
Abstract
Trappin-2/elafin is a novel innate immune factor that belongs to the serine protease inhibitor family and has known antibacterial, antifungal, and antiviral properties. In this study, we further investigated the anti-HIV activity of elafin using different cellular models and both X4– and R5–HIV-1 laboratory strains. We compared the antiviral activity of human recombinant elafin (rElafin) with three well-known antiretroviral drugs, AZT, tenofovir, and enfuvirtide. We have found that when the virus is pre-incubated with rElafin prior to the infection of the cells, HIV-1 replication is significantly inhibited. In target T cells and human peripheral blood mononuclear cells, maximal inhibition was achieved using submicromolar concentrations, and rElafin was found to be as potent as enfuvirtide, showing its potential for therapeutic application. We also show data on the mechanism of the antiviral activity of rElafin. We have demonstrated that rElafin neither binds to CD4, CXCR4, or CCR5 host cell receptors, nor to the viral glycoproteins gp120 and gp41. Furthermore, in our cell-to-cell fusion assays, in contrast to enfuvirtide, rElafin failed to block cell fusion. Altogether our results indicate that rElafin interferes with HIV replication at the early steps of its cycle but with a different mechanism of action than enfuvirtide. This study provides the first experimental evidence that elafin inhibits HIV replication in its natural target cells; therefore, elafin might have potential for its development as a new anti-HIV drug or microbicide.
Introduction
Elafin and its precursor protein trappin-2 are serine protease inhibitors members of the whey acidic protein (WAP) of the chelonianin family.1–5 They are produced by multiple cell types, secreted in mucosal secretions constitutively, and can be elevated in the presence of inflammatory stimuli.1,6–8 Trappin-2 possesses an N-terminal transglutaminase substrate-binding domain or cementoin domain and a C-terminal WAP domain that contains four disulfide bonds corresponding to elafin.9,10 Elafin, also named skin-derived antileukoprotease, was originally isolated as an elastase inhibitor from the skin of patients with psoriasis, 9 but is produced by epithelia, particularly in the lung.11,12
Trappin-2/elafin has been shown to display anti-inflammatory activity by inhibiting neutrophil serine proteases such as elastase and proteinase 3,9,13–15 antibacterial activities,2,3,16,17 antifungal activity,
18
and more recently, anti-HIV activity.19–21
We recently have identified elafin by tandem mass spectrometry and enzyme-linked immunosorbent assay (ELISA) in the human female reproductive tract and found that elafin was overexpressed in naturally HIV-resistant women compared with HIV-uninfected and -infected women, suggesting its possible role for
Materials and Methods
Materials
Analytical grade solvents and reagents were purchased from Sigma-Aldrich (Oakville, Canada) unless otherwise specified. Antibodies against CD4 and CXCR4 and anti-mouse Ig isotypes were purchased from BD Biosciences (Mississauga, Canada). Recombinant elafin (rElafin) and AZT were purchased from Sigma-Aldrich. P24 ELISA kits were purchased from Advance Bioscience Laboratories Inc. (Kensington, MD). T20 (Fuzeon) was obtained from Hoffmann-La Roche (Mississauga, Canada).
Cell cultures
The human epithelial cancer cervical cell line TZM-bl was obtained through the U.S. National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc. (Durham, NC).25,26 TZM-bl cells are a clone of HeLa cells that express human CD4 and the human chemokine receptors CXCR4 and CCR5. In addition, TZM-bl cells express β-galactosidase and luciferase under the control of HIV LTR, which is transactivated by the HIV Tat protein in relation to the level of virus replication. The human non-Hodgkin's T-cell lymphoma cell line Sup-T1 and monocytic THP-1 cells were purchased from the American Type Culture Collection. HeLa-
Infectivity and inhibition assays
To test the anti-HIV activity of elafin against HIV-1, we used two different virus strains, the X4 IIIB and R5 BaL strains, and four different cell models. HIV-1 IIIB viral stocks were prepared by infecting target cells with viral stock solutions, and viruses were collected from supernatants. Viral titration was performed in target cells to calculate the TCID50. Viral preparations were stored at −80°C. First, TZM-bl indicator cells were seeded at 6×103 cells per well in a 96-well microtiter plate and allowed to adhere overnight in a humidified CO2 incubator at 37°C. Varying doses of human rElafin or antiretroviral drugs in complete DMEM were incubated with HIV-1 IIIB for 1 h at 37°C in a final volume of 1 mL. Following incubation, the culture medium was aspirated from TZM-bl cells, and 100 μL of the mixture containing the virus at multiplicity of infection (MOI) 0.1 was added to the cells. Cells were then incubated for 24 h at 37°C with 5% CO2 in a humidified incubator. After 24 h, β-galactosidase activity was measured using the Beta-Glo assay (Promega, Madison, WI). Briefly, 100 μL of Beta-Glo reagent was added to each well, plates were incubated for 30 min at room temperature, and the light intensity of each well was measured using a luminometer (BioTek Synergy™ 2 instrument, Thermo Fisher Scientific, Nepean, ON, Canada). Uninfected cells were used to determine background luminescence. A subset of experiments were performed to determine possible post-infection inhibitory effects of elafin. In those experiments, TZM-bl cells were first infected with HIV-1 IIIB at MOI 0.1 for 1 h and then were rinsed with PBS to eliminate unbound viruses. Cells were then incubated with various concentrations of human rElafin or antiretroviral drugs for 24 h at 37°C with 5% CO2 in a humidified incubator. HIV-1 replication was measured by the Beta-Glo assay. Experiments were repeated three times with four replicates.
Other experiments were conducted using the Sup-T1 T cell line and PBMCs. Varying doses of rElafin or antiretroviral drugs in complete RPMI were incubated with HIV-1 IIIB for 1 h at 37°C in a final volume of 1 mL. After incubation, HIV-1 infection of Sup-T1 cells or PBMCs were carried out by incubating 2×105 cells with the mixture containing HIV-1 IIIB at MOI 0.1, for 2 h at 37°C in a humidified atmosphere of 5% CO2 in a total volume of 1 mL. After infection, cells were washed and plated in 96-well plates, at a density of 2×104 cells/well in 100 μL of complete RPMI without further addition of rElafin or drugs. At day 4, cells were split and drug-free fresh culture medium was added to the wells. At day 7, the supernatants were collected and the level of HIV-1 replication was determined by measuring the concentration of viral protein p24 (ELISA assay kit, Advance Bioscience Laboratories Inc.). Syncytium formation and cell viability were also analyzed 7 days post-infection in Sup-T1 experiments by microscopic observations and the MTT assay, respectively.
THP-1 cells were plated at 4×104 cells/well in 96-well plates and treated with 10 ng/mL PMA to allow them to differentiate. After 7 days of differentiation, PMA-treated cells were exposed to a mixture of HIV-1 BaL pre-incubated with rElafin or antiretroviral drugs for 1 h at 37°C in a humidified atmosphere of 5% CO2, at a MOI of 0.1. After 2 h, cells were rinsed and cultured in complete culture medium without further addition of rElafin or drugs. Every 3–4 days, supernatants were collected and drug-free fresh culture medium was added to the wells. HIV-1 replication was monitored by the viral p24 protein level in harvested supernatants. Additional experiments were conducted to determine the 50% inhibitory concentration (IC50) of rElafin or antiretroviral drugs in the different cell models. IC50 was calculated using regression analysis.
Each experimental condition consisted of quadruplicate samples and assays were repeated three times with Sup-T1 cells or twice with PBMCs and THP-1. In all experiments, untreated cells infected with HIV-1 virus only (positive control) and untreated or uninfected cells (negative control) were used.
Cytotoxicity assay
Sup-T1 cell viability was determined by the MTT assay. Cells were plated and treated as in the infectivity assays except that at day 4 when the cells were split, fresh culture medium containing rElafin or T20 was added. After 7 days of incubation with varying concentrations of rElafin or 1 μM T20, 50 μL of cell suspension was transferred to another 96-well plate. Then 8 μL of a solution of 4 mg/mL MTT was added to each well, and cells were incubated at 37°C in a humidified atmosphere of 5% CO2 for 2 h. Cells were lysed using isopropanol containing 1% HCl 12 N and 5% Triton X-100 and absorbance was measured at 570 nm on a microplate spectrophotometer reader (BioTek Synergy MX instrument, Fisher Scientific). Experiments were repeated twice with four replicates each.
Analysis of expression of cell surface proteins by flow cytometry
Effect of rElafin on cell surface protein expression was evaluated using two different cell models, Sup-T1 and TZM-bl expressing CD4 and CXCR4 receptors. Sup-T1 and TZM-bl cells were plated in six-well plates at a density of 1.5×106 cells/well in RPMI and DMEM, respectively, supplemented with 3% serum. Various concentrations of rElafin was added to the cells and at indicated times of incubation, cells were collected, washed, and stained with mouse anti-human monoclonal antibodies (BD Biosciences) for the cell-surface markers CD4 (allophycocyanin [APC], #340443), CXCR4-PE (phycoerythrin [PE], #555974), or goat anti-mouse Ig isotype controls (PE # 554680, APC #555576), for 60 min at 4°C in the dark. Cells were then washed again, resuspended in staining buffer (0.1% sodium azide and 1% fetal bovine serum in PBS, pH 7.4–7.6) and fixed with a solution of 2% formaldehyde in PBS. Cells were analyzed the same day using a FACS Canto II (Becton Dickinson, Franklin Lakes, NJ) and Diva software (Becton Dickinson Biosciences, Mississauga, Canada). Experiments were performed in duplicate and repeated at least twice. Data are expressed in terms of percentage of expression of cell surface markers compared to untreated control cells set at 100%.
Binding of rElafin to cell surface proteins
Binding of rElafin to cell surface receptors was evaluated using CD4+/CXCR4+ Sup-T1 cell line, whereas binding to viral glycoproteins gp120 and gp41 was evaluated using HeLa-
Cell-to-cell fusion assay
HeLa-
Statistics
Statistical analysis of differences between treatment groups was evaluated by Student's
Results
Inhibition of HIV infection in TZM-bl indicator cells
Inhibitory effects of human rElafin on viral infection was first determined by measuring the virus-induced β-galactosidase activity in HIV-1 IIIB–infected TZM-bl indicator cells and compared to potent clinically approved antiretroviral drugs (Fig. 1). When the virus was first pre-incubated with rElafin prior to infection of the cells, a concentration-dependent inhibition of HIV-1 replication was obtained, whereas no inhibition was observed when rElafin was added after viral infection. Same data were obtained when cells were treated with the antiretroviral drug T20 known to block HIV entry. In contrast, as expected, reverse transcriptase inhibitors tenofovir and AZT showed high levels of inhibition in the post-incubation experiments when the drugs were added 2 h after viral infection (Fig. 1). No significant cytotoxicity effect was observed with rElafin or any of the drugs at the doses used (data not shown). When compared to the infected but untreated TZM-bl control cells, rElafin was found to be 10-fold less potent than AZT (50% inhibitory concentration IC50 of 0.71 μM compared to 7.13 μM, respectively), 2-fold less potent than tenofovir (IC50 1.46 μM), and similarly potent as T20 (IC50 0.67 μM) to reduce β-galactosidase activity (Table 1). Our results in this HeLa cell model suggest that rElafin interferes at early steps of HIV-1 replication with comparable potency as the antiretroviral drugs evaluated since rElafin was able to affect viral replication only when viruses were still at the beginning of the infection cycle (not yet entered into host cells).
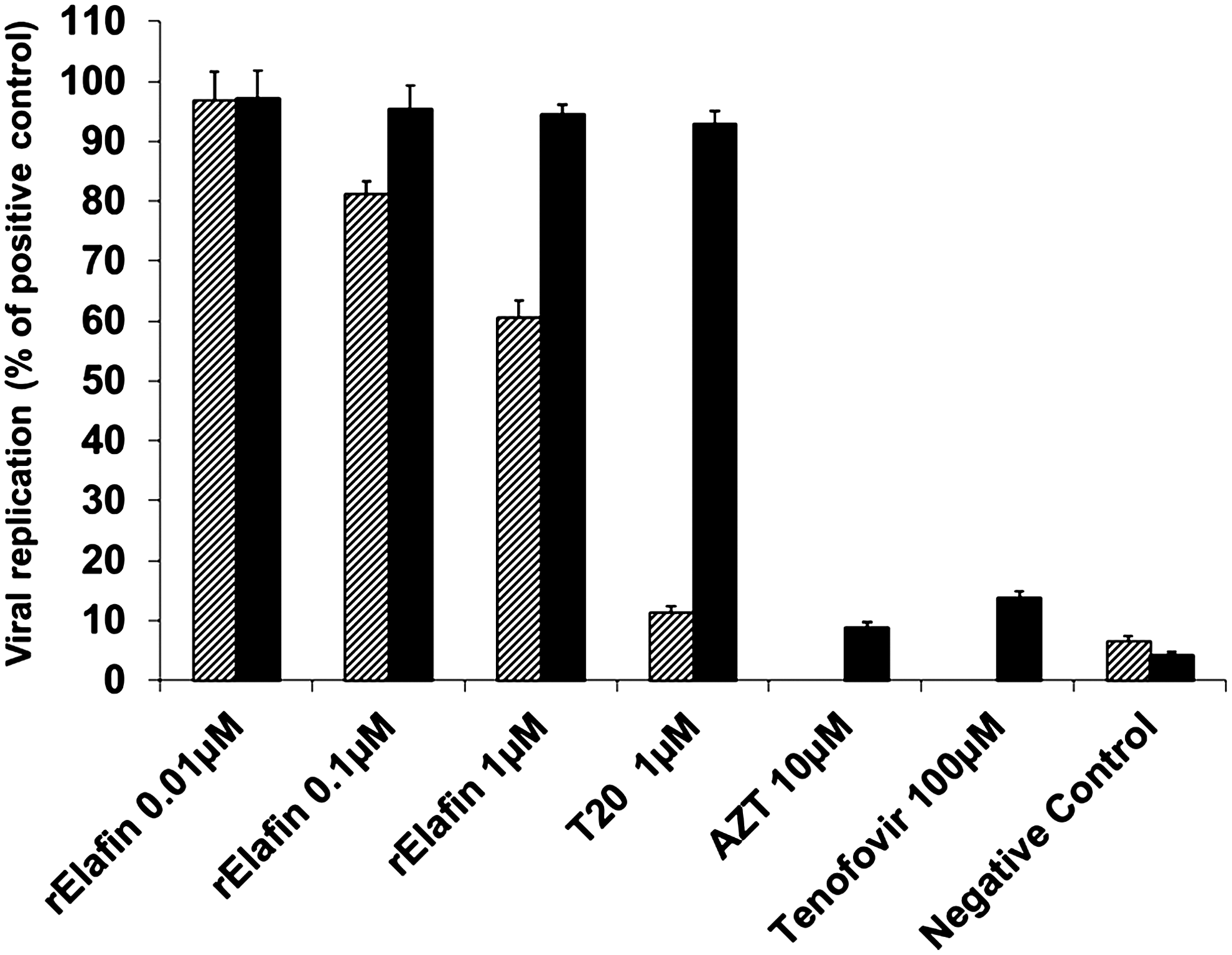
Anti-HIV-1 IIIB activity of recombinant elafin (rElafin) in TZM-bl cells. TZM-bl cells were infected with HIV-1 IIIB at multiplicity of infection (MOI) 0.1 for 2 h. HIV-1 virus was pre-incubated with rElafin or antiretroviral drugs for one hour before infecting TZM-bl cells (hatched box) or the antiretroviral drugs were added to the cells after infection (black box). Activity of β-galactosidase released by HIV infection of the cells was quantified by the Beta-Glo assay 24 h post-infection. Data are expressed in percentages compared with untreated/HIV-infected positive control cells which were set to 100%. Data are representative of three independent experiments. Values represent means of percentage±SEM (
Inhibitory Activity of rElafin in Different Cell Models
Cells were exposed to various concentrations of rElafin, and β-galactosidase activity (TZM-bl) or p24 protein level (SupT-1, PBMC, and THP-1) was determined, as described in Materials and Methods. The 50% inhibitory concentration (IC50) was determined in the different cell models using regression analysis. Experiments were repeated three times.
Inhibition of HIV infection in Sup-T1 cells
To further investigate the inhibitory effects of rElafin, we evaluated its potential to inhibit HIV-1 IIIB replication in target T cells, using Sup-T1 T cell line. The virus was first pre-incubated for 1 h with rElafin prior to infection, and then cells were infected with the mixture. After 2 h, the virus mixture was removed and fresh culture medium without drugs was added. Viral replication was quantified by measuring p24 protein production in supernatants after 7 days of infection. In target T cells, rElafin blocked HIV-1 replication in a concentration-dependent manner (Fig. 2A) and showed potent anti-HIV activity with an IC50 of 79 nM (Table 1). In uninfected and untreated cells (negative controls), p24 level was 2.62±1.12 ng/mL compared with 5022.12±365.69 ng/mL in infected and untreated cells (positive controls). As in the previous experiments with TZM-bl indicator cells, rElafin showed lower inhibitory effects when added to the cells 2 h after infection (IC50 of 3.32 μM in Sup-T1 cells; data not shown). All cell cultures treated with rElafin or T20 were found significantly different from the untreated/infected positive control cells by one-way ANOVA analysis (

Anti–HIV-1 IIIB activity of rElafin in T cells. HIV-1 IIIB virus was pre-incubated with rElafin or antiretroviral drugs for one hour prior to infection of Sup-T1 cells at MOI 0.1. After 2 h, cells were rinsed with phosphate-buffered solution (PBS) and plated in a 96-well plate with fresh complete RPMI. At day 7, cell-free supernatants were collected to quantify viral p24 protein using an enzyme-linked immunosorbent assay
HIV-1 infection of CD4+ human T-cell lines in culture leads to cytopathic effects characterized by formation of large, multinucleated giant cells called syncytia, followed by destruction and lysis of single cells. The efficacy of rElafin to protect the cells from the lytic effect of HIV-1 was evaluated in Sup-T1 cells infected with the syncytia-inducing X4 tropic HIV-1 IIIB virus using the MTT cell viability assay (Fig. 2C). The lytic effect of HIV-1 was significantly reduced in cells treated with rElafin in a dose-dependent manner in comparison to untreated/infected control cells. At such a low dose as 40 nM, rElafin was able to protect viability of Sup-T1 cells at 70% compared to untreated/uninfected negative control cultures (
Inhibition of HIV infection in PBMCs and THP-1 cells
Next we evaluated the anti-HIV activity of rElafin in HIV-infected PBMCs and the monocyte/macrophage THP-1 cell line. Prior to HIV infection, PBMCs were first stimulated for 24 h with 1 μg/mL PHA-P and THP-1 cells treated with 10 ng/mL PMA for 7 days, as described in Materials and Methods. HIV-1 IIIB (for PBMCs) or BaL (for THP-1) viruses were pre-incubated with rElafin for 1 h and then the cells were infected with their respective mixture. After 2 h, the virus mixture was removed and fresh culture medium without drugs was added. Viral replication was quantified by measuring p24 protein production in supernatants after 7 days of infection. As in previous cells models, rElafin blocked HIV-1 replication in a concentration-dependent manner (Fig. 3). In PBMCs and THP-1 cells, rElafin showed potent anti-HIV activity with an IC50 of 176 and 89 nM, respectively (Table 1). In PBMCs, the p24 level was 2.10±1.33 ng/mL in uninfected and untreated negative control cells but was not detected in negative controls in the THP-1 cell model. Because rElafin showed similar anti-HIV activity in PBMCs as in the established cell line Sup-T1, the Sup-T1 T cells were used for the next series of experiments to study the mechanism of action of rElafin.

Anti-HIV-1 activity of rElafin in peripheral blood mononuclear cells (PBMCs) and THP-1 cells. Prior to HIV-1 infection assays, PBMCs were stimulated with 1 μg/mL PHA-P for 24 h and THP-1 cells treated with 10 ng/mL phorbol 12-myristate 13-acetate (PMA) and allowed to differentiate into macrophages for 7 days. Cells were infected with a mixture of HIV-1 IIIB (--■--PBMC) or HIV-1 BaL (—▲—THP-1) pre-incubated with rElafin for 1 h, at a MOI of 0.1. After 2 h of infection, cells were rinsed and cultured in complete culture medium without further addition of rElafin. After 7 days, supernatants were collected and p24 level measured by ELISA. Values represent means±SEM (
Binding to host receptors or viral glycoproteins
Further experiments were performed to better understand the anti-HIV mechanism of rElafin. The first steps of the infection cycle of HIV-1 include binding of viral gp120 glycoprotein to its primary cell surface receptor CD4 and key co-receptors, CXCR4 or CCR5, for X4– and R5–tropism virus, respectively. The interactions between gp120 and cellular receptors trigger a series of conformational transitions in gp41 that ultimately lead to the fusion of viral and cellular membrane allowing virus entry into target cells. Entry of HIV-1 can therefore be blocked by interference with either the viral glycoproteins or host elements such as CD4 and co-receptors CXCR4 or CCR5.
To determine whether rElafin blocks HIV-1 at the entry steps, experiments were first conducted to investigate the possible inhibitory activity of rElafin on the binding of viral envelope to CD4 receptor and CXCR4 co-receptor. First we looked for the ability of rElafin to directly bind to the viral envelope then to CD4 and CXCR4 receptors using two different cellular models: HeLa-

Binding of rElafin to cell surface proteins. Sup-T1 cells or HeLa-
Effect on the expression of viral receptors at the surface of cells
To further confirm that rElafin does not interact with host cell receptors, we tested its effect on the expression of CD4 and CXCR4 proteins on the surface of Sup-T1, up to 72 h using flow cytometry analysis. As in the previous assay, no effect on the expression of both CD4 and CXCR4 proteins could be observed following incubation of Sup-T1 cells up to 72 h (Fig. 5) or TZM-bl cells (data not shown) with 330 nM of rElafin. Under these same conditions, as expected, AZT did not affect CD4 and CXCR4 expression on the surface of Sup-T1 or TZM-bl cells (data not shown).

Effect of rElafin on CXCR4 and CD4 protein expression on the surface of Sup-T1 cells. Cells were incubated with 330 nM of rElafin for 1 h up to 72 h and then rinsed and stained with anti-human CXCR4 or CD4 antibody. Level of expression of CXCR4 and CD4 proteins was detected by flow cytometry. Data are expressed in terms of percentage of expression of cell surface markers compared to untreated control cells set at 100% after deduction of nonspecific background fluorescence of control IgG. Values represent means±SEM of at least two independent experiments done in duplicate (
Blockage of cell-to-cell fusion
Next we investigated the ability of rElafin to interfere with membrane fusion using a cell-to-cell fusion assay. For this cell fusion experiment we used HeLa-

Cell-to-cell fusion assay. HeLa-
Discussion
Previous studies have reported the anti-HIV-1 activity of trappin-2/elafin,19–21
but to date there is still controversy about its role as an innate immune factor to protect from HIV-1 transmission and infection
To further confirm the anti-HIV activity of elafin and investigate its mechanism, we used the Sup-T1 T-cell line, the THP-1 monocyte/macrophage cell line, and human PBMCs, all natural target immune cells for HIV. TZM-bl cells are epithelial HeLa engineered cells expressing canonical HIV-1 receptor components, CD4, CXCR4, and CCR5, that allow them to be infected by HIV-1. In this cell model, viral replication is monitored by reporter assays. Infection of T cells, macrophages, and PBMCs with HIV-1 are cell models that more closely correlate with
Data from the present study and other studies suggest that trappin-2/elafin acts at early steps in the replication cycle of the virus. HIV-1 entry is initiated by the binding of viral gp120 glycoprotein to cell surface receptor CD4 molecule and a co-receptor (CXCR4/CCR5). Many molecules have been reported to have potent
Conclusions
Taken together, the data of this study provide important additional information on anti-HIV activity of the innate immune factor trappin-2/elafin. We have shown that rElafin significantly inhibited X4-tropic HIV-1IIIB replication in engineered genital epithelial cells, target T cells and primary immune cells, and R5-tropic HIV-1 BaL in monocytes/macrophages without binding to host cell receptors CD4, CXCR4, or CCR5 or to viral capsid proteins gp41 and gp120. Our current data suggest that trappin-2/elafin interferes with the early steps of HIV replication with a different mechanism than enfuvirtide (T20). The ability of elafin to act at an early step in the viral replication cycle to block infection of both X4- and R5-tropic virus strains in immune target cells as demonstrated in this study makes trappin-2/elafin a promising candidate for its development as an anti-HIV microbicide.
Footnotes
Acknowledgments
This work has been supported by the Public Health Agency of Canada. V.J.J. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for his postdoctoral fellowship. We are grateful to Michèle Bergeron and Tao Ding for their technical help with the flow cytometry analysis. We thank Germaine Fortier for her technical assistance with the cell cultures.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.