be responsible for observed recombination events. While it is possible to eliminate most sequence similarities between these components by ensuring that promoters are unique and that LTR sequences exist exclusively on the vector genome (i.e., LTRs are not used as promoters on other components), certain sequence similarities may be hard to avoid. For example, both the vector genome and the gag/pol transcripts contain the RRE that is required for nuclear export of these respective transcripts. Furthermore, the vector genome contains a portion of the gag gene that, together with the Ψ (psi) packaging signal, is required for packaging. Recombination is known to occur between the vector genome and the gag/pol transcripts due to both of them having identical partial gag sequences. Referred to as psi-gag recombination, this is a phenomenon known to occur even in third-generation LVs with split genomes. 9 Because the resultant psi-gag recombinant contains sequences that can facilitate packaging, these recombinants are potentially one step closer to forming RCL.
As part of an effort to increase the safety profile of LVs intended for in vivo applications, we constructed an HIV-1–derived LV that was designed to minimize sequence similarities between the vector genome and gag/pol transcripts. We assessed its function in antigen-specific immunotherapy and screened for genetic recombinants. Here we demonstrate that these design changes did not interfere with the ability to produce transducing LVs that are also capable of eliciting an immune response. We show that these changes resulted in complete elimination of detectable psi-gag recombination. Despite these changes, however, we observed recombination events that occurred outside regions of homology between the vector genome and gag/pol. We therefore present evidence that, although random recombination cannot be completely eliminated, a codon-optimized Rev-independent (RI) gag/pol is sufficient to prevent detectable psi-gag recombination in split genome LVs.
Materials and Methods
Vector components
Similar to third-generation LVs, the LVs used in this report have a split genome and contain a self-inactivating deletion in the 3′LTR (ΔU3). Additionally, the ΔU3 of the LV genome has an extended deletion of the usual self-inactivating deletion that has been reported to increase antigen expression. 15 LVs are generated via a four plasmid system in which the transfer vector (LV genome), a WT gag/pol or modified gag/pol transcript (RI gag/pol), accessory protein Rev from HIV-1, and VSV-G envelope are each encoded on separate plasmids. The LV genome contains HIV-1–derived cis elements that are important for packaging (psi-packaging signal) and for splicing (splice donor and acceptor sites), as well as the RRE. The genome is transcribed from a human cytomegalovirus (CMV) promoter and is flanked by a 5′LTR (R and U5) and a 3′LTR (ΔU3, R and U5). Downstream of the antigen insert is a modified version of the woodchuck hepatitis posttranscriptional element (WPRE) which is present to increase antigen expression. 16 The modified version of the WPRE prevents the translation of the open reading frame of the woodchuck hepatitis virus X protein, thereby eliminating chances of its expression in transduced cells. 17 Rev is included as an accessory protein to enable nuclear export of unspliced messages of the LV genome into the cytosol to be packaged into vector particles.11,12 The transgene promoter is either the CMV promoter or human ubiquitin C promoter.
RI gag/pol design
The design of RI gag/pol involved two changes to WT gag/pol, (i) removal of the RRE and (ii) codon optimization of the open reading frame (ORF) of the gag/pol gene, except for the frameshift region, to disrupt secondary structure elements that inhibit nuclear export of its transcript. The frameshift region forms a secondary RNA structure at the gag and pol junction that causes a −1 register shift of the ribosome during translation that is essential for translating pol gene products. 18 Based on the stem-loop structure of the frameshift reported in Watts et al. 18 we decided to not codon optimize a 282-bp region between base pairs 1228 and 1509 of the gag/pol ORF. This region starts at bp 1563 of the pNL4-3 sequence of WT HIV-1 which encodes lysine409 of the Gag protein and extends to include the stop codon of Gag. The remaining regions (bp 1–1228 and bp 1510–4307 of gag/pol) were codon-optimized based on the human codon table 19 and then manually edited to prevent regions of extremely high GC content. The complete ORF of RI gag/pol was synthesized at Genscript and was cloned in place of the ORF consisting of WT gag/pol and the RRE.
Expression of Gag protein
293T cells were plated in a six-well dish at 1×106 cells/well. Twenty-four hours later, cells were transfected with 0.5 μg of either WT gag/pol or RI gag/pol plasmids in the presence of either 0.5 μg of Rev plasmid or 0.5 μg of empty backbone plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Twenty-four hours later, cells were lysed with cell extraction buffer (Invitrogen, catalog no. FNN0011) and analyzed via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 4%–12% NuPAGE Bis-Tris precast gels (Invitrogen, catalog no. NP0321PK2) followed by transfer onto a nitrocellulose membrane. Blots were then probed with either anti-p24 antibody (Abcam, catalog no. ab9071) or anti-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA; catalog no. sc-130656).
Vector production
Vector was produced in either large scale (CF10) or small scale (10-cm dish). For large-scale production, 293T cells were seeded at 5×108 cells/1 L in a 10-layer cell factory (Nunc, catalog no. 140400) in Dulbecco's modified Eagle's medium (DMEM) containing 5% serum, L-glutamine, and antibiotics. Three days later, cells were transfected using polyethylenimine (PEI) (stock 1 mg/mL) and total plasmid DNA at a ratio of 3:1 (mL PEI:mg DNA). Per 10-layer cell factory, 1 mg of vector genome plasmid and 0.5 mg of remaining plasmids (gag/pol, Rev, and VSV-G) were used. Five hours later, medium was replaced with 1 L of serum-free medium (Transfx-293 media, Hyclone, Waltham, MA; catalog no. SH30860.LS). Vector was harvested 2 and 3 days after transfection. Harvests were clarified using a prefilter and 0.45-μm stericup filter (Millipore, Billerica, MA). Vector was concentrated by spinning in a 1-L centrifuge bottle at 16,000 g for 5 h. Pellet from each liter harvest was either resuspended in 1 mL of Hank's balanced salt solution (HBSS) and aliquoted for storage at −80°C, or was resuspended in 1 mL of buffer for benzonase treatment (50 mM Tris-HCl pH 7.5, 1 mM MgCl2, 5% v/v sucrose). Benzonase nuclease was added at a final of 250 U/mL and incubated overnight at 4°C in order to degrade any leftover plasmids from the transfection. Benzonase-treated vector preps were reconcentrated using a sucrose cushion (30% sucrose bottom, 5% sucrose top) and centrifuged at 116,000 g in an ultracentrifuge for 1.5 h at 4°C. Vector pellet was resuspended in 1 mL of HBSS, aliquoted and stored at −80°C. For small scale vector production, 293T cells were seeded in a 10-cm plate at 2.5×105 cells/plate and transfected the next day using PEI as already described, but with 6 μg of vector genome plasmid and 3 μg of remaining plasmids, except varying amounts of gag/pol plasmids for comparing vector production. Small-scale transfections were done in triplicate for accuracy. Five hours later, the medium was replaced with 4 mL of DMEM containing 5% serum, L-glutamine, and antibiotics. Vector was harvested 2 and 3 days after transfection and was clarified using a 0.45-μm filter. Vector was stored at −80°C.
Vector quantification
p24 assay
Quantification of p24 was performed using the HIV-1 p24 enzyme-linked immunosorbent assay kit by Advanced Bioscience Laboratories (Rockville, MD), following the manufacturer's directions.
GFU assay
293T cells were plated at 2×105 cells/well in a 12-well plate in 1 mL of DMEM containing 5% serum, L-glutamine, and antibiotics. Twenty-four hours later, cells in each well were transduced with twofold dilutions of vector encoding green fluorescent protein (GFP). Each amount of vector was prepared in a 1-mL final volume in complete DMEM. Five twofold serial dilutions of supernatant containing vector were prepared starting from 200 μL of vector per well. As a control to rule out pseudo-transduction, 10 μM of the reverse-transcriptase inhibitor nevirapine was used with the highest volume of vector in a parallel well. Forty-eight hours after transduction, cells were analyzed for GFP expression on a Guava machine (Guava Technologies, now Millipore). Green fluorescence units (GFU) per milliliter was calculated by using a best fit (least squares) linear regression model based on the volumes of vector and the resulting percent GFP values in order to predict the number of GFP-positive cells per milliliter of vector using the FORECAST function in EXCEL. Events that resulted in less than 1% of GFP positive cells were set as the limit of quantification.
Genomes assay
Genomic RNA was isolated from vector particles using the QIAamp Viral RNA Mini kit (Qiagen, Valencia, CA). To eliminate contaminating DNA, the extracted nucleic acid was then digested with DNAseI (Invitrogen) following the manufacturer's directions. Two dilutions of each DNAseI-treated RNA sample were then analyzed by quantitative reverse-transcription polymerase chain reaction (RT-PCR) using the RNA Ultrasense One-Step Quantitative RT-PCR System (Invitrogen) and previously described vector-specific primers and probe. 20 The RNA genome copy number was calculated in reference to a standard curve comprised of linearized plasmid DNA containing the target sequences, diluted over a 7-log range (10 copies to 1.0×107 copies). The genome titer as expressed here reflects the number of physical vector particles, calculated based on genomes, with each vector particle predicted to contain two single-stranded copies of genomic RNA.
Transduction units assay
Transduction units (TU) were determined using an assay in which transduction events in a target cell line are measured using a quantitative PCR (qPCR) assay that amplifies reverse-transcribed vector RNA sequences. Serial dilutions of test samples and reference material were incubated in duplicate in 96-well tissue culture plates in the presence of target 293T cells. The transduction step was performed both in the presence and absence of the reverse transcriptase inhibitor nevirapine as a means to assess background signal that may be contributed by residual plasmid DNA. At 1 day post-transduction, mock- or vector-transduced cells were lysed by the addition of a buffer containing sodium deoxycholate, Tween-20, SDS, and proteinase K. The cell lysates were then incubated sequentially at 37°C, 55°C, and 95°C to ensure proteolysis and DNA denaturation. Denatured cell lysates were then analyzed by qPCR using a primer–probe set that was designed to amplify a vector genome sequence of ∼400 bp located upstream of the antigen promoter (EXPRESS qPCR Supermix Universal, Life Technologies, Carlsbad, CA). The infectivity titer was calculated in reference to a standard curve comprised of linearized plasmid DNA containing the target sequences diluted over a 7-log range (5.3 copies to 5.3×106 copies).
Psi-gag recombination assay
293T cells were plated at 2×106 cells in a 10-cm plate. The next day cells were transduced with 1×1011 genomes of concentrated VSV-G pseudotyped vector made with either WT gag/pol or RI gag/pol. The titers for these vectors were 1.2×1013 genomes/mL and 1.5×1013 genomes/mL, respectively. Forty-eight hours after transduction, cells were harvested, and genomic DNA was isolated using a blood and cell culture DNA kit (Qiagen, catalog no. 13323). One hundred nanograms of genomic DNA was used as template for the first round of PCR using high-fidelity platinum taq polymerase (Invitrogen, catalog no. 11304-011) and the following cycling parameters: one cycle at 94°C for 2 min; 40 cycles at 94°C for 30 s, 55°C for 30 s, 68°C for 90 s; one cycle at 68°C for 5 min. One microliter (out of 50 μL) of the first PCR was used as template either undiluted (1:1) or 1:100 diluted or 1:1000 diluted for the nested PCR. The nested PCR cycling parameters were identical to that used in the first round. No primer controls were included for all reactions. Primers used for PCRs were 378 (TAA GGC CGA GTC TTA TGA GCA GC), 709 (AGG ACT CGG CTT GCT GAA G), 710 (AGC CTG TCT CTC AGT ACA ATC), 835 (TGT CTT ATG TCC AGA ATG CT), 863 (GCA CGG CAA GAG GCG AGG), and 864 (GCC GGA TGT CCA GGA TGC TG). Twenty-five microliters of total 50 μL of the nested PCR was visualized on a 1% agarose gel made with 1×TAE buffer and ethidium bromide. Bands were extracted and DNA was purified using a gel extraction kit (Qiagen, catalog no. 28704) followed by cloning into a TOPO-TA vector (Invitrogen, catalog no. K4500-02) and sequencing (Davis Sequencing, Davis, CA) using M13 forward and reverse primers.
Animals
C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed under specific-pathogen free conditions in a BSL-2 room in the Infectious Disease Research Institute (IDRI) animal facility. All procedures were approved by the IDRI Institutional Animal Care and Use Committee.
Immunizations
C57BL/6 mice were immunized subcutaneously in the tail base on day 0 with either 2×107, 1×108, or 5×108 TU of LV encoding LV1b, a polyepitope construct containing the OVA257 (SIINFEKL) H-2Kb-restricted epitope, or HBSS vehicle. Aliquots of LV stored at −80°C were thawed at room temperature and then kept on ice. Vector was serially diluted in cold sterile HBSS and transported to the animal facility for injection. Mice were placed in a conventional slotted restrainer with the tail base accessible. Vector was administered via 50-μL injection using a 29-gauge 0.3-mL insulin syringe (Becton Dickinson [BD], FranklinLakes, NJ) inserted subcutaneously on the right side of the tail base, ∼1 cm caudal to the anus, leading to minor but notable distension of the skin around the tail base.
Intracellular cytokine staining
Spleens were homogenized by pressing through a 70-μM nylon filter. Red blood cells were lysed by hypotonic shock by brief exposure to ice-cold ultrafiltered water followed by immediate isotonic restoration with 10×phosphate-buffered saline (PBS). For analysis of cytokines, cells were stimulated in a 96-well plate with peptides at a concentration of 1 μg/mL in complete RPMI (10% fetal calf serum [FCS], 10 mM HEPES, 2 μM β-mercaptoethanol, and L-glutamine) for 5 h at 37°C, 5% CO2. OVA257 (SIINFEKL) peptide was manufactured at 95% purity by AnaSpec (Fremont, CA). After stimulation, surface staining was carried out in FACS buffer (PBS, 1% FCS, 2 mM EDTA, 0.01% sodium azide) in the presence of FcR blocking antibody 2.4G2 and LIVE/DEAD® Fixable Near-IR (L/D NIR, Invitrogen). Antibodies used for surface staining in in vivo experiments included anti-mouse CD4-PerCP-Cy5.5 (eBioscience) or CD4-Alexa Fluor 700 (eBioscience, SanDiego, CA), CD8-Pacific Blue (eBioscience), and B220-V500 (BD). After surface staining, cells were fixed with Cytofix® (BD) and stored at 4°C overnight in FACS buffer. Cells were then permeabilized with Perm/Wash™ buffer (BD) containing 5% rat serum (Sigma Aldrich, St. Louis, MO). Antibodies for intracellular staining were diluted Perm/Wash buffer containing 5% rat serum and added to permeabilized cells. Antibodies included anti-mouse tumor necrosis factor-α–fluorescein isothiocyanate (eBioscience), interferon-γ–phycoerytherin (eBioscience), and interleukin-2-allophycocyanin (APC) (eBioscience). Cells were washed twice with Perm/Wash buffer and resuspended in FACS buffer and analyzed on a three-laser LSRFortessa with high-throughput sampler (BD). Data were analyzed using FlowJo software (TreeStar, Ashland, OR). Viable CD8 T cells were gated as follows: lymphocytes (FSCint, SSClo), single cells (SSC-A=SSC-H), live (L/D NIRlo), B220− CD4− CD8+. Cytokine gates were based on the 99.9th percentile (0.1% of positive events in unstimulated cells).
Vaccina virus challenge
C57BL/6 mice were immunized subcutaneously in the tail base on day 0 with 5×108 TU of vector encoding LV1b or HBSS vehicle. Four weeks later, mice were challenged intraperitoneally with 1×107 TCID50 recombinant vaccinia virus expressing OVA (rVV-OVA), 1×107 TCID50 WT vaccinia virus (VV-WT), or HBSS vehicle. Five days after challenge ovaries were harvested for quantitation of viral load by TCID50 assay.
Results
Design of a RI gag/pol plasmid
Of the four plasmids typically used to generate pseudotyped third-generation LVs, two of the plasmids contain sequences within their transcripts that have the potential of recombining. These are the transfer vector (referred to here as the vector genome) and the gag/pol plasmid. There are two regions of sequence homology between transcripts of the vector genome and gag/pol (Fig. 1). First, the vector genome has a partial gag sequence following the psi packaging signal that is comprised of 354 bp that are identical to the 5′ of the gag sequence in the gag/pol plasmid (highlighted in Fig. 1 with brackets around black bars). Recombination events that take place from this sequence overlap have been referred to as psi-gag recombination. Second, both the vector genome and gag/pol contain RRE, which is comprised of 234 bp that form a secondary RNA structure allowing Rev-dependent nuclear export of RRE-containing transcripts into the cytoplasm. We sought to eliminate these two homologous sequences by deleting the RRE from the gag/pol plasmid and by codon optimizing the gag/pol ORF, with the exception of a 282-bp frameshift region between gag and pol that is required for translation of pol protein products (Fig. 1). Deletion of the RRE is known to eliminate Rev-dependent export of gag/pol transcripts from the nucleus because the RNA secondary structure of gag/pol retains transcripts in the nucleus. 14 Therefore the codon optimization serves both to eliminate these retentive secondary structures and to minimize sequence homology with the partial gag in the LV genome. Due to the fact that these modifications hypothetically relieve the gag/pol transcript from requiring Rev, we refer to it as RI gag/pol, even though Rev is still required during vector production.
Nuclear export of RI gag/pol does not require Rev
In order to demonstrate that the RI gag/pol transcript is indeed Rev-independent, we transfected 293T cells with either the WT gag/pol or the RI gag/pol plasmids, in the presence or absence of a Rev encoding plasmid. Cell lysates were analyzed for expression of gag protein products using SDS-PAGE and Western blotting with anti-p24 antibody. The RI gag/pol plasmid was able to express p24 and its precursors whether or not Rev was present, whereas the WT gag/pol transcript required Rev for protein expression (Fig. 2). We also observed that the processing of p55 Gag protein appeared different between RI and WT gag/pol transcripts based on the ratio of p55:p24 protein suggesting effects of codon optimization on protein expression and/or processing. 21 These results indicate that transcripts can undergo nuclear export in the absence of Rev, confirming that design changes to the RI gag/pol construct do relieve the requirement for Rev.
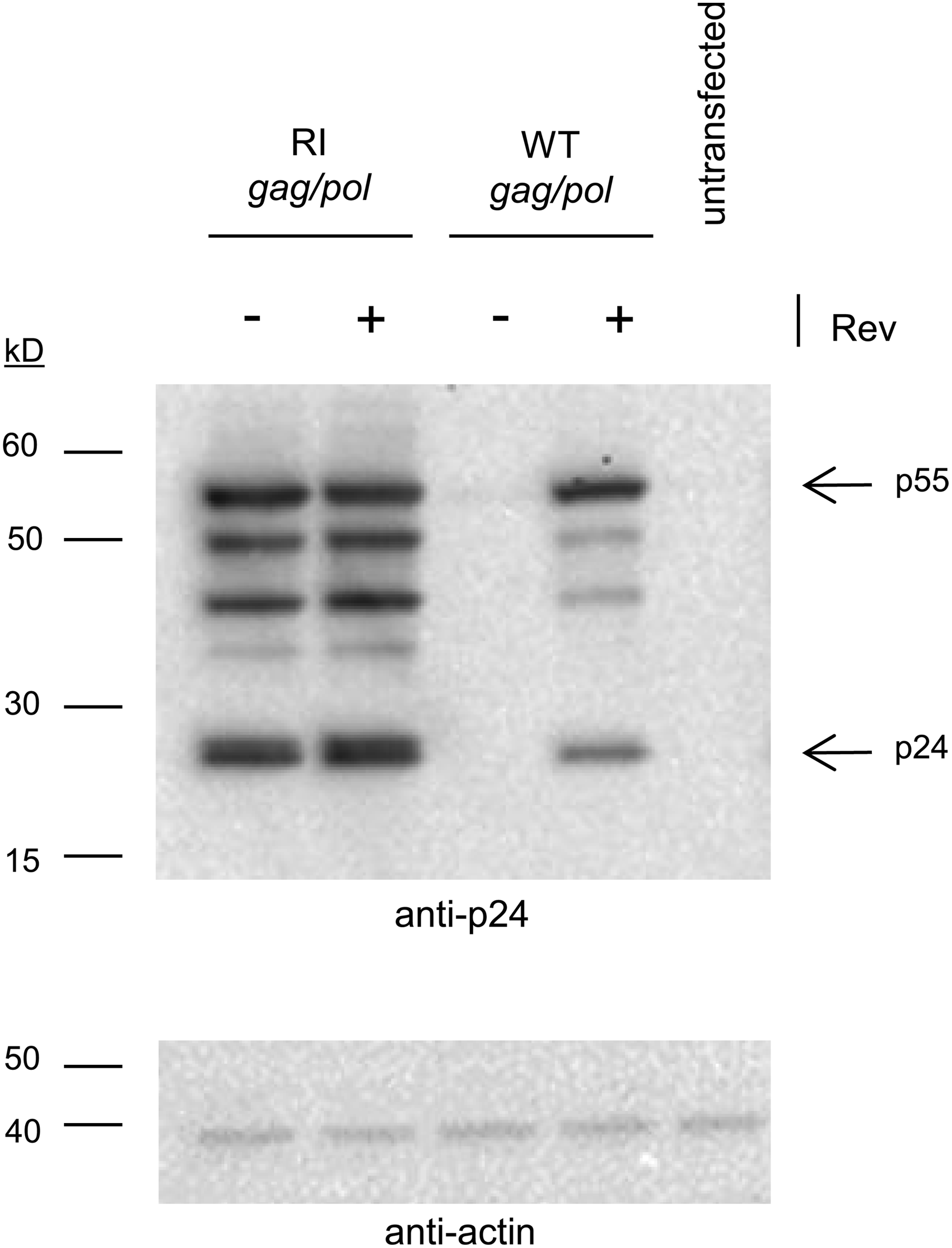
Nuclear export of RI gag/pol does not require Rev. 293T cells were transfected with either WT gag/pol or RI gag/pol plasmids in the presence of either Rev plasmid (+) or empty backbone plasmid (−). Twenty-four hours later cells were lysed and analyzed for expression of Gag proteins using anti-p24 antibody. Unprocessed Gag protein (p55) and p24 are indicated with arrows. Additional bands consist of processing intermediates such as p41. Lysate from untransfected cells was included as a control. Equal loading of wells was confirmed with anti-actin antibody.
RI gag/pol produces transducing vector with comparable titers to WT gag/pol
Previous studies have described reductions in titers of vectors produced in the absence of Rev.22,23 While we did not remove Rev from vector production, we still wanted to ensure that vector made with the RI gag/pol construct would generate particles with comparable titers and transduction capabilities to those of WT gag/pol. Two parallel vector preps pseudotyped with VSV-G were generated using a vector genome encoding GFP as a marker, and with two input DNA amounts of either the WT gag/pol or the RI gag/pol constructs (see Materials and Methods). Both vector preps were assayed for p24 and were shown to have comparable physical particle titers, regardless of the gag/pol plasmid used to produce them (Fig. 3A). These preps were then assayed for their ability to transduce target cells. When normalized by volume, RI gag/pol vector resulted in transduction events comparable to WT gag/pol for both of the amounts of input gag/pol plasmid (3 or 6 μg) used during vector production (Fig. 3B). These results indicate that design elements introduced to generate the RI gag/pol construct did not reduce the physical particle yield or transduction ability of the LV.
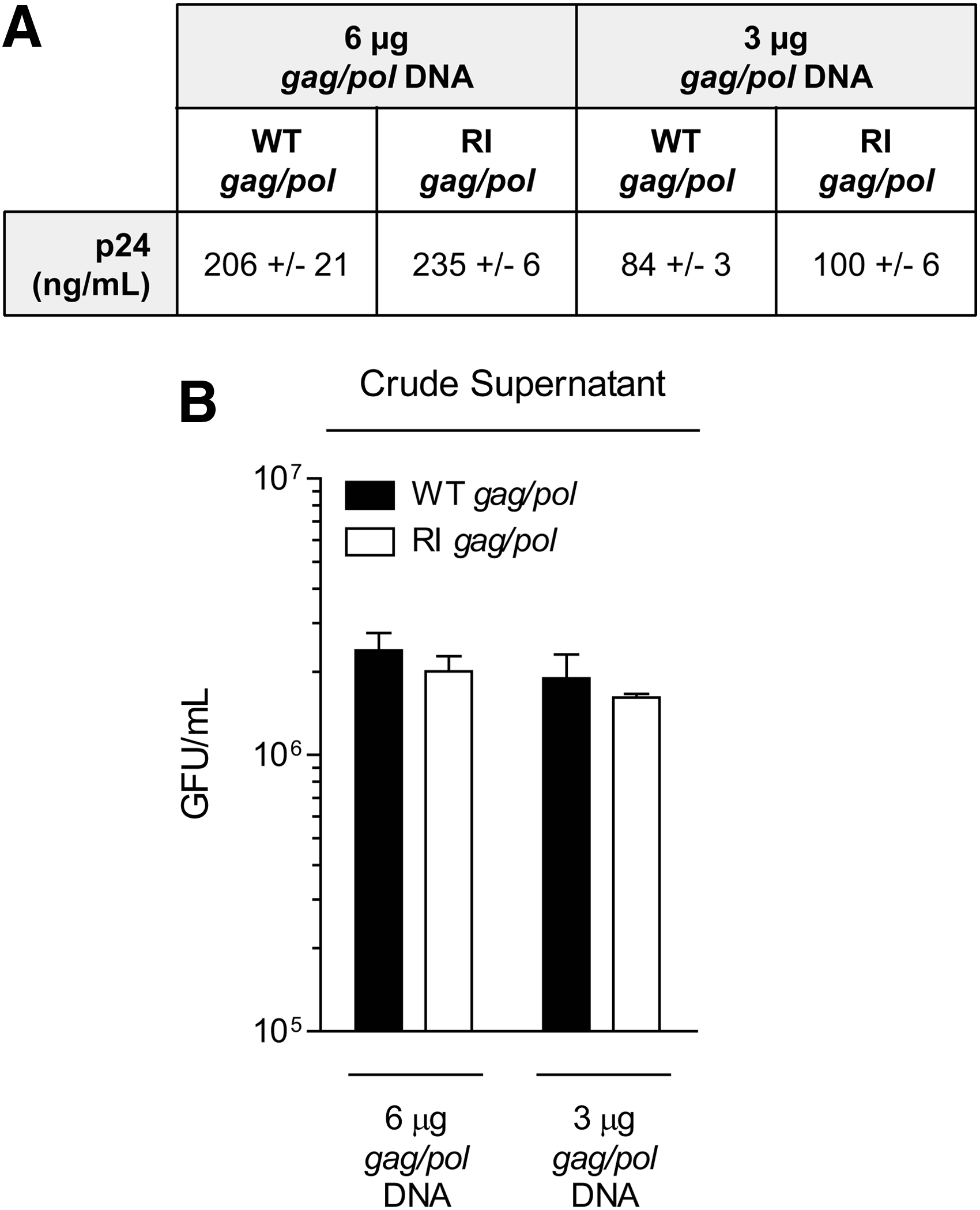
RI gag/pol vector titers and transduction are comparable to WT gag/pol.
RI and WT gag/pol vectors generate equivalent immune responses
LVs are commonly used to deliver protein-encoding nucleic acids to various cell types in vitro for research applications and in clinical settings. 24 However, directly injectable LVs are also being developed for both gene therapy and antigen-directed immunotherapy. To test whether a RI gag/pol vector would serve as an appropriate LV for immunotherapy applications, we evaluated immune responses generated against an antigen transgene encoded by the RI gag/pol vector. Mice were immunized with a dose range of WT or RI gag/pol vectors, containing either an integration-competent WT or integration-deficient D64V mutant integrase, encoding a polyepitope construct termed LV1b, which contains the OVA257 (SIINFEKL) H-2Kb-restricted CD8 T cell epitope. At 12 days postimmunization, OVA257-specific CD8 T-cell responses in the spleen were measured by intracellular cytokine staining for interferon-γ (Fig. 4). For both integration-competent and integration-deficient vectors, RI gag/pol and WT gag/pol vectors generated comparable CD8 T-cell responses, confirming that codon optimization of the gag/pol gene did not negatively impact LV function as an immunotherapy vehicle.

Lentiviral vectors (LVs) generated with an RI gag/pol induced primary CD8 T cell responses. C57BL/6 mice were immunized with the indicated doses (2×107, 1×108, or 5×108 transduction units [TU]) of LV encoding full-length OVA or HBSS vehicle alone. LVs were either integration-competent (INT+) or integration-deficient (INT−) and generated with either WT or RI gag/pol, as indicated. At day 12 postimmunization, the percentage of OVA257-specific splenic CD8 T cells was measured by intracellular cytokine staining for interferon (IFN)-γ after ex vivo peptide restimulation.
RI and WT gag/pol vectors both induce protective antiviral immunity
While we observed that primary LV-induced CD8 T-cell responses were equivalent with RI gag/pol vectors, we sought to establish whether functional immunity induced by these vectors was also similar. To address this, a recombinant live vaccinia virus challenge was employed as a model of viral infection. Mice were vaccinated with integration-deficient LVs encoding OVA257 with either a WT or RI gag/pol, and then challenged 4 weeks later with rVV-OVA or VV-WT as a control (Fig. 5). Mice vaccinated with RI gag/pol and WT gag/pol vectors both showed dramatic reductions in viral load after challenge with rVV-OVA. Confirming that protection was antigen specific, the viral load after VV-WT challenge was similar between vehicle- and LV-treated groups. These data indicate that RI gag/pol LVs can induce memory CD8 T cells that respond to viral challenge and provide functional immunity.

ID-LVs generated with a RI gag/pol induced CD8 T cells that provide protect against viral challenge. C57BL/6 mice (five per group) were immunized with 5×108 TU of integration-deficient (INT−) LV generated with either WT or RI gag/pol encoding a polyepitope antigen (LV1b) that contained the H-2b-restricted OVA257 CD8 T cell epitope and then challenged on day 28 postimmunization with 1×107 TCID50 WT WR-strain vaccine virus (VV-WT), WR-strain recombinant OVA vaccine virus (rVV-OVA), or left unchallenged. On day 33 (day 5 postchallenge) viral load in the ovaries of each animal was measured by TCID50 assay. ID, integration-deficient; WR, western reserve.
RI gag/pol modifications eliminate psi-gag recombination
The main purpose of the RI gag/pol design is to eliminate psi-gag recombination and thus further minimize the chances of RCL formation in third-generation LVs. We utilized a nested PCR–based approach to screen genomic DNA of cells transduced with integrating vector in order to detect psi-gag recombination. In this approach a first round of PCR is performed using a forward primer (709) that binds the vector genome 5′ of the psi packaging signal and a reverse primer (710) that binds within the frameshift region of both RI gag/pol and WT gag/pol (Fig. 1). The PCR product from this first round is then diluted and used as template for a second PCR using a nested forward primer (863) that binds the LV genome, and a nested reverse primer (835 or 864) that binds to the gag region within either the WT gag/pol or the RI gag/pol, respectively. These two reverse primers were designed to bind the same region in both constructs, with the only differences being due to codon optimization. The amplicon size from a hypothetical psi-gag recombinant would be 937 bp if using either primer pair 863 and 835 or primer pair 863 and 864. Vector preps encoding the LV1b polyepitope were generated with either the RI gag/pol or the WT gag/pol plasmids, and were used to transduce 293T cells. Forty-eight hours after transduction the genomic DNA was harvested and PCR was performed followed by analyzing on an agarose gel. First, we performed a positive control PCR using primers that bind only within the LV genome (primers 709 and 378). Cell cultures transduced with either WT gag/pol or RI gag/pol vector both yielded the expected amplicon size (1697 bp), thus confirming that transduction and integration of provirus are comparable for both vectors (Fig. 6A). Next, we performed the nested PCR for psi-gag recombination as described above. Genomic DNA from cells transduced with the WT gag/pol vector produced a band at the expected size of 937 bp, consistent with psi-gag recombination (left half, Fig. 6B). In contrast, genomic DNA from cells transduced with RI gag/pol did not yield any detectable band for a psi-gag recombinant, even when nested PCR was performed on 1 μL of undiluted (1:1) first-round PCR template (right half, Fig. 6B). Interestingly, a larger (1329 bp) but fainter band was detected only in the highest concentration of template used (1:1; right half, Fig. 6B). To determine the nature of the PCR bands, we extracted the 937-bp band from WT gag/pol as well as the 1329-bp band from the RI gag/pol and then cloned each band into TOPO-TA plasmids. As predicted, sequencing revealed that the WT gag/pol band was consistent with a psi-gag recombinant in that the first half of the sequence aligned with the vector genome and the second half aligned with the gag/pol transcript that extended beyond the partial gag sequence (Fig. 6C; data not shown). The fainter 1329-bp band from the RI gag/pol encoded a sequence that consisted of the first 1253 bp aligning with the LV genome but the last 77 bp aligning with a region of the RI gag/pol (described as “RI-recombinant” and discussed later; Fig. 6D and data not shown). In conclusion, the design of the RI gag/pol vector completely eliminated psi-gag recombination in cells transduced with integration-competent LV, and that no psi-gag recombination was detected even when nested PCR was performed on 1 μL of undiluted template.

Elimination of psi-gag recombination in RI gag/pol vectors.
Discussion
The generation of RCL constitutes the principal safety concern for use of LVs in human clinical protocols. 25 Third-generation split genome vectors serve to minimize the risk of RCL formation by separating vector components. 8 Although no RCL has been detected to date in limited clinical trials involving LVs, detection methods for RCL have technical challenges that may result in false-negative results. 26 Risks associated with RCL are greatest due to insertional mutagenesis 10 that has been observed with gammaretroviral vectors such as murine leukemia virus (MLV).27,28 Importantly, separation of vector components on MLV vectors 29 has not eliminated the generation of recombination-competent retrovirus that has resulted, though infrequently, either due to recombination among vector components30,31 or due to recombination between vector and endogenous retroviral sequences. 32
In this report we have provided evidence that a RI gag/pol construct completely eliminated detectable psi-gag recombination between the vector genome and gag/pol. The design elements were incorporated into the gag/pol plasmid and consisted of codon optimization of the gag/pol sequence outside of the frameshift and the deletion of the RRE downstream of the gag/pol ORF. These changes resulted in the abrogation of the requirement for Rev in expression of Gag protein, hence the resulting gag/pol construct being Rev-independent. However, LV production still requires Rev due to the inability to eliminate the RRE from the vector genome without affecting vector titers.22,23 Nevertheless, the overall p24 titers were comparable between the two vectors. Although previous iterations of gag/pol have been generated for increased safety using different codon-optimized sequences and/or deletion of the RRE,33,34 to our knowledge we have for the first time demonstrated that such design changes can eliminate detectable psi-gag recombination.
Psi-gag recombination has previously been detected with a single-round PCR in transduced cells that were passaged for 21 consecutive days in order to allow amplification of any putative RCL. 9 Developing and/or performing a RCL assay and showing the lack of a putative RCL is a necessary part of any preclinical investigational new drug (IND)-enabling activities. For the purposes of this study presented here, we are focusing on the detection of genetic recombinants and not RCL. Recombination between vector components can arise at numerous stages of vector production and/or vector transduction. However, recombination is most likely to occur during reverse transcription, due to strand switching between vector genome and other nonspecifically packaged transcripts. 35 Using a more sensitive nested PCR approach we have been able to confirm, as early as 2 days after transduction, psi-gag recombination in cells transduced with WT gag/pol vector but not with RI gag/pol vector. Further work would need to be done on whether or not elimination of psi-gag recombination has any functional significance. Since to date no RCL has been reported, instead an in vitro assay such as marker capture (e.g., antibiotic resistance gene) may be devised to assess the functional significance of an RI gag/pol vector design.
Surprisingly, RI gag/pol produced a recombinant (described here as RI-recombinant) that was not dependent on any sequence homology between the vector genome and gag/pol. While we cannot elucidate the mechanism for this recombination event or cannot even rule out recombination occurring at other stages, we can say that it occurs at a lower frequency than psi-gag recombination and that it is a rare enough event that was detectable only when nested PCR was performed on the highest concentration of template PCR product (1 μL of undiluted template), but not detected at other more relevant template amounts (such as 1 μL of 1:1000 dilutions of template PCR). While data suggest that the RI recombinant is a rare nonhomologous recombination event, further studies would need to be performed to determine whether or not the RI recombinant is real and not just a PCR artifact.
Since the origin of third-generation LVs, additional attempts have been made at enhancing the predictable safety profiles of vectors for example either by separating gag/pol components 36 or by conditional removal of the packaging signal from proviral DNA. 37 We are developing integration-deficient LVs for use as vaccines in immunotherapy. We were able to demonstrate that design changes resulting in the RI gag/pol vector did not affect its in vivo potency compared with WT gag/pol vector for both integration-competent and integration-deficient versions. We propose that design elements introduced here with the RI gag/pol vector provide a significant advantage to preventing measurable psi-gag recombination in third-generation LVs.
Footnotes
Acknowledgment
Work at TRIA Bioscience Corp. (TRIA) was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health Grant 1R43AI087444-01.
Author Disclosure Statement
One or more of the authors is a current employee of Immune Design Corp. (IDC), a holder of stock and/or options in IDC, or an inventor on an issued patent or patent application for technology discussed in this article which has been assigned to or licensed by IDC. All the presented work was designed by or for IDC at TRIA.