
Abstract
In addition to being a part of the metabolic fatty acid fuel cycle, butyrate is also capable of inducing growth arrest in a variety of normal cell types and senescence-like phenotypes in gynecological cancer cells, inhibiting DNA synthesis and cell growth in colonic tumor cell lines, suppressing hTERT mRNA expression and telomerase activity in human prostate cancer cells, and inducing stem cell differentiation and apoptosis by DNA fragmentation. It regulates gene expression by inhibiting histone deacetylases (HDACs), enhances memory recovery and formation in mice, stimulates neurogenesis in the ischemic brain, promotes osteoblast formation, selectively blocks cell replication in transformed cells (compared to healthy cells), and can prevent and treat diet-induced obesity and insulin resistance in mouse models of obesity, as well as stimulate fetal hemoglobin expression in individuals with hematologic diseases such as the thalassemias and sickle-cell disease, in addition to a multitude of other biochemical effects in vivo. However, efforts to exploit the potential of butyrate in the clinical treatment of cancer and other medical disorders are thwarted by its poor pharmacological properties (short half-life and first-pass hepatic clearance) and the multigram doses needed to achieve therapeutic concentrations in vivo. Herein, we review some of the methods used to overcome these difficulties with an emphasis on HDAC inhibition.
Introduction
In a seminal article published in 1990, a Japanese group 1 reported that (R)-trichostatin A (a fungal antibiotic, Fig. 1) is a potent and specific inhibitor of mammalian histone deacetylase (HDAC) activity in vivo, and that this inhibition strongly induced tumor cell apoptosis, a process that can be mediated epigenetically by regulating the histone function. Since that time, research into the roles HDAC enzymes play in cancer development, progression, and survival has led to the identification of other histone deacetylase inhibitors (HDACi),2–5 and several of these have been evaluated in clinical trials, primarily for the treatment of hematologic malignancies.6–8 HDACi are also being investigated for synergistic cytotoxicity in combination therapies with conventional chemotherapy and targeted agents against solid tumors.9–12 One of the combination strategies fuses the cytotoxic platinum DNA-binding potential of platinum agents and HDAC inhibitory activity into a single molecular entity. 13 In 2006, vorinostat (SAHA, Zolinza®, Fig. 1) became the first HDAC inhibitor to be clinically approved by the Food and Drug Administration 14 for use in the treatment of cutaneous T-cell lymphoma (a rare form of non-Hodgkin's lymphoma that localizes to the skin). In clinical trials, vorinostat has failed as a monotherapy in the treatment of metastatic breast cancer (and other solid tumors). However, in combination with paclitaxel and bevacizumab as a first-line therapy, the results are more promising. 15
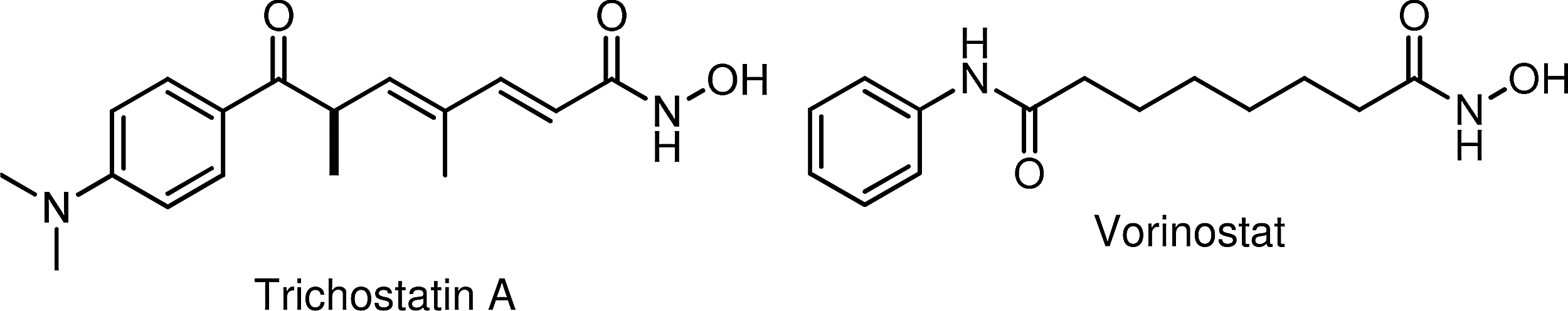
Hydroxamic acid HDACi: trichostatin A and vorinostat. HDACi, histone deacetylase inhibitors.
Mammalian HDAC Isoforms
The mammalian HDAC family of enzymes consists of at least 18 isoforms that are grouped into four principal class types according to their expression patterns and sequence homology relative to the yeast HDAC proteins. 16 Class I (HDACs 1, 2, 3, and 8) proteins are found predominantly in the nucleus; Class IIa (HDACs 4, 5, 7, and 9) and Class IIb (HDACs 6 and 10) shuttle between the nucleus and the cytoplasm; Class III enzymes, known as the sirtuins (Sirt1–7), are a silent information regulator (Sir)2 family of proteins17,18 that are NAD+-dependent deacetylases inhibited by nicotinamide;18–20 and Class IV (which contains only HDAC 11) has properties of both Class I and Class II HDACs, and like the Class I and Class IIa/b isoforms, uses a lysine residue to bind to Zn2+ in the active state.3,18,21
In addition to the histone proteins, some nonhistone proteins are also targeted by histone acetyltransferases (referred to as HATs22–24 in keeping with the historical histone relationship) and deacetylases (referred to as HDACs), and as a consequence, acetylation and deacetylation processes figure prominently in mitigating cellular function and activity as well as transcriptional and chromatin-related processes.18,25 Choudhary et al. 26 undertook a mass-spectral-based study designed to analyze sites of lysine-specific HDACs (KDACs) and concluded that protein modification by acetylation in vivo could be as common as an event as phosphorylation27–29 —a hypothesis predicted a decade earlier by Kouzarides. 30 Acetyl is the primary acyl group exchanged by the HATs and HDACs, although propionyl and butyryl may also be substrates.20,31–33 Typically, histone acetylation is associated with the activation of gene transcription, whereas deacetylation is associated with transcriptional repression. 34 Similarly, methylation and demethylation are alternative pathways cells utilize to effect epigenetic changes.23,35,36 Strahl and Allis 37 suggest that combinatorial sequences of epigenetic changes represent a histone language sometimes referred to as a histone code that proteins could read, write, erase, and modify. 38 Importantly, since the energy needed to support epigenetic transformations in a cell is provided by its mitochondria, nuclear and mitochondrial genomic interactions in the cell are coordinated. 18
DNA Accessibility
In mammalian cells, the genome is tightly packed into chromatin units called nucleosomes, which consist of ∼147-base-pair segments of DNA wrapped around a core of eight histones (two each of H2A, H2B, H3, and H4).39,40 Electrostatic forces between positive-charged lysine residues on the histone proteins and negative-charged phosphates on the DNA backbone allow the nucleosome to adopt a highly condensed three-dimensional structure that limits access to the DNA segment by transcription factors and other DNA-seeking chemicals. For example, for a segment of DNA to be read by the cell's transcriptional machinery, the DNA must first be made accessible. One means by which this is achieved in the cell is through acetylation by HAT 36 of the lysine residues on the histone tails protruding from the nucleosome cores.24,40 This neutralizes the positive charges on the histone tails (relieving electrostatic forces that keep the histone–DNA pair bound closely together) and exposes the DNA. 39 Once transcription has been completed, HDAC enzymes remove the N-acetyl groups from the acetylated lysine residues, which restores positive charge to the histone and draws the DNA back into its protected, less-accessible tertiary structure. 5
Consequently, inhibiting HDACs can cause gene activation to be initiated or prolonged—an effect that has a therapeutic application across a wide-spectrum of disease phenotypes, as well as in the ex vivo production of stem cells by induced pluripotency (reprogramming),41,42 for use in the emerging field of regenerative therapy.43,44 As a result, much effort is directed toward identifying druggable HDACi with therapeutic potential, particularly of the small-molecule type.7,45–51
n-Butyric Acid
n-Butyric acid, a short-chain naturally occurring fatty acid, is produced in transient amounts during the natural synthesis and breakdown of longer-chain fatty acids in vivo. A significant source in the diet comes from dairy products (such as Greek feta cheese) containing lamb rennet.52,53 However, n-butyric acid (butyrate) is also endogenously made in the human body by anaerobic bacterial fermentation of carbohydrates (derived from dietary fiber) in the colon,54–56 but in addition to being a part of the metabolic fatty acid fuel cycle,57,58 butyrate is also capable of inducing growth arrest in a variety of normal cell types and senescence-like phenotypes in gynecological cancer cells,59,60 inhibiting DNA synthesis and cell growth in colonic tumor cell lines,61–64 suppressing hTERT mRNA expression and telomerase activity in human prostate cancer cells, 65 and inducing stem cell differentiation66–71 and apoptosis by DNA fragmentation. 72 It regulates gene expression by inhibiting HDACs,73,74 enhances memory recovery and formation in mice, 75 stimulates neurogenesis in the ischemic brain,70,76,77 promotes osteoblast formation, 78 selectively blocks cell replication in transformed cells (compared to healthy cells),79–81 and can prevent and treat diet-induced obesity and insulin resistance in mouse models of obesity, 82 as well as stimulate fetal hemoglobin expression in individuals with hematologic diseases such as the thalassemias and sickle-cell disease,83–85 in addition to a multitude of other biochemical effects in vivo.86–88
However, efforts to exploit the potential of butyrate in the clinical treatment of cancer and other medical disorders are thwarted by its poor pharmacological properties (short half-life and first-pass hepatic clearance) and the multigram doses needed to achieve therapeutic concentrations in vivo.39,79,89–95 Prodrugs of butyric acid96,97 such as Pivanex98–100 and tributyrin58,94,101 (Fig. 2) help mitigate the impediments, but have not been viable as therapeutic agents. Nonetheless, the arginine salt of butyrate has been utilized successfully in clinical studies for the therapy of sickle-cell disease,102,103 thalassemia, 104 Epstein-Barr Virus-related malignancies, 6 and chronic, non-healing wounds, 105 clearly demonstrating the proof of principle for butyrate-based epigenetic therapeutic approaches to these diseases.

Butyrate HDACi: butyric acid, Pivanex, and tributyrin.
Short-Chain Fatty Acids
Although not as potent as butyric acid, other short-chain fatty acids (SCFAs) like valproic acid and 4-phenylbutyrate (Fig. 3) also have HDAC inhibitory activity.106,107 However, a structure–activity relationship study carried out by Gilbert et al. 108 found that nonbranching SCFAs having three to five carbon atoms in length (Fig. 4) are the best inhibitors of HDACs. According to Gilbert et al. 108 branching decreases the inhibition of HDACs relative to that observed with n-propionate, n-butyrate, or n-pentanoate. Among the branched-chain acids, valproate exhibited the greatest activity with 2,2-dimethylbutyric acid109,110 and 2-ethylbutyric acid exhibiting only half the activity of valproate. The observations are consistent with the Lu et al.111,112 model of the active site of HDACs being a narrow tube-like pocket, spanning a length equivalent to a straight chain of four to six carbon atoms, with a Zn2+-chelating moiety positioned near the bottom to help facilitate the deacetylation catalysis.113–116 Interestingly, pyruvate, the three-carbon chain end-product of glycolysis, is an endogenous HDAC inhibitor, whereas lactate is not. 117 Thus, the tumor-associated cytoplasmic diversion of pyruvate into lactate (the Warburg effect) may serve not only to induce a state of apoptosis resistance by suppressing mitochondrial activity in the tumor cell 118 but also as a means for the tumor cell to avoid HDAC inhibition, a potential alternate and suicidal pathway.

Short-chain fatty acid HDACi: valproic acid and phenylbutyric acid.
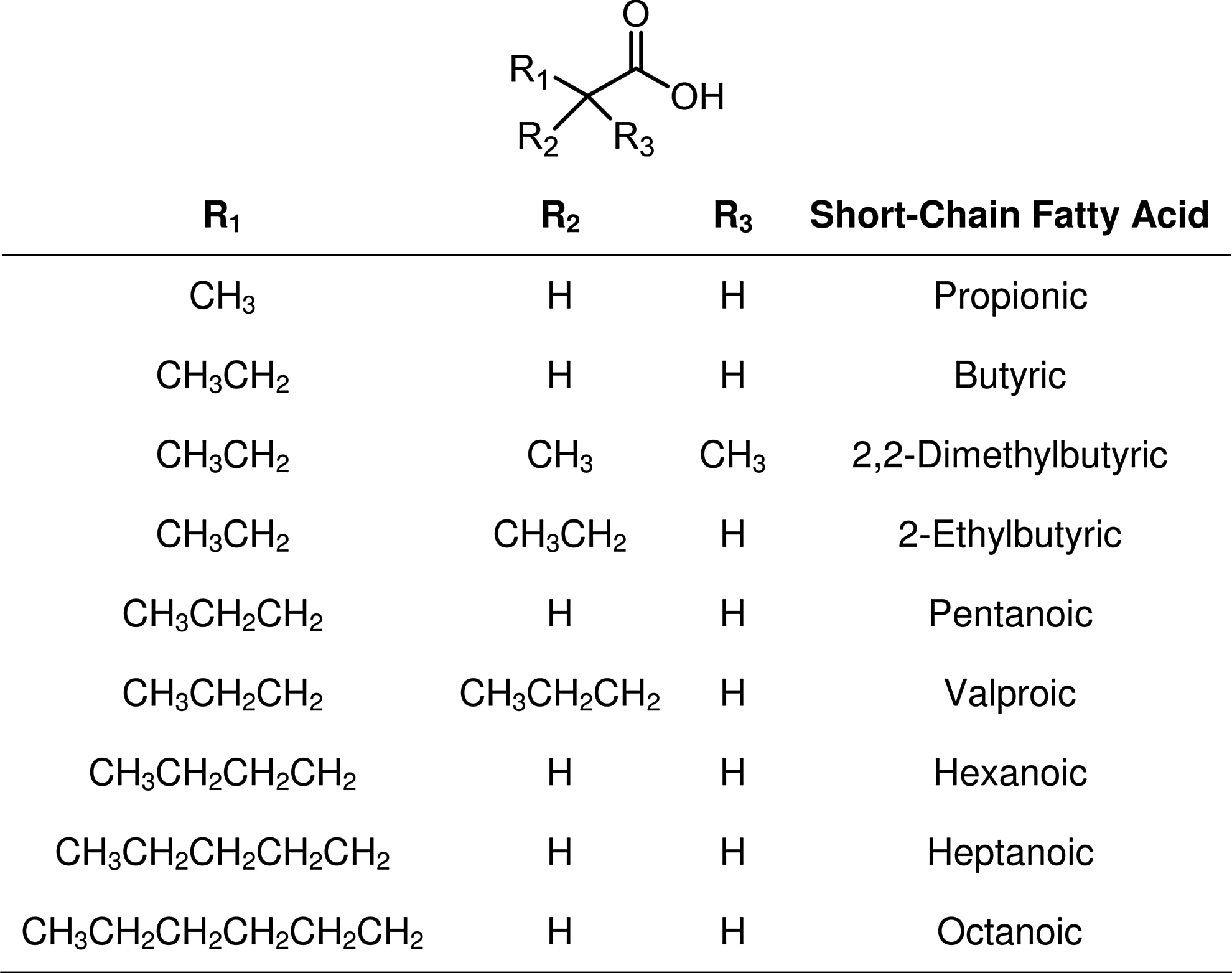
Variable short-chain fatty acid HDACi.
Cellular Uptake by Transport Proteins
While prodrugs can facilitate cell penetration and help prolong bioavailability, their metabolic cleavage does not necessarily result in therapeutic concentrations of the drug being delivered where needed. Cellular uptake of fatty acids, butyric acid included, is achieved with the aid of specific transporters119,120 and cell surface receptors.120,121 Likewise, cellular entry of carbohydrates is also tightly regulated by carbohydrate-specific transporters and cell surface receptors.120,122,123 The most sensitive clinical test for detecting occult metastases is the enhanced uptake of 2-deoxy-2-[18F]fluoro-d-glucose in vivo by cancer cells compared to normal cells as determined by positron-emission tomography 124 —implying that glucose (and other natural carbohydrate) scaffolds may be used to carry (cytotoxic) substrates selectively into cancer cells.125–127 This construct was applied to butyrate and other SCFAs,33,79,128–131 for example, Bu4ManNAc and 3,4,6-O-Bu3GlcNAc (Fig. 5).125,130 However, though families of transporters exist for the common dietary sugars such as glucose, they are refractory to modified derivatives (substituted carbohydrates) or analogs, and without assisted cell membrane crossing, cellular uptake of non-natural sugars is limited. 122

Anticancer sugar-based HDACi butyrates.
Butyryl-l-Carnitine Esters
An alternative delivery method we118,132,133 and others134–138 have found to be an effective transporter of modified SCFA substrates in vivo utilizes the cell's natural carnitine–acylcarnitine transport machinery. More than 100 years ago, Knoop 139 published a seminal study on the metabolism of omega-phenyl-substituted fatty acids that he fed to dogs. When the dogs were fed odd-chain-substituted fatty acids, hippuric acid (2-benzamidoacetic acid) was found in their urine, and when fed even-chain-substituted fatty acids, phenylaceturic acid [2-(2-phenylacetamido)acetic acid] was the result. 140 To account for these findings, he proposed that the metabolism proceeds by the successive removal of two carbon units via the existence of a mitochondrial fatty acid β-oxidation pathway—a type of carbon oxidation having no reported examples in organic chemistry at the time. 140
However, to enter the mitochondrial inner matrix, where β-oxidation takes place, fatty acids (presenting as acylcarnitines) 140 are passaged through the mitochondrial membrane via the carnitine acyltransferase pathway. 141 Long-chain fatty acids are obligated to be processed this way, but SCFAs can also use this pathway too. 142 Thus, as Srinivas et al. 135 reported, butyryl-l-carnitine can act as a prodrug for delivering butyrate into cells in vivo, and our own studies (article in preparation) found that butyryl-l-carnitines, PMX™ 550B and PMX 550D (Fig. 6), are more potent HDACi than butyrate itself.
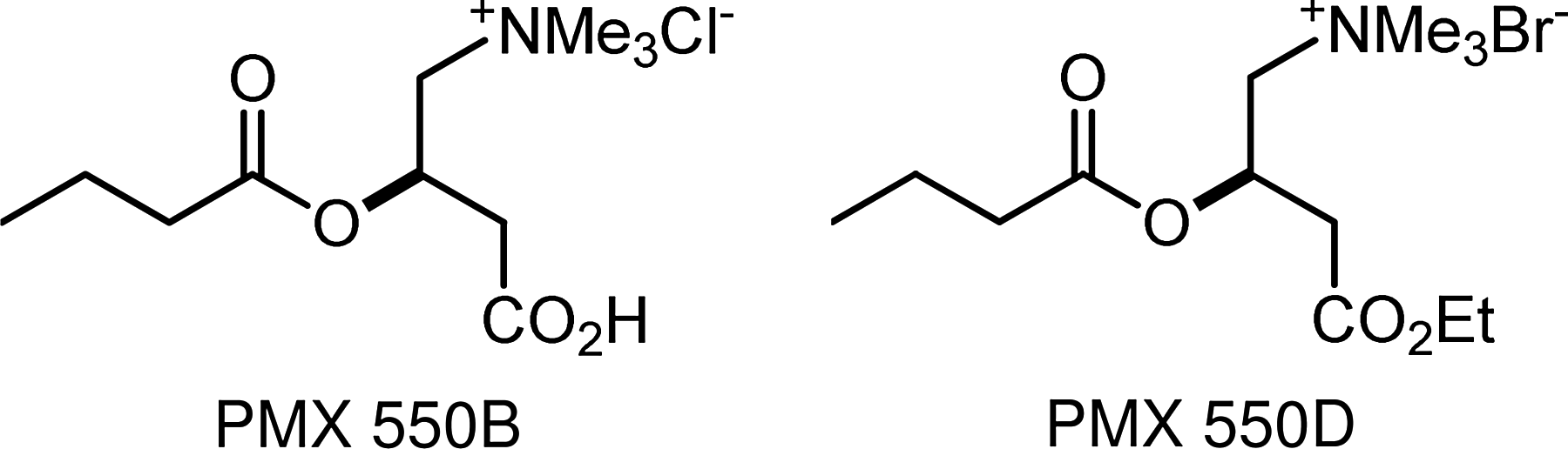
Butyryl-l-carnitines.
Conclusions
Unlike the sugar transporters, which highly discriminate against synthetically modified sugars, the acylcarnitine transporters appear to tolerate a range of acyl-substrate variations 118 that can traverse both the plasma118,135,140 and blood–brain barriers.133,134 PMX 550B and PMX 550D are strong butyrate HDACi, and their potential for oral delivery and mitigation of short-half life may lead to more effective epigenetic therapeutics for treating the thalassemias, sickle-cell disease, neurological disorders, and cancer.
Footnotes
Acknowledgments
The authors gratefully acknowledge support from the Foundation for a Cure for Mitochondrial Disease (MitoCure) and from the Karin Grunebaum Cancer Research Foundation, and by the grants: DK-R01-052962, R41 HL-110727, CA-153474, and T32 HL007501 from the National Institutes of Health, and by a grant from the V Foundation.
Authors' Contributions
The article was written through contributions of all authors. All authors have given approval to the final version of the article.
Author Disclosure Statement
K.S. owns shares in PhenoMatriX. No competing financial interests exist.