
Abstract
Target gene delivery is needed to induce cellular differentiation or a specific therapeutic effect. Electroporation is a relatively safe and simple technique to deliver nucleic acids to the cell that acts by rendering cells transiently permeable using short periods of high voltage. In stem cell research, human dental pulp stem cells (hDPSCS) are highly accessible, and they exhibit broad differentiation potential. Until now, no studies have attempted to optimize electroporation parameters for DPSCs with respect to transfection efficiency and viability. In this study, we aimed to optimize transfection of DPSCs through varying different electroporation parameters, including voltage, mode of pulsation, and the number of pulses. As positive control, we used commonly utilized the chemical transfection reagents Lipofectamine 2000 and FuGene 6. In addition, we used our newly optimized transfection conditions to transfect hDPSCs with a functional chondrogenic transgene. We obtained higher transfection efficiency and cell viability with these electroporation conditions compared to controls. The highest transfection efficiency (63.81±4.72%) was achieved with 100 V, 20 msec, one-pulse square-wave condition. Among chemical transfection groups, FuGene 6 showed the highest cell viability at all tested transfection ratios, while Lipofectamine 2000 showed the highest transfection efficiency (19.23±3.19%) using 1:1 DNA (μg):Lipofectamine (μL). Transfected DPSCs functionally expressed the transforming growth factor β-3 chondrogenic transgene on the mRNA level as detected by real-time polymerase chain reaction and on the protein level as detected by Western blot analysis. An increase in various chondrogenic markers was also found when studying mRNA expression in transfected cells. In conclusion, the results of our study demonstrate optimal electroporation and chemical transfection reagent conditions for hDPSCs, and, subsequently, we provide proof of concept for expression of a functional gene using those conditions. These results demonstrate a widened scope for use of DPSCs in various tissue engineering applications.
Introduction
Cell-based therapy is currently at the forefront of tissue engineering strategies and is expected to dominate the field in the future. The use of autologous cells has several advantages, including avoidance of adverse immune response and infection risk problems associated with allogenic or xenogenic grafts. Furthermore, because they can be cultured ex vivo, autologous cells provide an alternative for repair of large defects when enough healthy autologous grafts are not feasible or sufficient.1,2 Mesenchymal stem cells offer several advantages over typical cell-based therapy and are readily available in several tissues in the body. 3 They are able to differentiate into several lineages and thus offer the possibility of repairing complex tissues. 2
Adult stem cells have been isolated from dental pulp, and their multilineage differentiation ability has been demonstrated. Unlike other commonly used stem cell sources, dental pulp stem cells (DPSCs) have a neural crest cell embryonic origin, thus they may therefore show differences in subsequent cellular differentiation.4–7
Differentiating mesenchymal stem cells towards different lineages usually requires the use of signaling factors in the form of recombinant proteins or the induction of functional genes using gene therapy. Viral vectors are highly efficient in transducing various cells and expressing the desired functional trans-genes over prolonged times.8,9 However, several biological safety issues are associated with the use of virus-based vectors. 10 Alternatively, chemical methods for transfection are simpler to use and relatively safe compared with viral vectors. However, they show low transfection efficiency as well as marked cytotoxicity when used with primary cell lines. 11
In 1982, Neumann et al. 12 introduced the “electroporation” technique for transfection of DNA to animal cells. Electroporation is a physical transfection method, in which short high-voltage electrical pulses are used to transiently permeabilize the cell membrane and allow trans-genetic material into the cells.12–14 Several variables are involved in the electroporation technique, such as the electroporation buffer, DNA concentration, mode of electric pulse, number of pulses, and voltage, all of which require optimization for different cell types to achieve proper transfection efficiency with reasonable viability.11,15
In our knowledge, no study to date has performed optimization of electroporation parameters for DPSCs, although optimization has been described for other mesenchymal stem cells. 16 Although electroporation has been reported to transfect dental pulp cells without optimization, those cells were porcine not human. 17
In this study we aimed to optimize electroporation parameters for the transfection of human DPSCs (hDPSCs), with cellular viability taken into account. Other chemical transfection agents were used for comparison, and their transfection efficiency and relevant cell viability were assessed. We also used our newly optimized transfection parameters to transfect DPSCs with a transgene and assessed its expression in the transduced cells.
Materials and Methods
DPSC isolation
Ethical approval was obtained from the Institutional Review Board of the University of Hong Kong to ensure the research complied with Declaration of Helsinki and was in accordance with international conference on harmonisation (ICH) good clinical practise (GCP) guidelines (IRB:UW 09-318). After written informed consent and medical history were obtained from patients, dental pulp was collected from sound, healthy premolars extracted for orthodontic treatment purposes at the Prince Philip dental hospital at the University of Hong Kong. Collected pulp tissue was washed twice using antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin), rinsed with phosphate-buffered solution (PBS), and dissected into 2-mm pieces. Pulp fragments were predigested in collagenaise/dispase enzyme mixture for a period of 15 minutes, transferred to culture plate supplied with α-MEM culture medium, supplemented with 10% fetal bovine serum and antibiotics, and then incubated at 37°C and 5% CO2.
DPSCs selection
Cells obtained from explants were subcultured until a sufficient number (approximately 1×107 cells) was reached as determined by hemocytometer. Afterwards, cells were isolated using immune magnetic bead selection mouse IgG kit (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol. Briefly, detached cells were incubated with antibody reactive to CD146 (OJ79c MUC 18, sc-53369; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h on ice on a shaker, washed twice with PBS/1% bovine serum albumin, and then incubated with anti-mouse IgG-conjugated magnetic Dynabeads (Invitrogen) for 40 min on a rotary mixer at 4°C. Cells binding to beads were removed using Magnet (Dynal, MPC, Invitrogen), then released from binding beads using DNase buffer. 18
DPSCs characterization
After isolation and culturing of DPSCs, cells were trypsinized and fixed using 4% paraformaldehyde in PBS (phosphate-buffered saline) then stained for CD146 (OJ79c MUC 18, sc-53369, Santa Cruz Biotechnology) or Stro-1 (sc-47733, Santa Cruz Biotechnology) mouse monoclonal primary antibodies in 4°C overnight, followed by fluorescein isothiocyanate–conjugated secondary antibody for 2 h. Matched isotype controls and nonselected cells were used as controls. Positive cells were assessed using confocal microscopy and flow cytometric analysis. Characterization was performed in triplicates for five independent donor samples.
Construction of (hTGF-β3) pcDNA3.1/V5-His-TOPO expression plasmid
The coding sequence for human transforming growth factor β3 (hTGF-β3) was obtained by PCR from (hTGF-β3)-pCMV6-XL5 plasmid DNA (Origene Technologies, Rockville, MD) and was kindly provided by Dr. Wang (Nanyang Technological University, Singapore). 19 The coding sequence was then amplified via reverse transcription PCR, using AccuPrimeTaq DNA Polymerase High Fidelity reverse transcription enzyme (Invitrogen). PCR products were checked by agarose gel electrophoresis, and fresh PCR products were used for cloning reaction.
Fresh Taq polymerase–amplified PCR products possess a single deoxyadenosine (A) at their 3′ end, which enables direct ligation to the single thymidine (T) overhang that the linear pcDNA3.1/V5-His-TOPO (Invitrogen) possesses at its 3′ end.
Ligation components were gently mixed and incubated at room temperature for 30 min. The reaction mix was then placed on ice until transformation. Two microliters of the reaction mix was gently mixed with one vial of DH5-α Escherichia coli and then incubated on ice for 30 min. Cells were then heat shocked for 60 sec in a 42°C water bath, after which they were incubated again on ice for 5 min. The transformation mix was then incubated in 0.9 mL of room temperature SOC medium and shaken at 225 rpm (∼30 g) in a 37°C incubator for 1 h. Transformed competent DH5-α was plated on selective LB plates with ampicillin, then incubated at 37°C overnight. To ensure proper insert ligation and orientation, colonies were picked out of each transformation and screened using PCR and XbaI restriction enzyme. Purification of target plasmid from positive colonies was done using Miniprep kit (Qiagen, Hilden, Germany).
Sequencing extracted plasmid
The inserted gene coding sequence was verified to ensure proper insertion and absence of site mutations in the sequence. Sequencing was done at The University of Hong Kong Genome Research Center using T7 and BGH primers to provide bidirectional sequencing.
Transfection of DPSCs with eGFP or pcDNA3.1-hTGF-β3 using electroporation
Cells were combined with nucleic acid in an electroporation buffer and exposed to high voltage pulsation, which causes transient pores in the cellular membrane thus allowing the intake of the nucleic acid into the cell.13,14 DPSCs were transfected with either eGFP plasmid or pcDNA3.1-hTGF-β3 plasmid depending on the experiment.
Optimization of transfection efficiency of DPSCs
For electroporation, 70–80% confluent DPSCs of passages 3–4 were washed with PBS, trypsinized, and then centrifuged for 5 min at 1200 rpm (∼400 g). Cells were then resuspended in a serum- and antibiotic-free isotonic pulsing buffer at 1×106 cells/mL. A total of 100 μL of DPSCs were mixed with 5 μg of enhanced green fluorescent plasmid (eGFP) in 0.2-mm cuvettes and electroporated using Gene Pulser Xcell electroporation system (Bio-Rad, Hercules, CA). 11
Electroporation was done with varied parameters: voltage, time of pulse, mode of pulse, and number of pulses. Transfected cells were then analyzed for transfection efficiency and cell viability. Chemical transfection was used as a positive control.
For chemical transfection, Lipofectamine 2000 transfection reagent (Invitrogen) or Fujene6 transfection reagent (Roche, Basel, Switzerland) was added to the cells. Cells were combined with eGFP plasmid with different transfection reagent/DNA ratios according to the respective manufacturer's recommendations. Experiments were performed in triplicate for each transfection condition.
Analysis of transfection efficiency
Forty-eight hours after transfection, cells were checked using fluorescent microscopy, then trypsinized and washed for flow cytometric analysis. Cells were gated and counted according to their positivity and negativity for eGFP.
Cell viability analysis
Cells transfected using variable electroporation parameters and chemical transfection reagents were transferred into a 96-well plate and incubated in humidified 5% CO2. Cell viability was measured at 24 and 72 h using a CCK-8 cell counting kit (Dojindo Molecular Technologies, Rockville, MD). Non-transfected DPSCs were used as control. Experiment was done in triplicate.
Transfection of DPSCs with pcDNA3.1-hTGF-β3
Optimal electroporation parameters were applied to transfect DPSCs with pcDNA3.1-hTGF-β3. Confluent cells (70–80%) were trypsinized, collected by centrifugation, and resuspended in a pulsing buffer without serum and antibiotic at 1×106 cells/mL. A total of 100 μL of DPSCs was mixed with 5 μg of pcDNA3.1-hTGF-β3 in 0.2-mm cuvettes and electroporated using Gene Pulser Xcell (Bio-Rad).
hTGF-β3 in vitro expression level in transfected cells
Real-time PCR was used to evaluate the expression level of hTGF-β3 of DPSCs transfected with pcDNA3.1-hTGF-β3 using non-transfected and eGFP-transfected DPSCs as negative and positive controls, respectively.
Western blot analysis for hTGF-β3 expression
Human TGF-β3 protein expression was investigated in nonreduced medium collected from cultured transfected cells 72 h after transfection using Western blotting. The protein amount in each sample was measured using BCA protein assay reagent (Pierce, Rockford, IL). Detection was done using rabbit anti-human polyclonal TGF-b3 (ab15537, Abcam, Cambridge, United Kingdom) primary antibody and goat anti-rabbit IgG-horseradish peroxidase (Santa Cruz Biotechnology) secondary antibody.
Statistical analysis
One-way ANOVA was used to compare the cell viability, transfection efficiency, and real-time PCR data among the groups. Differences between experimental groups were considered to be significant at (p<0.05). All statistical analyses were done using SPSS (version 19; IBM, Chicago, IL).
Results
Isolated DPSC characterization
Migration of the cells from the explants to the culture plate usually occurred within the first week of culture. The cells appear as elongated adherent cells that expanded and proliferated rapidly (Fig. 1).
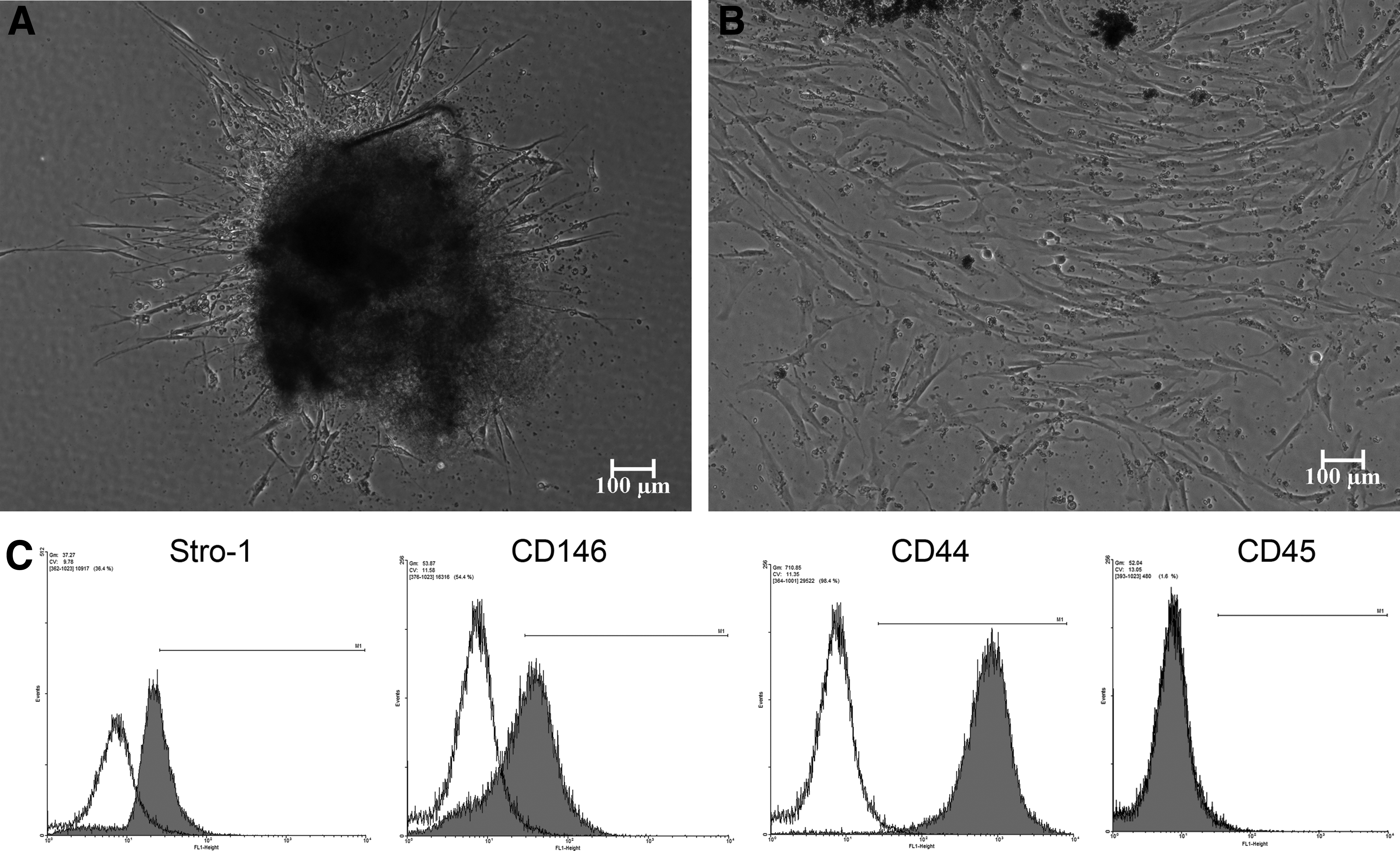
Flow cytometric analysis clearly showed the stromal origin of the selected cell population. The isolated cell population showed high expression of Stro-1 (mean=34.35±5.06%) and CD146 (mean=42.885±4.27%) surface markers, demonstrated entire positivity to CD44 (stromal cell marker; mean=98.6±0.9%), and negative expression of CD45 (mean=0.4±1.22%), a hematopoietic progenitor and stem cell marker.20–22
Optimization of transfection efficiency of DPSCs.
DPSCs transfected with FuGene 6 chemical transfection reagent showed a statistically significant higher cell viability percentage (p<0.001) compared with cells transfected with Lipofectamine transfection agent at all tested transfection agent/DNA ratios. Among the different FuGene 6/DNA ratios groups, no statistically significant differences were found. The groups showed a mean cell viability of 87.797±3.027% at day 1, which reached the viability of the non-transfected control group by day 3.
Among the different DNA/Lipofectamine ratio groups, statistically significant differences were found between the three tested ratios. The 1:1 DNA/Lipofectamine ratio showed the highest cell viability percentage at both day 1 and day 3, while the 1:5 ratio showed the lowest cell viability percentage at both day 1 and day 3. Notably, the DNA/Lipofectamine 1:1 ratio group was the only group in which the cells at day 3 revived to normal viability levels. This was not the case for the 1:2.5 group in which the viability was only slightly recovered, and the 1:5 group even showed a decrease in viability at day 3 compared to day 1 (Fig. 2).
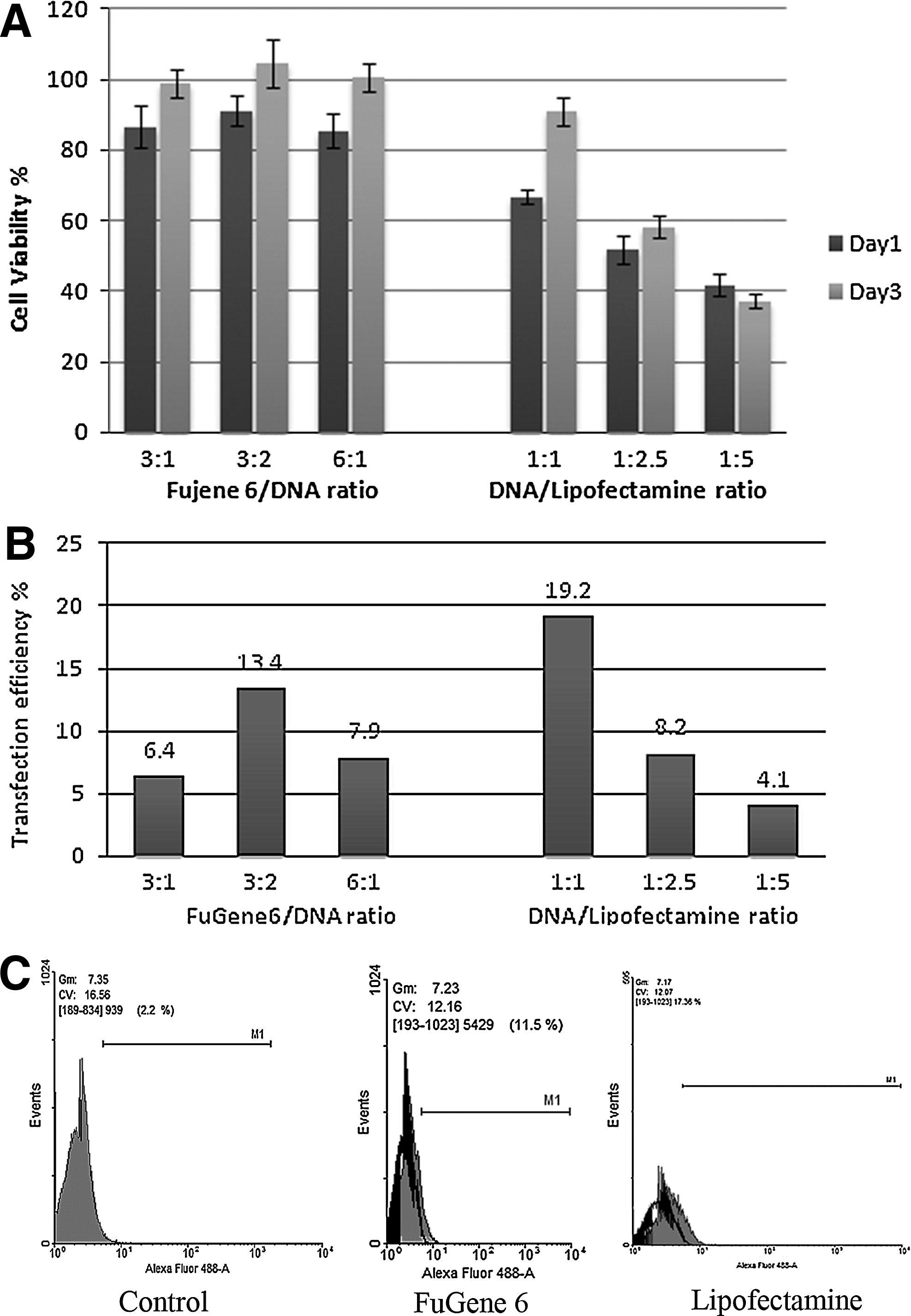
FuGene6 and Lipofectamine chemical reagents transfection with variable reagents to DNA ratios.
The electroporation groups showed variable range of viabilities depending upon the pulsing mode, voltage, and the number of pulses (Fig. 3).

Electroporation transfection.
Analysis of transfection efficiency
As a transfection reference for using electroporation, FuGene6, and Lipofectamine 2000, the 293 cell line was used. Quantified by flow cytometric analysis, optimal transfection efficiencies were achieved for this cell line by using respective manufacturers' optimized transfection protocols.
For the chemical transfection methods, the highest transfection efficiency achieved with the FuGene6 transfection agent (13.44±2.03%) was by the 3:2 FuGene6 (μL) to DNA (μg) ratio, while for the Lipofectamine 2000 transfection agent the highest transfection efficiency (19.23±3.19%) was achieved by the 1:1 DNA (μg) to Lipofectamine (μL; Fig. 2).
Flow cytometric results showed higher transfection efficiencies when comparing the following electroporation groups: square wave 100 V (one pulse), 210 V (three pulses), exponential 120 V–500 μF capacitance, and 160 V–300 μF capacitance, to chemical transfection reagents, as shown by the percentage of positive GFP-expressing cells.
Among the electroporation groups, the highest transfection efficiency (63.81±4.72%) was achieved using 100 V, 20 msec, one-pulse square-wave electroporation condition. Comparable transfection efficiency (59.91±5.03%) was achieved by the 210 V, 0.999 msec, three-pulse square-wave electroporation condition. As for the exponential electroporation mode, the 120 V–500 μF capacitance showed the highest tranfection efficiency (29.46±1.99 %; Fig. 3).
Chemical and electroporation transfection profiles were confirmed using fluorescent microscopy (Fig. 4).

Transfection efficiency of electroporation and chemical transfection reagents examined by fluorescent microscopy for cells expressing eGFP fluorescent protein. 293 cell line used as internal control.
Transfection of DPSCs with pcDNA3.1-hTGF-β3
Real-time PCR showed a statistically significant higher expression level of hTGF-β3 for DPSCs transfected with pcDNA3.1-hTGF-β3 relative to non-transfected and eGFP-transfected DPSCs.
Human TGF-β3 protein expression was also investigated in the medium collected from cultured transfected cells to investigate its secretion via Western blot analysis (Fig. 5).

Human transforming growth factor β-3 (hTGF-β3) expression by transfected DPSCs using quantitative reverse-transcription PCR; the relative expression of the TGFβ-3 gene was assessed in transfected DPSCs and compared with cells transfected with eGFP plasmid
Real-time PCR showed an increase in chondrogenic gene relative expression levels in transfected cells (Fig. 6).

Chondrogenic genes relative expression levels in TGF-β3 transfected cells (p<0.05, one-way ANOVA).
Discussion
The main aim of this study was to establish the electroporation parameters needed to optimally transfect hDPSCs. In electroporation, an electric high voltage pulse is applied to the cells to be transfected, thus rendering those cells transiently permeable to the target nucleic acid. Although the theory behind electroporation is fairly simple, the application of the technique is far from simple because of the combinations of variables that need to be honed in order to achieve an acceptable transfection level. In our study we optimized electroporation parameters for DPSCs. We were able to achieve high transfection efficiencies, together with better cell viability using the electroporation technique, in comparison to our controls, commonly used chemical transfection reagents. As a proof of concept, we used our optimized transfection conditions to study expression and the effect of a commonly used functional chondrogenic transgene (TGF-β3) in transfected DPSCs.
Dental pulp has proved to be a valuable source for mesenchymal stem cells. 23 However, in most of the studies, differentiation of DPSCs into a specific lineage is done using recombinant proteins or growth factors directly added to the culture medium.4,24,25 The short half life, high cost, and difficulty to deliver in vivo are all downsides to the direct use of signaling and recombinant proteins. 19
As far as we know, the only study reporting transfection optimization for dental tissues driven cells was the study by Yalvac et al., 11 in which optimization of chemical methods and electroporation for human dental follicle cells were investigated. The highest transfection efficiency for chemical methods in their study was achieved by FuGene HD, which yielded 19% transfection efficiency with approximately 80% cell viability. Notably those results were very comparable to our chemical transfection results in which a transfection efficiency of 19.23±3.19% was achieved with 1:1 DNA (μg):Lipofectamine (μL), with a 66.58% cell viability. As for the electroporation parameters optimization, the authors only explored the exponential decay wave mode for electroporation, in which the target voltage exponentially decays overtime. They reported the optimal parameter for transfection of human dental follicle cells to be 300 V exponential decay wave, which resulted in 39% transfection efficiency and 69% cell viability. Our exponential decay wave results showed that the highest transfection efficiency of 29.46±1.99% was for the 120-V group, a result that approximates the results achieved by Yalvac et al., 11 but with a much lower voltage and cell viability (26.89%).
The large difference in voltage used between our study and the previous study is due to the difference in the cuvette width, which denotes the gap distance between the cuvette electrodes. In the previous study, 0.4-mm cuvettes were used; in our study, we used 0.2-mm cuvettes. The field strength, in which the voltage is divided by the cuvette gap, is doubled by using 0.2-mm cuvette distance compared with the 0.4-mm cuvette distance. This should thus be accounted for through voltage adjustment. 26 Using the exponential decay wave electroporation, much lower cell viability was attained in our study compared to the Yalvac et al. study, which may be an indication that the hDPSCs are more sensitive to the exponential decay wave mode compared to human dental follicle cells.
Unlike the exponential decay mode, the square-wave mode can control both the voltage and the pulse time simultaneously, which can be more efficient for sensitive cells. In our study, the 100 V, 20 msec, one-pulse square-wave electroporation parameter offered the highest transfection efficiency (63.81±4.72%), with a 31.83% cell viability that soon attained a 70% cell viability at day 3.
In one of the very few studies using electroporation to transfect dental pulp cells, Nakashima et al. 17 used 210 V, 0.999 msec, three-pulse square-wave electroporation to transfect porcine dental pulp cells with growth/differentiation factor 11 (Gdf11). They achieved a transfection efficiency of 69±5% and a cell viability of 85%. In our study, using the same square wave electroporation parameters, the 210 V indeed gave the highest transfection efficiency (59.91±5.03%), which was comparable to the findings of Nakashima et al., which had much lower cell viability. This can be attributed to the differences in the time and method of assessment of viability; they assessed the viability directly after electroporation using trypan blue, while we assessed the 24-h viability using the cell counting kit and a relative control. Another factor could be the species difference; they examined porcine DPSCs while we examined hDPSCs. Aside from the population difference, there was no further selection to the cells they used.
The electroporation method gave much higher tranfection efficiency, while simultaneously avoiding the marked cell toxicity compared with chemical transfection methods. In our study, Lipofectamine chemical reagent to DNA ratios higher than 1:1 showed marked cytotoxicity, with very low transfection efficiencies.
The study provides optimized parameters for electroporation for DPSCs, as well as optimized transfection ratios for two commonly used transfection reagents. This could provide valuable information that could help in various gene therapy and tissue engineering applications that using hDPSCs.
Footnotes
Acknowledgments
This research was funded using Prof. Rabie's research account. The authors wish to thank Dr. Wang Dong-an, PhD, for kindly providing us with (hTGF-b3)-pCMV6-XL5 plasmid, Mr. Raymond Tong (Oral Biosciences Technician, HKU) for guidance in transfection and flow cytometric analysis, and Dr. Finn Geoghegan, MOrth, for help in reviewing and editing the manuscript.
Author Disclosure Statement
No competing financial interests exist.