
Abstract
Motility is widely distributed across the tree of life and can be recognized by microscopy regardless of phylogenetic affiliation, biochemical composition, or mechanism. Microscopy has thus been proposed as a potential tool for detection of biosignatures for extraterrestrial life; however, traditional light microscopy is poorly suited for this purpose, as it requires sample preparation, involves fragile moving parts, and has a limited volume of view. In this study, we deployed a field-portable digital holographic microscope (DHM) to explore microbial motility in Badwater Spring, a saline spring in Death Valley National Park, and complemented DHM imaging with 16S rRNA gene amplicon sequencing and shotgun metagenomics. The DHM identified diverse morphologies and distinguished run-reverse-flick and run-reverse types of flagellar motility. PICRUSt2- and literature-based predictions based on 16S rRNA gene amplicons were used to predict motility genotypes/phenotypes for 36.0–60.1% of identified taxa, with the predicted motile taxa being dominated by members of Burkholderiaceae and Spirochaetota. A shotgun metagenome confirmed the abundance of genes encoding flagellar motility, and a Ralstonia metagenome-assembled genome encoded a full flagellar gene cluster. This study demonstrates the potential of DHM for planetary life detection, presents the first microbial census of Badwater Spring and brine pool, and confirms the abundance of mobile microbial taxa in an extreme environment.
1. Introduction
Extant life elsewhere in our solar system, if it exists, is likely to be entirely microbial. Although the invention of the light microscope led to the discovery of prokaryotic life on Earth (Leeuwenhoek, 1677), standard light microscopes are of limited utility for in situ planetary life detection because they have many moving parts that are hard to miniaturize and ruggedize, and they can only inspect tiny volumes. Limits of detection for prokaryotes in typical light microscopy experiments are ∼105 cells/mL, which is higher than the cell density observed in many oligotrophic environments on Earth (Bedrossian et al., 2017).
For this reason, and because of the ambiguity of morphology for life detection, there has traditionally been little interest in microscopy-based life detection (Ruiz et al., 2022). A consensus is emerging, however, that microscopy can be a powerful tool for life detection and that development of new microscopy techniques for in situ use is needed (Neveu et al., 2018). The Europa Science Definition Team specifically called for a microscope capable of detecting microorganisms down to 0.3 μm in diameter at densities down to 103 cells/mL (Hand et al., 2017). This represents the worst-case scenario of small cell size and sparsity and presents a formidable challenge to instrument developers.
Digital holographic microscopy has the potential to meet this challenge. Digital holographic microscopes (DHMs) require no moving parts, compound objective lenses, or focusing (reviewed in Wu and Ozcan, 2018). The volumetric nature of the images, which may be digitally dissected after collection, makes this approach ideal for autonomous operation. When a microscopic particle comes within the field of view of the camera of the DHM, it creates an interference pattern that is recorded as a hologram. The hologram is used to reconstruct the image of the particle based on a selected algorithm. The resulting instantaneous depth of field is at least 100 times greater than for traditional light microscopy (Dubois et al., 1999; Kim, 2011). Capturing whole volumes with no moving parts allows DHM to be robust enough to survive deployment in harsh environments; user input for data acquisition is optional.
The sample chamber can be emptied and refilled with new samples or by continuous flow. However, trade-offs between interrogated volume, flight worthiness, and optical performance must be made. Field-deployable DHM instruments lacking compound objectives have low resolution and are unable to resolve fine cellular structures required to definitively identify microbial life (Wallace et al., 2015). Yet, at the present technical readiness level, fieldable DHMs are still useful for detecting life by detecting motile microorganisms, as microbial motility has characteristics that distinguish it from the passive movements of inanimate particles. The current study was enabled by the recent development of a field-deployable DHM (Fig. 1A–C) (Lindensmith et al., 2016; Wallace et al., 2015), which uses 405 nm laser illumination to achieve ∼0.8 μm resolution in a volume of view (XYZ) of 0.365 × 0.365 × 1.0 mm. These parameters were chosen to optimize characterization of microbial swimming motility and to use only flight-compatible components.

DHM instrument, Badwater Spring, and sampling locations.
Motility is a widespread feature of microbial life on Earth. Not all bacteria, eukarya, and archaea are motile, but all three domains have many motile members (Miyata et al., 2020). Motility improves chances of survival as it enables active movement to sources of nutrients and away from toxins, instead of relying solely on diffusion (Stocker et al., 2008; Taylor and Buckling, 2013; Taylor and Stocker, 2011). The mechanisms of motility in different life-forms vary drastically, from flagella in prokaryotes and flagellates, to cilia in ciliates, to propagating kinks in filamentous bacteria, to polysaccharide pili secretion in filamentous cyanobacteria and pennate diatoms, and many more (Bayless et al., 2019; Bondoc et al., 2016; Khayatan et al., 2015; Merz et al., 2000; Nakamura and Minamino, 2019; Palma et al., 2022; Shaevitz et al., 2005; Thornton et al., 2020). These mechanisms manifest in different forms of motility that are often characterized as swimming, sliding, gliding, twitching, and swarming (Henrichsen, 1972; Wadhwa and Berg, 2022).
Each motility form can display sets of unique motility patterns; for example, studied swimming patterns include run-tumble, run-pause, run-reverse-flick, and run-reverse, which describe motions that alternatingly propel cells forward and then actively or passively reorient them (Berg, 2004; Hintsche et al., 2017; Son et al., 2013). Distinguishing these active forms of motility from drift and Brownian motion is mathematically straightforward (Rouzie et al., 2021). Drift can be calculated by taking the average of uniform motion, which may then be subtracted. Brownian motion may be distinguished from swimming as it shows root mean square displacements that scale as the square root of time. While we can quantitatively characterize swimming patterns and distinguish motility from passive motion, it is much easier and still unambiguous to identify extant microbial life through qualitative observation, as was performed by Leeuwenhoek (1677) when prokaryotes were first discovered.
A separate approach for life detection that has been widely discussed is the detection and study of macromolecules that are universal in known Earth life (Neveu et al., 2018). On Earth, the availability of low-cost, rapid DNA sequencing technologies has led to a shift from culture- and microscopy-based approaches to studies of community DNA sequence data, based on either amplicons or shotgun metagenomes.
Such cultivation-independent approaches have paid huge dividends in the study of microbial life on Earth because they have greatly expanded our view of microbial diversity and supported hypothesis testing about the functions of yet-uncultivated microorganisms that dominate most biomes (Jiao et al., 2020; Nayfach et al., 2021). While these DNA-based approaches are extremely informative and can effectively complement more incisive experimentation on microbial activities that are important for growth and survival in situ (i.e., phenotypes), the success of DNA-based approaches for life detection depends on a similar biochemistry; therefore, the scope of DNA-based life detection approaches beyond Earth is inherently limited.
The goal of this study was to complement the results obtained from in situ microscopy via DHM with community DNA sequencing in an extreme environment to guide development of instrument suites for missions. Our study focused on Badwater Spring and brine pool, a hypersaline environment that is analogous to cryovolcanoes on Europa (Steinbrügge et al., 2020) and recurrent slope lineae on Mars (Chevrier and Rivera-Valentin, 2012), and which is teeming with microbial life. We identify distinct patterns of motility both in situ and in the laboratory with returned samples and validate the microscopic observations with observations of the high incidence of prokaryotes predicted to encode flagellar genes based on 16S rRNA genes and the construction of a complete flagellar gene cluster in a metagenome-assembled genome (MAG) from one of the most abundant bacteria in the spring.
2. Materials and Methods
2.1. Study site and physicochemical measurements
To quantify the incidence of motility via microscopy and molecular techniques, we sampled water and benthic microbial communities in Badwater Spring, CA, which is sourced from the Amargosa River and discharges at the lowest point in North America in Badwater Basin within Death Valley National Park (Fig. 1D). pH, water temperature, dissolved oxygen, and specific conductance were recorded on site with a YSI Professional Plus (Quatro) multiparameter probe. Spring water was collected for hydrogeochemical measurements directly from the source of Badwater Spring with a Geopump peristaltic pump (Geotech, Denver, CO) using autoclaved Masterflex platinum-cured silicone tubing (Cole-Parmer, Vernon Hills, IL) and filtered using 0.2 μm polyethersulfone membrane Sterivex-GP pressure filters (Millipore Sigma, Burlington, MA).
Filtered water was collected in pre-rinsed 250 mL high-density polyethylene bottles and refrigerated until being sent to the New Mexico Bureau of Geology and Mineral Resources Chemistry Laboratory for analysis of major cations and anions, alkalinity, and total dissolved solids. Cations were measured using inductively coupled plasma optical emission spectrometry in accordance with EPA 200.7; anions were measured using ion chromatography in accordance with EPA 300.0. For every 10th sample, a duplicate was run. Alkalinity was measured by titration in accordance with EPA 310.1. Physicochemical data and reporting limits are shown in Supplementary Table S1.
2.2. Microscopy and data processing
A field-portable DHM was used on April 10–12, 2017, in coordination with sampling for 16S rRNA gene surveys and metagenomics, described below. A water sample (DHM1) and two benthic samples (DHM2, DHM3) for DHM were collected near the site labeled BW.B3. Sample DHM2 was composed mostly of sediment and water (i.e., sediment slurry) with little mat material. Sample DHM3 was composed of fluffy orange mat similar to that in Fig. 1E with water (i.e., mat slurry).
The DHM used is a common-path off-axis holographic microscope with illumination at 405 nm and lateral spatial resolution of ∼0.8 μm. The sample chamber consists of two parallel channels, an empty reference and a sample channel, with a volume of view of 365 × 365 × 0.8 mm. 2048 × 2048-pixel images were generated using an Allied Prosilica GT camera with an acquisition speed of 15 frames per second.
Data processing and analysis workflows aimed to reconstruct holograms, apply filters, identify particles, and link particles into tracks. Holograms were reconstructed into amplitude and phase images using the angular spectrum method with our FIJI plug-in described previously (Cohoe et al., 2019). We used a reference hologram during reconstruction of phase images to reduce noise (Colomb et al., 2006) and a median subtraction filter via another FIJI plug-in to reduce noise in amplitude. Amplitude and phase data sets represent all four dimensions of space-time. The z-spacing, 2.5 μm, is a discrete value chosen to reflect the values of the cells in the sample and is also the theoretical axial resolution of the instrument (Wallace et al., 2015). After reconstruction and filtering, identification of particles was performed manually to characterize the particles that were likely microbes, and describe their concentration, size distribution, and morphology.
Taking advantage of the Gouy phase anomaly to localize particles in z, we applied a z-derivative to the pixel values in the phase images (Gibson et al., 2021). With these images, we applied a threshold filter that increased the signal-to-noise ratio by isolating specific swimming microbes while removing all other pixel values from the images. Particle and motile organism identification and tracking were then performed using FIJI. Two organisms were classified as moving with distinct swimming patterns. The rest of the motile organisms were qualitatively determined to be extant life-forms. The instantaneous speed of tracked particles in one dimension was calculated by applying Eq. 1. The speed of the particles in two dimensions in the XY-plane was calculated using Eq. 2.
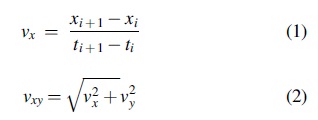
Data summarizing all particles are shown in Supplementary Table S2. Laboratory microscopy was also conducted on an Olympus BX51 phase-contrast microscope to document major morphologies and modes of motility in selected samples.
2.3. Microbial sample collection, DNA extraction, 16S rRNA gene amplicon Illumina sequencing, and taxonomy
Microbial community samples for 16S rRNA gene amplicon sequencing were collected on April 12, 2017. A single sample of the microbial community in the water was sampled by pumping bulk spring water (>2 L total) with a Geopump peristaltic pump (Geotech) using autoclaved Masterflex platinum-cured silicone tubing (Cole-Parmer) onto 0.2 μm polyethersulfone membrane Sterivex-GP pressure filters. Before sample storage, excess water was cleared from each filter using a sterile syringe. In addition, four benthic microbial communities were sampled by collecting the upper ∼1 cm of microbial mat or sediment using a sterile shovel. The sampling locations of the benthic samples were chosen based on the substrate diversity to maximize the sampling of unique benthic habitats within the spring. Sampling locations within Badwater Spring are shown in Fig. 1E.
Briefly, sample BW.B1 consisted of a fluffy orange benthic mat, representative of the dominant benthic morphology in the spring at the time of sampling (Fig. 1F); sample BW.B2 consisted of a fluffy orange benthic mat, similar to BW.B1, but with an overlying gelatinous layer, possibly composed of decaying organic matter; BW.B3 consisted of fluffy phototrophic growth and a lower green gelatinous layer; BW.B4 consisted of, from top to bottom, fluffy phototrophic growth, a lower green gelatinous layer, and a lower red layer.
After collection, all samples were frozen immediately on dry ice in the field and kept frozen in a −80°C freezer until DNA extraction. DNA was extracted using the FastDNA™ SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA) with two modifications to the manufacturer's instructions. First, the samples were homogenized three times total for 30 s each time at a speed setting of 4.5 using a FastPrep FP120 instrument (Thermo Fisher Scientific, Waltham, MA). Second, the supernatant from the Protein Precipitation Solution step was added to 500 μL of Binding Matrix suspension in a 1.5 mL tube.
Microbial communities were characterized by sequencing the V4 region of the 16S rRNA gene using the updated Earth Microbiome Project primers 515F (GTGYCAGCMGCCGCGGTAA) and 806R (GGACTACNVGGGTWTCTAAT) (Apprill et al., 2015; Parada et al., 2016). Sequencing was performed using the Illumina MiSeq platform at Argonne National Laboratory. All sequence-based analyses were performed in QIIME 2-version-2019.10 (Bolyen et al., 2019). Raw Illumina reads were demultiplexed using sample-specific barcodes and denoised using the dada2-denoise-paired plug-in to remove low-quality, chimeric, and artifactual sequences. Forward and reverse reads were truncated to 150 and 140 bp, respectively; in addition, 13 bases were trimmed from the 5′ end of all reads during the denoising step.
This resulted in a total of 176,627 high-quality sequences from the 5 samples from Badwater Spring and brine pool. These sequences were then dereplicated into 1899 amplicon sequence variants (ASVs). Taxonomy was assigned to each ASV using QIIMEs feature-classifier plug-in and the “Silva 132 99% OTUs full-length sequences” database (Quast et al., 2020). ASVs assigned to mitochondria, chloroplasts, eukarya, or not otherwise identified as bacterial or archaeal were excluded from further analysis. Alpha diversity indices (Observed ASVs, Shannon H′, Gini-Simpson Index, and InvSimpson) were calculated using unrarefied data in R packages phyloseq version 1.28.0 (McMurdie and Holmes, 2013) and picante version 1.8 (Kembel et al., 2010).
2.4. Prediction of flagellar motility based on mapping 16S rRNA gene sequences to the closest related genomes
PICRUSt2 (Douglas et al., 2020) was used to assess the potential to synthesize a functional flagellum based on the most closely related sequenced genome. Briefly, the representative sequence for each ASV was inserted into a reference tree containing 20,000 16S rRNA gene sequences from genomes in the Integrated Microbial Genomes and Microbiomes database. Next, a genome from the nearest genome-sequenced taxon for each ASV was identified and used to predict the gene families present in the ASV. The abundance of each gene family was normalized for each ASV in each sample based on the 16S rRNA gene copy number of the most closely related genome for each ASV. KEGG orthologs for each ASV were then assessed for 22 motility-associated genes and ASVs with ≥15 of those genes were predicted to be motile.
Using this approach, the genomes had a bimodal distribution, with the majority of genomes with ≥15 or <5 motility-associated genes, supporting the basis for the cutoff based on ≥15 motility-associated genes (Supplementary Table S3). This cutoff was validated by searching ∼100 ASVs assigned to cultivated and validly named taxa with each predicted motility assignment (motile or nonmotile) against our literature searches (Supplementary Table S4) (described below), which revealed near-perfect (>95%) agreement between genomically predicted flagellar motility with observed motility phenotypes as documented in the literature. As a conservative measure, figures were prepared only considering predictions for ASVs that could be assigned at the family or genus level.
2.5. Motility prediction based on the 16S rRNA gene classification and literature with precise taxonomy
16S rRNA gene ASVs assigned to the family or genus level were also used as a basis for predicting motility based on the literature by using a hierarchical search strategy by two separate authors (G.S. and B.P.H.). The search strategy consisted of: (i) searching for chapters within Bergey's Manual of Systematics of Archaea and Bacteria, 2015; (ii) searching the List of Prokaryotic Names with Standing in Nomenclature (Parte et al, 2020), followed by searching the effective publications describing all correct child taxa (i.e., avoiding misassigned child taxa); and (iii) for Cyanobacteria, also searching AlgaeBase. If any species within the taxon were observed to be motile, the type of motility was noted and was considered feasible for the taxon. Cell morphology and size, where distinctive, were also noted. For the most abundant ASVs, modes of flagellar motility were also noted, where known. All information and supporting references are summarized in Supplementary Table S4.
Initial searches revealed 89% agreement between the two annotators. The 11% of ASVs that were not initially agreed upon were reviewed by both annotators to resolve the differences. The resulting data file (Supplementary Table S4) contains the motility predictions, notes on morphology, and references for all the literature used to support the predictions.
2.6. Metagenomic DNA sequencing, assembly, and generation of MAGs
DNA from benthic sample BW.B4 were also used for shotgun metagenome sequencing. DNA extraction was performed as detailed by Urdiain et al (2008). DNA sequencing libraries were prepared using the Illumina Nextera Flex protocol, and libraries were sequenced using a NextSeq 150PE (150 × 2 bp) instrument. 18 Gbp of raw reads were imported into KBase (Arkin et al., 2018) where they were trimmed with Trimmomatic (v0.36) using default settings and put through six assembly/binning pipelines, including two assembly methods and three binning methods. Assembly was conducted with both metaSPAdes (v3.13.0) (Nurk et al., 2017) and MEGAHIT (sensitive, v1.2.9) (Li et al., 2015), using default settings and with a minimum contig length of 2 kb.
The assembly size for metaSPAdes was 27.1 Mbp, and the assembly size of MEGAHIT assembly was 26.6 Mbp (Supplementary Table S5). The contigs from each assembly were then binned individually, using default settings, with the following: MetaBAT2 (v1.7 min contig 2.5 kb) (Kang et al, 2019), MaxBin2 (v2.2.4 min contig 2 kb) (Wu et al., 2016), and CONCOCT (v1.1 min contig 2.5 kb) (Alneberg et al., 2014). The resulting MAGs from the six assembly/binning pipelines (six combinations consisting of two assembly tools and three binning tools) were then checked for estimated completeness, contamination, and heterogeneity using CheckM (v1.018) (Parks et al., 2015), and their phylogenetic position was estimated using GTDB-tk (v1.1.0) (Chaumeil et al., 2019).
MAGs with similar classifications between the different pipelines were compared with a pairwise average nucleotide identity (ANI) (
2.7. Prediction of flagellar motility in metagenomic contigs and MAGs
Motility was independently assessed in the metagenomic contigs with and without binning. Unbinned contigs from the metaSPAdes assembly and the individual MAGs were submitted to RASTtk (v1.073 genetic code 11, domain Bacteria) (Brettin et al., 2015) within KBase for annotation. Unbinned metagenomic contigs from the SEED functional categories motility and chemotaxis were analyzed with BLASTN against the GenBank NR database to determine a potential taxonomy by using an E-value of 10−50 (Supplementary Table S7). Annotations for MAGs classified with GTDB-tk were examined against the 22 core flagellar gene set to assess possible flagellar motility.
2.8. Nucleotide accession numbers
Associated data files are available in the NCBI under BioProject ID PRJNA807719 for 16S rRNA genes, metagenomic reads, and MAGs.
3. Results
3.1. In situ and laboratory microscopy
Analysis of three separate samples from Badwater Spring was performed on DHM recordings. These samples consisted of water, sediment slurry, and mat slurry. We identified the total number of objects consistent with size and morphology of bacteria or archaea at a single time point for each recording; a total 698 of these objects were identified throughout all 5 recordings. The number of these objects per recording ranged from 26 to 193 (120.7 ± 82.1, mean ± SD) (Supplementary Table S2). This equates roughly to a density of 960 prokaryotic cells per microliter. Most objects in the field of view were consistent with microbes; occasionally objects with sharpened edges and high-contrast artifacts were observed that appeared to likely be minerals. Of the 698 putative prokaryotes, 18 were obviously motile. This analysis estimated ∼25 motile prokaryotes per microliter, or ∼2.6% of the total cells.
Two of the 18 motile prokaryotes, both in the sediment slurry sample (DHM2), were identified as likely having distinct swimming patterns of run-reverse-flick and run-reverse with swimming speeds of 54.8 ± 22.7 and 61.4 ± 19.7 μm/s, respectively. Images of these two organisms and their swimming patterns are shown (Figs. 2 and 3). Considering all tracked motile microbes, average swimming speeds ranged from 5.3 ± 3.3 to 267.5 ± 60.6 μm/s. A histogram of instantaneous speeds between time points is shown in Supplementary Fig. S1A–D. The acquisition frame rate used seemed well suited to capture turn angles indicative of reversal events for the two motile organisms shown in Figs. 2 and 3. This is supported by comparing the number of reversal events in Figs. 2A and 3A with the number of turn angles above 120° in Figs. 2C, D and 3C, D, respectively. Previous work indicates that the flick mode manifests as a broad distribution of turns around 90 degrees (Xie et al., 2011).
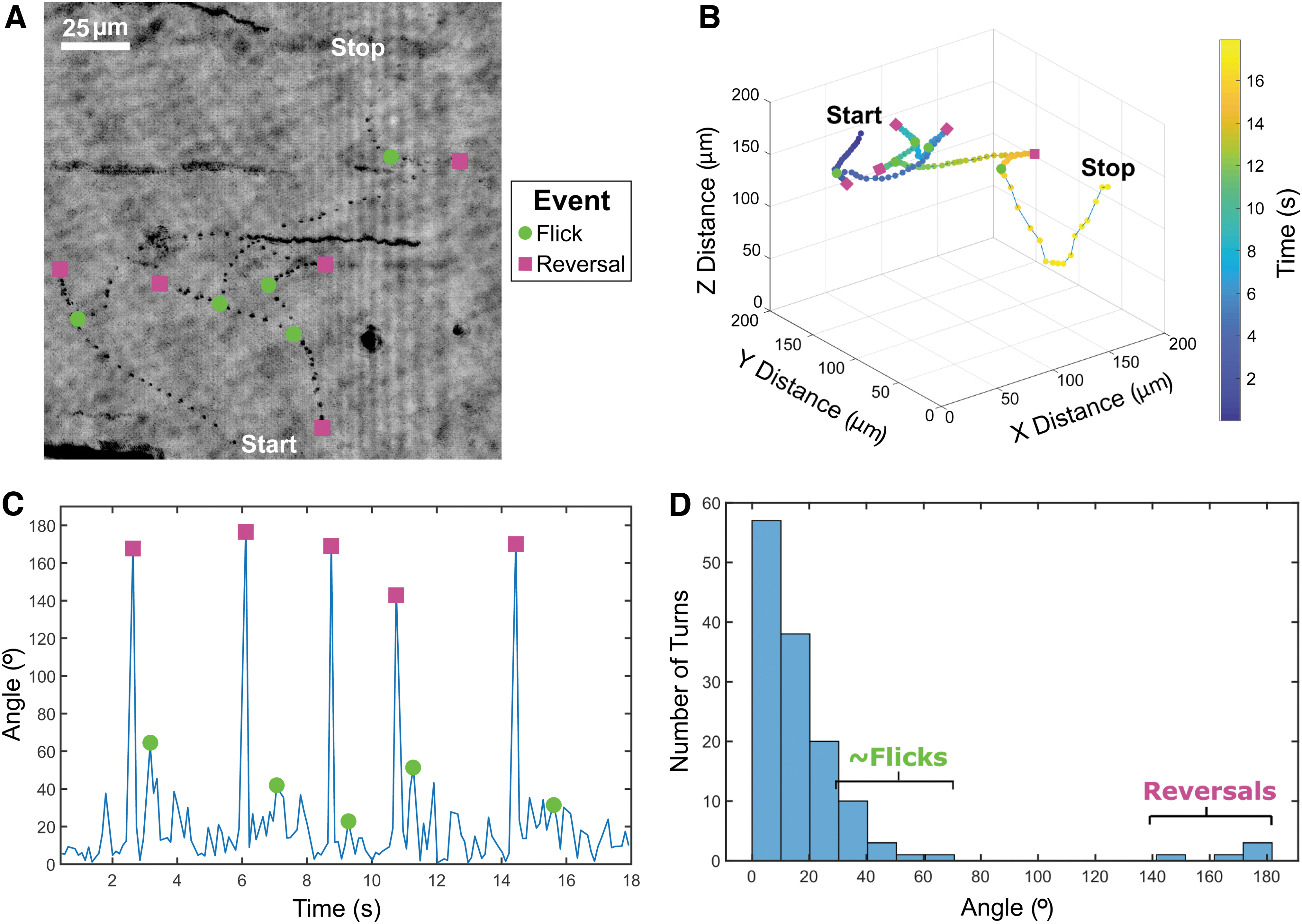
Cell with run-reverse-flick flagellar motility.
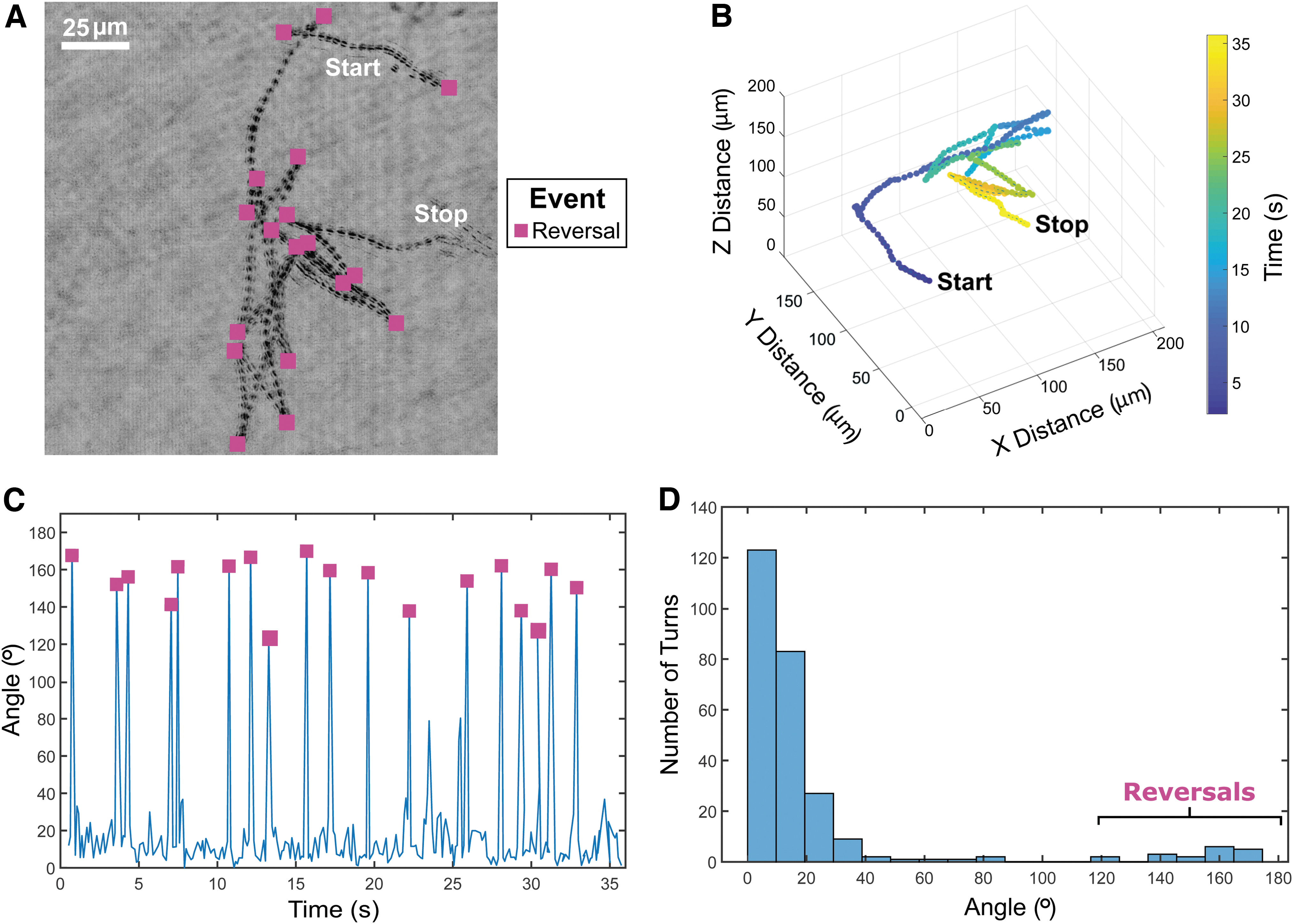
Cell with run-reverse motility.
Here, the flicking mode was determined via qualitative assessment of the overall tracks, but they rarely exceeded 60°. The qualitative assessment of flicks was performed by considering the angle between two path length vectors on either side of the moments where the turning occurred. These path lengths would be associated with moments where the particle appears to be traveling straight for at least several time points prior and post to the turning event. The other 16 motile prokaryotes did not have clear swimming patterns, likely due to the short duration of the recordings, hydrodynamics within the sample chambers, complex or incomplete swimming patterns, incomplete understanding of microbial motility swimming patterns, or a combination of the aforementioned reasons.
The DHM used can also identify unique prokaryotic morphologies, such as diplococci and tetrads, as shown in Supplementary Fig. S2. These unique morphologies can only be discerned when microbes are sufficiently elongated or above a diameter of ∼1 μm. Therefore, the resolution limit of the microscope allowed the accurate distinction between two general morphology types. One type of particle that made up ∼95% of the putative microbes present consisted of round and slightly elongated morphologies. At the resolution provided by this DHM, these particles could be small cocci, bacilli, diplococci, or short spiral morphologies. Examples are shown in Supplementary Fig. S2A and C. The other type, comprising the remaining 5% of putative microbes present, consisted of elongated morphologies, which could be streptococci, filaments, or longer spiral morphologies, as shown in Supplementary Fig. S2B.
The two motile microbes characterized in Figs. 2 and 3 appear to be rod-shaped prokaryotes. Videos of the DHM-imaged cells with run-reverse-flick and run-reverse swimming patterns are shown in Supplementary Videos S1 and S2. Supplementary Video S3 shows DHM video of a motile eukaryote and several motile and nonmotile prokaryotes. Supplementary Video S4 shows DHM video of motile prokaryotes and drift across the field of view. Representative examples of putative flagellar motility and spirochaetes in Badwater Spring samples captured by phase-contrast microscopy are shown in Supplementary Videos S5–S7 for reference.
3.2. Prediction of motility based on 16S rRNA genes
16S rRNA gene amplicons and shotgun metagenomic data were used to supplement microscopy to census the microbial community in Badwater Spring (Supplementary Figs. S2–S4; Supplementary Table S8; Supplementary Data). Two approaches to predict flagellar motility of the 1899 ASVs yielded similar results (Fig. 4). The closest genome prediction and literature prediction approaches predicted flagellar motility in 41.6–60.1% (51.3 ± 7.2, mean ± SD) and 36.0–48.7% (42.9 ± 5.1, mean ± SD) of the ASVs, respectively. The higher percentage of motile ASVs using the closest genome prediction approach may be due to the underreporting of motility in the literature and to genome-based predictions for taxa that are unavailable in culture. These effects could be exacerbated for taxa that are poorly studied in the laboratory due to poor culturability.
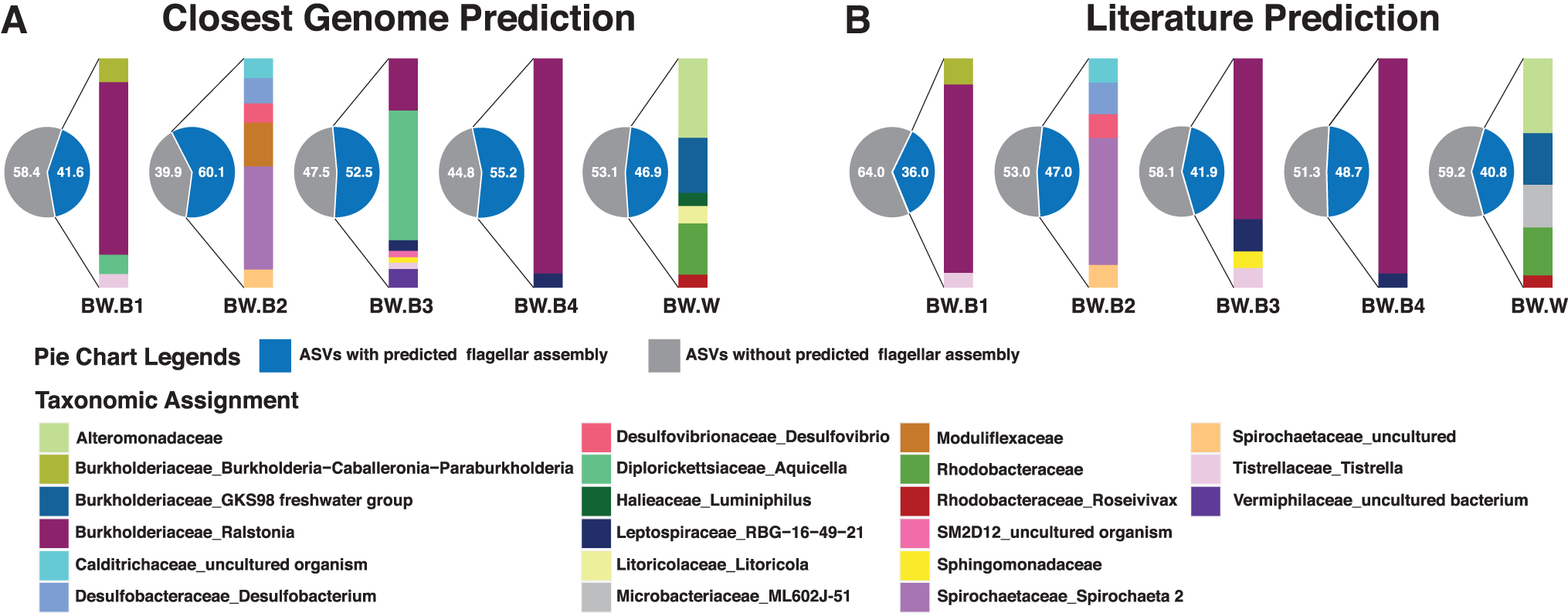
Flagellar motility predictions based on 16S rRNA gene data.
In the water sample, both approaches predicted the most abundant taxa with flagellar motility within the Alteromonadaceae, Burkholderiaceae (GKS98 freshwater group), Litoricolaceae (genus Litoricola), and Rhodobacteraceae, with the latter including the genus Roseivivax and ASVs unassigned at the genus level. The closest genome approach also predicted motility in the genus Luminiphilus, although flagellar motility has not been described in the single cultivated strain of this genus (Spring et al., 2013). On the contrary, flagellar motility was considered feasible by the literature prediction for Microbacteriaceae because flagellar motility has been observed in Microbacterium and Curtobacterium (Evtushenko and Takeuchi, 2006), although abundant planktonic members of this family have not been reported to be motile (Hahn, 2009).
In the benthic samples, both approaches predicted the most abundant motile organisms as Ralstonia, Tistrella, Calditrichaceae, Desulfobacterium, Desulfovibrio, Spirochaeta, Leptospiraceae, Sphingomonadaceae, Vermiphilaceae, and the Burkholderia–Caballeronia–Paraburkholderia complex, the latter of which is indistinguishable via 16S rRNA gene tags. In three of the benthic samples, BW.B1, BW.B3, and BW.B4, Ralstonia was by far the most abundant motile taxon, as predicted by one or both prediction approaches. The few differences in the predictions were again justifiable based on poor culturability and thus differences between genomic data and phenotypic observations.
In BW.B2, the Moduliflexaceae was predicted to be motile based on the closest genome prediction; however, no members of the family have been cultivated and in situ-studied Moduliflexaceae in wastewater do not contain flagellar genes (Sekiguchi et al., 2015). Similarly, Aquicella was predicted to be motile based on the closest genome prediction, but flagella were not observed in either of the two isolated species (Santos et al., 2003).
3.3. Prediction of motility genotype based on metagenomic contigs and MAGs
Metagenomic contigs from the BW.B4 metagenome were annotated using RAST, and those within “motility” or “chemotaxis” SEED categories were taxonomically assigned using BlastN. All contigs containing genes for flagellar biosynthesis or chemotaxis were assigned to the genus Ralstonia (53 genes) or had low-confidence taxonomic assignments (E-value >10−50; 7 genes), consistent with the high abundance of Ralstonia in the samples (Fig. 5A). Together, these genes account for the synthesis of MS, P, and L rings, MotA/B, hook, filament, and cap, and multiple methyl-accepting chemotaxis systems (Fig. 5B). Contigs containing the twitching motility genes pilTGHJ were also annotated for Ralstonia, consistent with its known twitching motility phenotype. Several contigs with chemotaxis (cheR/B), flagellar hook length (fliK), or gliding motility (e.g., mglA) genes with low-confidence taxonomic assignments were assigned to various members of the Chloroflexi likely deriving from Candidatus Chlorothrix.
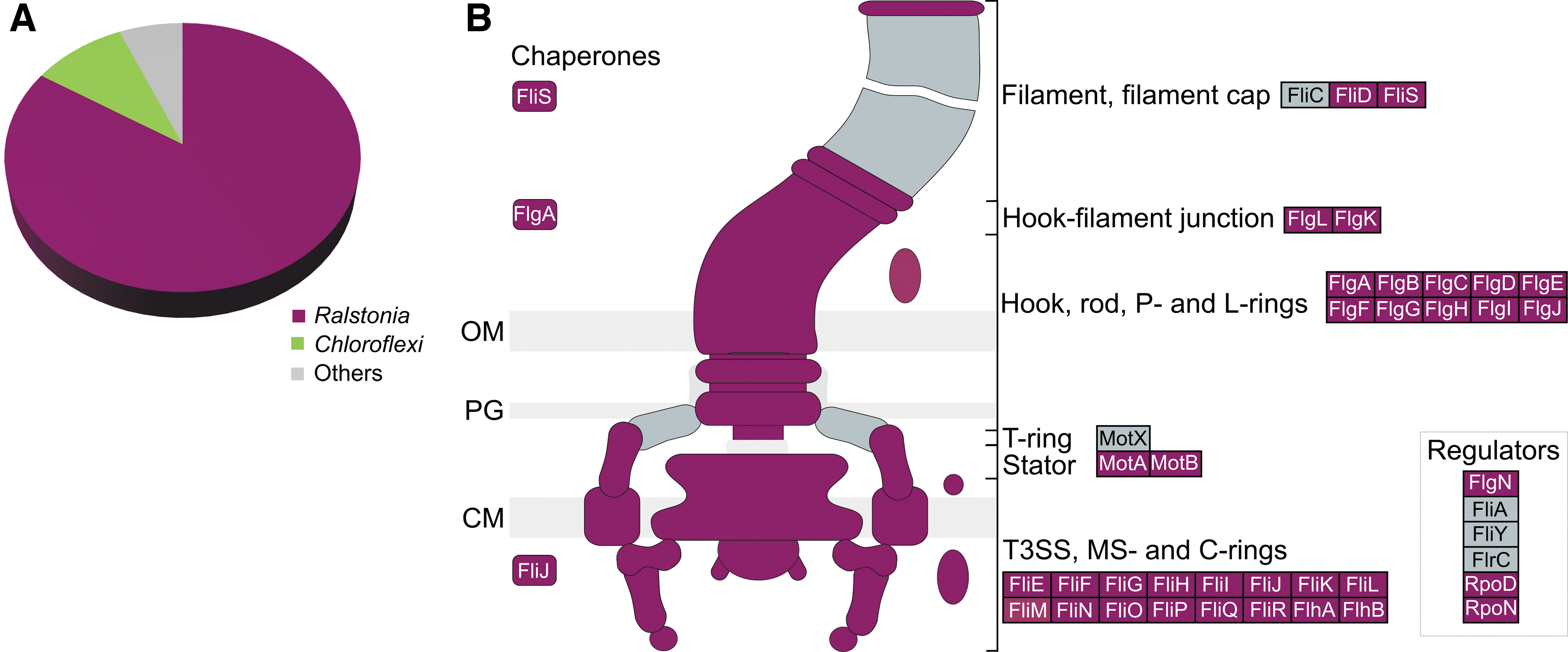
Motility predictions based on full metagenome and MAG approaches.
Separately, the three MAGs assembled from the BW.B4 metagenome were annotated using RAST. A high-quality MAG (CheckM2 estimated completeness 99.98% and contamination 2.16%) assigned to Ralstonia pickettii_B per GTDB-tk was sequenced at 7x coverage and contained a full complement of flagellar genes and twitching motility genes (Supplementary Table S4) that corresponded 1:1 with Ralstonia-assigned genes from the analysis of unbinned metagenomic contigs. A high-quality MAG (CheckM2 estimated completeness 96.86% and contamination 0.77%) sequenced at 123x coverage was assigned to Chloroflexaceae, most likely representing Ca. Chlorothrix, and was not predicted to have flagellar motility, but was predicted to be capable of gliding motility via the same genes identified in the unbinned contig analysis.
A MAG assigned to the Patescibacteria (CheckM2 estimated completeness 91.94% and contamination 0.18%) was sequenced at 5 × coverage and was not predicted to be motile by any mechanism. At 95% nucleotide identity, the percentage of reads mapped to the Ralstonia, Chloroflexaceae, and Patescibacteria MAGs was 0.3%, 3.4% and 0.1%, respectively. Recruitment plots for the MAGs are shown in Supplementary Figs. S4–S6, respectively.
4. Discussion
To say that it is a challenge to detect microbial life on another planet is an understatement. This challenge may very well follow a similar trajectory to how microorganisms were first detected on Earth, although it is complicated by long spans of time between missions, limited sample access, and unknown biochemistry. On Earth, the discovery of microbes first took place by optical observation via the light microscope, followed by laboratory cultivation and, recently, molecular methods (Borgosian and Bourneuf, 2001; Emerson et al., 2017; Leeuwenhoek, 1677). The presence of directed motion is a compelling biosignature that, combined with other methods such as chemical analysis by mass spectrometry, can provide unambiguous evidence not just of life, but of something alive, independent of evolutionary history or biochemistry.
The observation of motile microorganisms in most environments here on Earth supports the case that microbial motility is a compelling biosignature target for future planetary exploration missions. In every environment we have studied (Clarke et al., 2010; Jericho et al., 2010; Kühn et al., 2014; Rogers et al., 2010), motile microorganisms are always present, although sometimes as a small fraction. In this study, we combined DHM and genetic data to characterize the microbial community inhabiting Badwater Spring and brine pool, located near the lowest elevation point in North America and the highest ambient temperatures measured on Earth. We were able to obtain both phenotypic and genotypic data on this community and demonstrate a proof of concept of the use of DHM for life detection.
In this environment, DHM identified a small minority of motile cells in situ, only ∼2.6% of the total cells, similar to the lowest estimates of motility in a coastal marine system (Grossart et al., 2001). No evidence of other modes of motility (e.g., twitching or gliding) were noted, although they require solid surfaces and are more difficult to unambiguously distinguish from Brownian motion or directional movement due to hydrodynamic flow compared with flagellar motility (Henrichsen, 1972). The ∼2.6% of swimming cells identified by the DHM is a lower boundary for the actual percent of swimming cells in situ due to our limited ability to accurately distinguish living prokaryotic cells from dead cells and abiotic particles, which would lead us to over count the total number of cells in situ. This gap could be bridged in future studies by correcting total DHM particle counts with data from fluorescence microscopy or fluorescence-activated cell sorting that include viability estimates based on live/dead stains.
We also performed one of the first comparisons of observed in situ motility with genetic indicators of flagella. Biosynthesis of complexes required for flagellar motility and their regulation requires many genes that are best characterized for Gram-negative bacteria such as Escherichia coli and Salmonella typhimurium (Soutourina and Bertin, 2003). Interestingly, we found that there was a clear separation of genomes into those with fewer than five flagellar motility-related genes versus those with more than 15. Despite this dichotomy, finding literature confirmation of motility in the identified families/genera that were predicted to be motile based on genomic databases was not always consistent, likely due to limitations in genomic coverage and cultivability. The closest genome and literature-predicted approaches used here estimated flagellar motility in 36.0–60.1% of the total ASVs. Adjusted for abundance, these account for 14.2–57.1% of the cells present, which is considerably higher than the ∼2.6% of motile microbes identified by DHM in situ.
The ability to predict genotypes or phenotypes from amplicon sequence data is notoriously difficult due to the incomplete genomic coverage across the prokaryotic tree of life, the incomplete and uneven distribution of cultivated and phenotypically characterized microorganisms, and the dynamic nature of microbial pangenomes. The closest genome approach employed PICRUSt2 and resulted in the highest estimates of flagellar motility genotypes, with abundance-weighted estimates of 23–57% with flagellar motility genotypes. In comparison, our literature-based approach yielded abundance-weighted estimates of flagellar motility phenotypes of 14.1–29.3%. It is worthwhile to note that PICRUSt2 does not rely on taxonomic assignments because ASVs are placed onto a phylogeny, whereas our literature-based approach did rely on taxonomic assignments called by Silva.
Thus, our literature-based approach could suffer somewhat by lack of accuracy of taxonomic assignments, although this is likely to be only a minor problem because we only considered high-confidence taxonomic assignments to families that contain cultivated organisms. Additional factors that could lead to differences between the PICRUSt2 and literature-based estimates include: (i) PICRUSt2 adjusts for rRNA copy number, whereas our literature-based approach did not; (ii) PICRUSt2 only considers genotypes, whereas our literature-based approach only considered phenotypes; and (iii) PICRUSt2 only considers the single most closely related genome, whereas our literature-based approach considered motility phenotypes feasible if observed in any member of the family. Ultimately, the ability to predict traits from ASV data derived from diverse and poorly characterized natural microbial communities remains a formidable task.
A more definitive but less sensitive approach to identify genotypes is through shotgun metagenomics, particularly for traits with well-characterized systems such as flagellar motility. In this study, all flagellar genes in a single shotgun metagenome from benthic sample BW.B4 mapped to the genus Ralstonia. These same genes were also contained within a single high-quality MAG encoding a full flagellar gene cluster. However, reads mapping to this MAG accounted for only 0.3% of the total quality-filtered reads, which is slightly lower than ∼2.6% of motile microbes observed by DHM. Ultimately, the accuracy of any prediction of in situ motility based on genomic potential is prone to overestimation because motility is expensive and therefore tightly regulated. In E. coli, the expression of flagellar motility is complex and may occur under nutrient-rich or nutrient-poor conditions (Honda et al., 2022; Thomason et al., 2012); however, swarming motility phenotypes are only expressed under high-nutrient conditions in many microorganisms (Kearns, 2010).
These responses pose an interesting opportunity to increase the likelihood of observing motile organisms by altering the local environment to stimulate motility, which may prove critical for the success of future life-detection missions. Stimuli such as heat, light, and chemicals are good candidates for such missions because terran microorganisms show tactic responses to these physical and chemical factors. Stimuli deployed in such a manner may be non-Earth-centric. For example, in the case of water-based worlds, a panel of
Future work will develop onboard ecological experiments designed to relate microbial motility to the natural environment and ecology. This will involve development of specialized sample chambers for delivery of stimuli and identification of taxis and could be coupled with metagenomics, as done here, or with metatranscriptomics or metaproteomics to provide tighter links between motility phenotypes identified by the DHM to the identity of specific organisms and motility systems. On Earth, DHM has been used to study chemotaxis, biofilm formation, and predator/prey interactions (Qi et al., 2017; Wang et al., 2020; Yuan et al., 2021). The development of experiments to observe complex microbial behaviors on other worlds would provide not only biosignatures but also possible insights into the biology of the organisms discovered.
Ultimately, because this DHM instrument can identify submicron motile organisms throughout the volume of view of the instrument without sample preparation or fragile moving parts, we contend that the large-scale deployment of DHMs would be an excellent strategy to detect extant microbial life in any aqueous setting, particularly on water worlds. Such devices could be programmed to record and send video feeds when mathematically defined (Rouzie et al., 2021) motile organisms are detected. This technology should be advanced and benchmarked in aquatic environments on Earth as a prelude to exploration of aquatic microorganisms across the solar system.
Footnotes
Acknowledgments
We also thank Josh Hoines, Genne Nelson, Kevin Wilson, and David West from the US National Park Service for permission and guidance to conduct sampling at Badwater Spring. Sampling was conducted under permits DEVA-2017-SCI-0023 and DEVA-2016-SCI-0039. R.R.-M. research was supported by the Spanish Ministry of Science, Innovation and Universities projects PGC2018-096956-B-C41 and PRX18/00048, both also supported with European Regional Development Fund (FEDER) funds.
Authors' Contributions
J.N., A.D.F., and B.P.H. participated in the design of the study and conducted the field work. C.S., A.D.F., and T.V. performed all wet laboratory experiments and processed the data. A.D.F., J.P.C., and S.B. conducted the bioinformatic analyses. G.S. and B.P.H. performed the literature searches on microbial motility to support literature predictions. T.V. and R.R.-M. performed metagenomic sequencing. J.P.C. and K.T.K. aligned, annotated, and curated the metagenomic data and MAGs. J.N. and B.P.H. wrote the initial drafts of the article. All authors edited and critically revised the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the NASA EPSCoR program (award 80NSSC18M0027) and the National Science Foundation (EAR 15-16680 and EAR 2038420).
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
Supplementary Video S4
Supplementary Video S5
Supplementary Video S6
Supplementary Video S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Abbreviations Used
Associate Editor: Don Cowan