
Abstract
Metal complexes of porphyrins and porphyrin-type compounds are ubiquitous in all three domains of life, with hemes and chlorophylls being the best-known examples. Their diagenetic transformation products are found as geoporphyrins, in which the characteristic porphyrin core structure is retained and which can be up to 1.1 billion years old. Because of this, and their relative ease of detection, metalloporphyrins appear attractive as chemical biosignatures in the search for extraterrestrial life. In this study, we investigated the stability of solid chlorido(2,3,7,8,12,13,17,18-octaethylporphyrinato)iron(III) [FeCl(oep)], which served as a model for heme-like molecules and iron geoporphyrins. [FeCl(oep)] was exposed to a variety of astrobiologically relevant extreme conditions, namely: aqueous acids and bases, oxidants, heat, and radiation. Key results are: (1) the [Fe(oep)]+ core is stable over the pH range 0.0–13.5 even at 80°C; (2) the oxidizing power follows the order ClO− > H2O2 > ClO3 − > HNO3 > ClO4 −; (3) in an inert atmosphere, the iron porphyrin is thermally stable to near 250°C; (4) at high temperatures, carbon dioxide gas is not inert but acts as an oxidant, forming carbon monoxide; (5) a decomposition layer is formed on ultraviolet irradiation and protects the [FeCl(oep)] underneath; (6) an NaCl/NaHCO3 salt mixture has a protective effect against X-rays; and (7) no such effect is observed when [FeCl(oep)] is exposed to iron ion particle radiation. The relevance to potential iron porphyrin biosignatures on Mars, Europa, and Enceladus is discussed.
1. Introduction
The Non Sequitur “Stability”
Chemists are often sloppy in their thinking when they refer to a compound as “stable” or “unstable.” Even when the context in which the word is used is obvious, it is strictly meaningful to use the word only in reference to a particular kind of reaction. In defense of current usage, however, compounds that do not readily decompose at room temperature are referred to as “stable,” and as long as this meaning is understood we can live with the usage.
(Purcell and Kotz, 1977)
“Stability” is a multi-faceted concept referring to aspects that range from thermodynamics—where stability can be expressed quantitatively by relative molar Gibbs energies (IUPAC, 1997)—to qualitative properties such as “constancy” (a system remains unchanged), “robustness” (a system remains unchanged when perturbed), and “resilience” (a system returns to or close to its original state after being perturbed) (Hansson and Helgesson, 2003).
Thus, when talking about stability, it is appropriate to first define the way in which the term is being used. In this article, “stability” refers to the resistance of a compound to decomposition under given conditions.
One of the major themes in astrobiology is biosignatures (e.g., Chan et al., 2019). A biosignature can be broadly defined as “any substance, group of substances, or phenomenon that provides evidence of life” (Catling et al., 2018). More narrowly defined, biosignatures are chemical, isotopic, and morphological traces of life preserved in minerals, sediments, and rocks (Westall and Cavalazzi, 2011). Cady et al. (2003) grouped microbial biosignatures into three categories: microfossils, sedimentary structures influenced by microorganisms, and chemofossils.
The latter include diagenetically altered organic matter as well as primary, unaltered biomolecules (e.g., Parnell et al., 2007; Summons et al., 2008). Summons et al. (2011) categorized biogenic organic molecules as highly definitive biosignatures. However, it must be kept in mind that the unambiguous identification of chemical biosignatures is frequently not straightforward (Fox and Strasdeit, 2017).
Finding a chemical biosignature depends, among other things, on the state of preservation, which is crucially determined by the stability of the biosignature. Therefore, knowledge about the stabilities is important when it comes to deciding which biosignatures should be searched for in future space missions, for example to Mars, Enceladus, or Europa. Extraterrestrial biosignature candidates are usually chosen in an Earth-centric manner, based on the assumption that life elsewhere is similar to life on Earth. Therefore, organic molecules that are abundant and widespread in living organisms are typically considered as biosignatures.
Molecular biosignatures can be destroyed by both biological recycling and abiotic processes. In this work, we focus exclusively on the effects of abiotic factors, such as radiation and high temperature, and use a representative member of the porphyrin family as a model compound. Porphyrins are unique molecules whose properties make them promising biosignature candidates (Suo et al., 2007). Later, the term “porphyrin-type compounds” (PTCs) is used to include not only porphyrins (i.e., derivatives of porphine with various side chains), but also contracted porphyrins (e.g., corrins) and partially reduced ones (e.g., chlorins, bacteriochlorins, and corphins).
Metal complexes of PTCs are ubiquitous and perform a variety of functions in living organisms (Kaim et al., 2013). For example, the magnesium-containing chlorophylls and bacteriochlorophylls are central to photosynthesis, and the iron-containing hemes (Fig. 1a) are involved in one-electron transfer, oxygen transport, and oxygen activation. Other well-known PTCs are the cobalamins (cobalt complexes), which include vitamin B12, and the methanogenic cofactor F430 (a nickel complex). There are also less common PTCs with no known metabolic function, such as the copper porphyrins turacin and turacoverdin, which are avian pigments (Dyck, 1992), and the nickel porphyrin tunichlorin of some marine invertebrates (Küpper et al., 2006). Because of their ubiquity and intense colors, PTCs have been called “the colors of life” (Milgrom, 1997) and “the pigments of life” (Battersby, 2000).
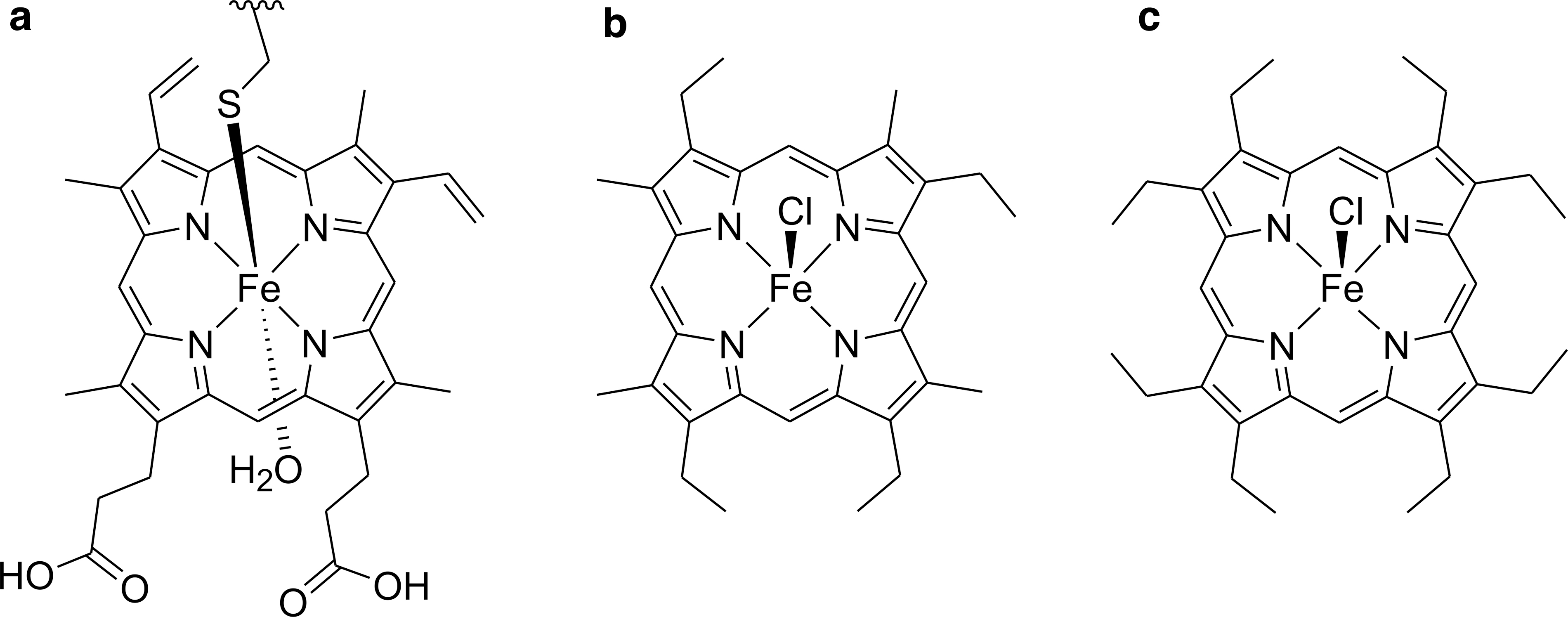
The PTC molecules have a highly persistent core structure, which may be preserved as geoporphyrins for at least 1.1 billion years (Gueneli et al., 2018). Most geoporphyrins derive from chlorophylls and hemes, which are altered during diagenesis to a variety of free base porphyrins and metalloporphyrins, such as metalloetioporphyrins (Fig. 1b) (Callot and Ocampo, 2000). Transmetalation occurs frequently, resulting in vanadyl and nickel porphyrins usually being the most abundant metallogeoporphyrins (Filby and Van Berkel, 1987).
However, iron porphyrins can be present in comparable concentrations (e.g., Eckardt et al., 1989; Zheng et al., 2018). It is also worth mentioning that a crystalline nickel geoporphyrin mineral exists (Mason et al., 1989; Hummer et al., 2017).
Among the biological PTCs, iron complexes play an outstanding role. The heme groups of the cytochromes, for example, are widely involved in electron transfer and the cellular production of energy (Kaim et al., 2013). Even organisms that rely on thermal energy, such as hyperthermophiles near hydrothermal vents, and chemoautotrophic species use cytochromes (e.g., Pihl et al., 1992; Lovley et al., 2004). These proteins are found in all three domains of life (bacteria, archaea, and eukaryotes). A virus genome that encodes cytochrome P450 has been described (Lamb et al., 2009). Further, cytochrome P450 enzymes could have existed for more than 3.5 billion years (Nelson et al., 1993).
The great evolutionary age, abiotic formation pathways, the involvement in basic biochemical processes, and the ubiquitous occurrence suggest that iron porphyrins might also be used by extraterrestrial life. Heme- and cytochrome-like molecules are considered targets for the search for life on Mars (Haezeleer et al., 2019).
Suo et al. (2007) suggested porphyrins as ideal molecular biosignatures, partly because no plausible abiotic synthesis was known at that time. Since then, abiotic porphyrin and metalloporphyrin syntheses have been discovered that could occur in aqueous environments on terrestrial planets (Lindsey et al., 2009, 2011; Soares et al., 2012a, 2012b; Taniguchi et al., 2012; Fox and Strasdeit, 2013; Soares et al., 2013a, 2013b; Alexy et al., 2015; Pleyer et al., 2018). Thus, we now know that PTCs entail the risk of being false positive biosignatures (Fox and Strasdeit, 2017). However, the problem of a possible abiotic origin also exists for other potential biosignatures, such as lipids, amino acids, and nucleobases (e.g., McCollom et al., 1999; Burton et al., 2012). In contrast to many other chemical biosignatures, PTCs have the advantages of high stability (at least of the core structure) and ease of detection.
In this study, we use chlorido(2,3,7,8,12,13,17,18-octaethylporphyrinato)iron(III) [FeCl(oep)] (Fig. 1c) as a model for heme-like molecules. In terms of the iron coordination, [FeCl(oep)] is closely related to hemin, which is a natural oxidation product of heme (Umbreit, 2007). Both [FeCl(oep)] and hemin contain iron(III) in a square-pyramidal ClN4 environment. However, the two molecules differ in the polarity of the porphyrin.
Protoporphyrin IX, which is present as the dianion in hemin, has vinyl and carboxyl groups and is therefore amphiphilic, whereas octaethylporphyrin, the proligand of [FeCl(oep)], is hydrophobic (Taniguchi et al., 2012). However, this difference is not relevant for our study because (1) we focus only on the solid state, and (2) during diagenesis, reduction of the vinyl groups and decarboxylation frequently occur, both producing ethyl groups (compare a and b in Fig. 1) (Callot and Ocampo, 2000).
[FeCl(oep)] was also chosen because, like hemes and chlorophylls, it is substituted at the β-positions. The location of the substituents affects the chemical stability of a porphyrin. Free meso-positions are the most reactive positions for nucleophilic as well as electrophilic attack (Fuhrhop, 1978). Also, cleavage of the porphyrin ring starts there (Ortiz de Montellano, 1998; St Claire and Balch, 1999). Therefore, when it comes to stability studies, the meso-substituted chlorido(5,10,15,20-tetraphenylporphyrinato)iron(III) ([FeCl(tpp)], see below) is not the best choice as a model compound for biological PTCs.
By contrast, the meso-positions of [FeCl(oep)] are unblocked. In addition, there is much experimental data on [FeCl(oep)], including the crystal structure (Senge, 2005; Kohnhorst and Haller, 2014), making this complex very suitable for model studies.
In this study, the stability of solid [FeCl(oep)] is investigated under various harsh conditions that are relevant to astrobiological questions. The majority of PTCs are aromatic systems. Therefore, the macrocylic core structures of PTCs are generally regarded as very stable. However, so far, relatively few studies have addressed the stability of solid PTCs, in contrast to, for example, solid amino acids whose resistance to ultraviolet (UV) (e.g., Ten Kate et al., 2005) and ionizing radiation (e.g., Cataldo et al., 2011), high temperatures (e.g., Fox et al., 2015), and multiple harsh conditions (e.g., perchlorates and energetic electrons) (Góbi et al., 2016) was studied.
Metalloporphyrins were divided into five classes according to their susceptibility to demetallation by Brønsted acids: (I) not completely demetallated by 100% sulfuric acid, (II) demetallated by 100% sulfuric acid, (III) demetallated by hydrochloric acid/dichloromethane, (IV) demetallated by 100% acetic acid, and (V) demetallated by water/dichloromethane at 25°C within 2 h (Falk, 1964; Buchler, 1975). [FeCl(oep)] belongs to class II, whereas the corresponding vanadyl complex is categorized as class I and the magnesium complex as class IV.
This is consistent with the observation that geoporphyrins often occur as vanadyl and sometimes as iron complexes but never as magnesium complexes. Some solid metal complexes of 5,10,15,20-tetraphenylporphyrinate(2–) (tpp) were studied by thermogravimetry (Allan et al., 1989; Gokakakar and Salker, 2010). It was found, for example, that [FeCl(tpp)] was stable in air up to ∼380°C. Also, radiation experiments were conducted, for example, on the photostability of [Zn(tpp)] in solution (Sobbi et al., 1993), the effect of γ-irradiation on thin films of [FeCl(tpp)] (El-Nahass et al., 2017), and the γ-radiation resistance of copper, nickel, and vanadyl porphyrins in the context of geoporphyrins (Dunning and Moore, 1959). However, none of these works used [FeCl(oep)].
In an astrobiologically motivated study, thin films of [FeCl(tpp)] were exposed to radiation in a low Earth orbit on the O/OREOS satellite and in the laboratory (Cook et al., 2014). The iron complex was photodecomposed in all microenvironments used (dry argon, humid argon, and CO2/O2/Ar atmosphere), with the decomposition being fastest in the humid environment. However, as explained earlier, meso-substituted metalloporphyrins such as [FeCl(tpp)] may not be ideal models for naturally occurring PTCs.
Therefore, in the present study, we used the β-substituted complex [FeCl(oep)] to evaluate the resistance of iron(III) porphyrins to UV and ionizing particle radiation (He and Fe). [FeCl(oep)] was also exposed to high temperatures, acids, bases, and oxidizing agents to obtain a more comprehensive picture of its stability. To facilitate the interpretation of the results, we investigated the potentially destructive factors individually; it should be kept in mind, however, that factors may act differently in combination than they do separately. The study was conducted against the background of the search for life in the Solar System, for example, on Mars and Enceladus. The results may help to identify locations where the search for PTC biosignatures is most promising—because PTCs can withstand local conditions—and those where the probability of finding PTCs is low.
A knowledge of the stability of solid PTCs is important not only in the context of biosignatures, but possibly also for prebiotic chemistry and origin-of-life research. Lipophilic, water-insoluble alkylporphyrinato iron(III) complexes, such as the one used in this study, may have formed abiotically on primordial volcanic islands (Fox and Strasdeit, 2013; Pleyer et al., 2018). As with many prebiotic reactions, the yields were probably small. However, if the metalloporphyrins were sufficiently stable to withstand the harsh conditions that occurred locally and temporarily on volcanic islands (e.g., high temperatures, short-wavelength UV radiation, and low pH), they could have accumulated over time.
Thus, metalloporphyrins may have been available to (hypothetical) protometabolisms and to early organisms, for example as UV-shielding pigments or (photo)catalysts.
2. Materials and Methods
2.1. Chemicals
The following chemicals were used as received: 2,3,7,8,12,13,17,18-octaethylporphyrin [H2(oep), ≥98%; PorphyChem], chlorido(2,3,7,8,12,13,17,18-octaethylporphyrinato)iron(III) ([FeCl(oep)], ≥98%; PorphyChem), sodium chloride (99.99%, “Suprapur”; Merck), sodium carbonate (>99.5%, p.a.; Fluka), sodium hydrogen carbonate (≥99.0%, p.a.; Merck), sodium hydroxide (≥99%, p.a.; Roth), sodium chlorate (≥99.0%, ACS reagent grade; Alfa Aesar), calcium hypochlorite (“for synthesis”; Merck), magnesium perchlorate hexahydrate (99%; ABCR), hydrogen peroxide (30% solution in water, ACS reagent grade, unstabilized; Acros Organics), hydrochloric acid (37%, p.a.; Roth), nitric acid (69%, p.a.; Merck), and sulfuric acid (96%, p.a.; Roth).
All organic solvents were HPLC grade. μ-Oxido-bis[2,3,7,8,12,13,17,18-octaethylporphyrinatoiron(III)] ([{Fe(oep)}2(μ-O)]) was prepared as described by Dolphin et al. (1978). Double distilled water from a BD 50 quartz glass distillation apparatus (Westdeutsche Glasschmelze) was used throughout. Synthetic air (20.5% vol % O2, remainder N2), nitrogen gas (99.999 vol %, 3 vol ppm O2 content), and carbon dioxide (99.995 vol %, 45 vol ppm N2 and O2 content) were purchased from Westfalen AG (Westfalen AG, 2019a, 2019b, 2019c).
2.2. Analytical instrumentation
Infrared (IR) spectra were recorded on a Nicolet 5700 FT-IR spectrometer (Thermo Scientific) either in attenuated total reflection (ATR) mode by using a Smart Orbit accessory or in transmission mode. For quantitative transmission measurements, a tightly sealed 1 mm NaCl cell was used. All spectra were recorded with a resolution of 2 cm−1 and averaged over 64 scans. UV-visible absorption spectroscopy was performed by using a SPECORD 210 spectrometer (Analytik Jena) and 10 mm quartz glass cuvettes.
The cuvettes were gas-tight to prevent evaporation during the measurements. Instrument settings were as follows: wavelength range 250–700 nm, slit width 0.5 nm, resolution 1 nm, and scan speed 1 nm/s. A TA Instruments TGA 550 was used for thermogravimetric (TG) and differential thermal analyses of [FeCl(oep)] in three different atmospheres: synthetic air, pure nitrogen, and pure carbon dioxide. The measurements were performed from 50°C to 900°C at a heating rate of 5°C/min.
X-ray powder diffraction patterns were recorded on a Bruker D8 Focus diffractometer equipped with a Sol-X energy dispersive detector. CuKα radiation was used for these measurements. The diffraction patterns were analyzed with the Bruker DIFFRAC.EVA software (version 5.0) by using reference patterns from the PDF-2 2019 database (International Centre for Diffraction Data, version 2.1901).
Spectral irradiance measurements were performed by using a calibrated StellarNet BLACK-Comet C spectroradiometer equipped with an F600-UV-VIS-SR fiber optic cable, a CR2 cosine receptor, and a CR2 aperture with 90% attenuation. Measurement conditions were as follows: spectral range 190–800 nm, slit width 50 μm, resolution 1.5 nm, and 20 scans per spectrum. Particle size distribution was determined in dispersion by using a Mastersizer 2000 equipped with a Hydro 2000 SM small-volume sample dispersion unit (Malvern Instruments). Three individual subsamples of the bulk sample were measured (10 cycles, 15 s per cycle, and dispersion unit set at 1300 rpm).
2.3. Stability experiments using acids, bases, and oxidizing agents
Safety note: Special care should be taken when handling corrosive substances, such as concentrated acids and hydrogen peroxide solutions. The same is true for the oxidizing salts used in this study. For example, sodium chlorate, calcium hypochlorite, and magnesium perchlorate hexahydrate may cause a fire or an explosion when in contact with combustible materials. Hypochlorite can react with acids to release toxic chlorine gas.
Aqueous solutions of hydrochloric acid, nitric acid, sulfuric acid, sodium hydroxide, sodium carbonate, sodium chlorate, calcium hypochlorite, magnesium perchlorate, and hydrogen peroxide were used; five working solutions of the following concentrations were prepared for each of the compounds: 1000, 100, 10, 1, and 0.1 mmol/L. The pH of the solutions was measured with a calibrated pH meter. As solid calcium hypochlorite slowly decomposed in air (Bibby and Milestone, 1984), a saturated stock solution of the salt was prepared, filtered, and its hypochlorite concentration was determined by iodometric titration (Williams, 1979). The calcium hypochlorite working solutions were immediately prepared by dilution of this stock solution and used on the same day.
Typically, 1 mg (1.6 μmol) of [FeCl(oep)] was placed in a 2 mL Eppendorf polypropylene microcentrifuge tube, 1 mL of the respective working solution was added, and the tube was tightly closed. The samples were then shaken in a thermoshaker at 1°C, 25°C, or 80°C for 24 h. Subsequently, each sample was extracted 10 times with 750 μL of dichloromethane. Before extraction, the lids of the hydrogen peroxide-containing tubes were pierced with a fine cannula to relieve potential overpressure.
Each extraction step consisted of vigorous mixing for at least 30 s on a vortex mixer, centrifugation to accelerate phase separation, and collection of the dichloromethane phase. The dichloromethane phases were combined, and the solvent and other volatiles were removed in vacuo. The residue obtained was again dissolved in dichloromethane, and the final volume was made up to 10 mL. This extract was used for quantification purposes and UV-visible spectroscopy. After this first extraction, the aqueous phase was neutralized with hydrochloric acid or sodium hydrogencarbonate solution, and a second extraction was performed as described earlier. This additional extraction was necessary to determine whether water-soluble porphyrin anions or cations had formed after demetallation of [FeCl(oep)] during the experiment, which could not be extracted in the first extraction. All experiments were performed in duplicate.
2.4. Thermal stability experiments
The general thermal stability of [FeCl(oep)] was investigated by using thermogravimetric analysis (TGA) up to 900°C. Measurements were performed in air, nitrogen, or carbon dioxide (see above) to determine the influence of different gases. A platinum pan with the sample (typically ∼10 mg, 16 μmol) was loaded into the thermobalance. Then, the system was purged with the respective gas for 60 min, and the measurement was started.
In the experiments with CO2, colorimetric carbon monoxide gas detection tubes H-10-102-20 (extended range: 2.5–200 vol ppm), H-10-102-30 (extended range: 10–1000 vol ppm), and H-10-102-45 (extended range: 0.1–8%) (Honeywell, Inc., 2019) were used for the semi-quantitative determination of CO in the gas from the outlet of the TGA instrument. The gas was collected in a 250 mL flask closed with a rubber septum. The septum was pierced with two cannulae of 1 mm inner diameter: a long one, which reached the bottom of the flask and was connected to the gas outlet of the instrument, and a short one, which ended in the upper part of the flask and was left open.
The flask was filled with the gas from the outlet for 5 min. Then, the long cannula was disconnected from the instrument, a CO detection tube was attached to one cannula, and 100 or 200 mL of the sampled gas was sucked through the tube. After 2 min, the result was evaluated by comparing the length of the brown coloration with the imprinted scale. From this result and the known volume, the quantity of CO present could be calculated. A total sampling time of 5 min was chosen so that at a gas flow of 60 mL/min, the 250 mL flask and the connection tubing were completely filled.
The volume of the furnace was also considered. The flask was attached to the gas outlet 30 s before the actual sampling started; then, a 4 min sampling period in the desired temperature range followed; and finally, after an additional 30 s, the flask was disconnected from the gas outlet. With this sampling method, it is believed that the flask contained the time-averaged gas composition from a temperature interval of 20°C (corresponding to a 4 min sampling period at a heating rate of 5°C/min). Eight samples were taken: one during the purge step and one each at 200–220°C, 400–420°C, 500–520°C, 600–620°C, 700–720°C, 800–820°C, and 880–900°C.
Based on the TG results, a subsequent experiment with 200 mg (320 μmol) of [FeCl(oep)] was performed at a constant temperature of 500°C under a nitrogen atmosphere. A specially designed apparatus was used, which has been described in detail elsewhere (Fox and Strasdeit, 2013; Dalai et al., 2017). In brief, the setup consisted of a quartz glass tube (40 mm inner diameter, 1200 mm length) that was heated by a Carbolite CTF 12/75/700 tube furnace. The apparatus was purged with nitrogen for 24 h before the experiment was started.
The nitrogen flow had a velocity of 10 cm/min inside the tube and was maintained until the end of the experiment. During purging, the [FeCl(oep)] sample was situated in a quartz glass container outside the heating zone. After purging, the furnace was preheated to 500°C, and the sample container was pushed into the center of the heating zone where it remained for 24 h. The residue obtained was analyzed by elemental analysis (C, H, N, Cl, Fe; Mikroanalytisches Labor Pascher, Remagen, Germany), IR spectroscopy, and X-ray powder diffractometry. In addition to the residue inside the sample container, a sublimate that formed on the cooler parts of the inner tube wall was analyzed by IR spectroscopy.
In another series of tube furnace experiments, the TGA experiments in air, nitrogen, and carbon dioxide were repeated on a larger scale. Again, 200 mg (320 μmol) of [FeCl(oep)] was used. As in the TGA experiments, the gas flow was adjusted to 60 mL/min and maintained at this value throughout the experiment. The apparatus with the sample inside was purged with the respective gas for 24 h before heating; in contrast to the tube furnace experiment described earlier, the sample was in the heating zone from the beginning.
Then, the temperature was increased from room temperature to 900°C at the same heating rate used in the TGA measurements (5°C/min). The furnace was switched off at the final temperature, and the sample was quickly pushed out of the heating zone and allowed to cool to room temperature in the respective atmosphere. Again, the residues obtained were analyzed by elemental analysis (see above), IR spectroscopy, and X-ray powder diffractometry.
To prevent the sublimation of [FeCl(oep)] during heating, additional experiments were conducted with the compound sealed in glass ampoules. Exactly 1 mL of a dichloromethane solution of [FeCl(oep)] was transferred to a 2 mL ampoule (borosilicate glass 3.3, outer diameter 9 mm, wall thickness 1 mm). The solution had a concentration of 1.000 mg/mL. Thus, each ampoule contained exactly 1 mg of the complex. The dichloromethane was carefully vacuum removed. Then, the ampoules were sealed under vacuum.
Two ampoules each were kept at room temperature, 100°C, 200°C, 250°C, 275°C, 300°C, 325°C, 350°C, 400°C, and 500°C, respectively, in a Carbolite CWF 11/13 muffle furnace for 24 h. For safety reasons the ampoules were wrapped in aluminum foil before heating; however, none of the ampoules burst during the experiments. After the experiments, the ampoules were allowed to cool to room temperature and opened. The contents of each ampoule were extracted 10 times with 750 μL of dichloromethane, and the combined extracts were made up to 10 mL with dichloromethane.
The resulting solution was used for quantitative IR measurements both before and after demetallation (Section 2.8) and for UV-visible spectroscopy. All experiments were performed in duplicate.
2.5. UV-visible irradiation experiments
The [FeCl(oep)] particles used had to be small enough to be penetrated by a radiation of 380 nm, which corresponds to the Soret band wavelength. Therefore, first the transmittance was estimated as a function of particle size (Supplementary Fig. S1 and Supplementary Data). It was found that, for example, 5% of the radiation will penetrate to the center of a particle with 300 nm diameter, and 1% will reach the center of a 460 nm particle.
Particles of suitable size range were then prepared by grinding 200 mg of [FeCl(oep)] in a Retsch PM 100 planetary ball mill (120 h at 350 rpm; 12 mL stainless steel grinding jar with five 10 mm stainless steel grinding balls). A fine powder with mean and maximum particle diameters of 132 and ∼450 nm, respectively, was obtained (for particle size distribution, see Supplementary Fig. S2).
Typically, 2 mg (3.2 μmol) of the [FeCl(oep)] powder was placed in a small quartz glass tube (length 60 mm, outer diameter 9.5 mm, wall thickness 1.25 mm, conical ground socket). To achieve better mixing during the experiment, ∼60 mg of quartz glass fragments (diameter range 0.71–1.00 mm) was added. The tube was thoroughly flushed with nitrogen, tightly stoppered, and then connected to an overhead stirrer RZR 0 (Heidolph). The rotation speed was fixed at 240 rpm. Rotation of the tube, together with the quartz glass fragments, ensured a good mixing and a uniform irradiation.
The rotating tube was typically positioned 12 cm from the center of the light source. A thermal sensor was attached next to the tube to continuously monitor the temperature. Only slight temperature variations occurred (Supplementary Fig. S3). The temperature was typically 35°C ± 2°C and varied from 33°C to 39°C between different experiments.
A 150 W xenon short-arc lamp (Osram XBO), which was placed in a Spindler & Hoyer lamp housing, was used for irradiation. The experimental setup is shown in Supplementary Fig. S4. Before each experiment, the emission spectrum of the light source and the spectral irradiance in the range of 190–800 nm were measured (Supplementary Fig. S5). The measurements were made through one half of a lengthwise-cut quartz glass tube to closely approximate the radiation conditions in the tubes.
There was virtually no intensity below 230 nm. Typically, the total irradiance between 230 and 800 nm was 40 W/m2, of which 0.9 W/m2 were in the UVC, 1.9 W/m2 in the UVB, and 6.8 W/m2 in the UVA region. If necessary, the total irradiance was adjusted to 40 W/m2 by varying the distance to the light source. Samples were irradiated for 95, 190, 380, and 570 h. In dark control experiments, aluminum foil-wrapped tubes containing [FeCl(oep)] and quartz glass fragments were kept rotating for 570 h at 30–40°C (mean ∼32°C). All experiments were performed in duplicate.
After irradiation, the contents of each tube were extracted 10 times with 750 μL of dichloromethane, and the combined extracts were made up to 10 mL with dichloromethane. The resulting solution was used to determine the degree of [FeCl(oep)] decomposition and for UV-visible spectroscopy.
2.6. X-ray irradiation experiments
The X-ray experiments were performed at the Institute of Aerospace Medicine of the German Aerospace Center (DLR) in Cologne, Germany. An RS225 irradiation cabinet (Xstrahl) was used that was equipped with a tungsten X-ray tube operating at 200 kV and 15 mA. Soft X-rays were eliminated by a 0.5 mm copper filter (for a calculated spectrum, see Supplementary Fig. S6). The linear energy transfer (LET) was 0.3–3.0 keV/μm (Moeller et al., 2017). The dose rate was 44 Gy/min, and the maximum dose was 10 kGy. Samples were irradiated in air at ambient temperature.
Duplicates of each sample were prepared. Typically, 4 mg (6.4 μmol) of [FeCl(oep)] or an intimate mixture of 4 mg of [FeCl(oep)] and 100 mg of a salt mixture were placed in a thin-walled 0.2 mL polymerase chain reaction (PCR) tube (Eppendorf). The salt mixture consisted of NaCl and NaHCO3 in a 4:1 molar ratio and served to simulate the salt composition of the Enceladus plumes (Postberg et al., 2009). After X-irradiation, samples were stored at 4°C in the dark for no longer than 30 days before analysis.
Transport controls were sent with the other samples to Cologne and returned without being irradiated. In addition, as a laboratory control, identical samples were kept at room temperature in the dark.
The work-up of the samples was performed as follows. As the PCR tubes with the salt mixture were almost completely filled, the content was transferred to a 2 mL microcentrifuge tube for extraction. The content of the microcentrifuge tube was then extracted 10 times with 800 μL of dichloromethane, and the residue in the PCR tube was extracted 10 times with 100 μL. Each extraction stage was followed by centrifugation at 14,100g for 3 min (microcentrifuge tubes) and at 6000g for 12 min (PCR tubes), respectively, to remove particulate material.
The extracts were combined and made up to 10 mL with dichloromethane. To ensure consistency, this extraction procedure was also used for the PCR tubes containing [FeCl(oep)] only. The final solutions were analyzed by IR and UV-visible spectroscopy and used for quantification of [FeCl(oep)] (Section 2.8).
2.7. Ion bombardment experiments
Experiments with helium and iron ions were performed at the Heavy Ion Medical Accelerator in Chiba (HIMAC) facility of the National Institute of Radiological Sciences (NIRS) in Japan. The He ions had an energy of 145.2 MeV, an LET of 2.24 keV/μm, and a range in water of 147.8 mm. The dose rate used was 4.2 Gy/min, and the maximum dose was 1000 Gy. Particle and energy fluxes of He ions were calculated as 1.528 × 1012 particles/(m2·s) and 2.219 × 1014 MeV/(m2·s), respectively. The Fe ions had an energy of 418.3 MeV, an LET of 197.6 keV/μm, and a range in water of 74.4 mm. The dose rate used was 12.1 Gy/min, and the maximum dose was 3000 Gy. Particle and energy fluxes of Fe ions were calculated as 3.183 × 1010 particles/(m2·s) and 1.331 × 1013 MeV/(m2·s), respectively. For both ion types, the diameter of the homogeneous beam was 100 mm.
Two replicates (He irradiation experiments) or four replicates (Fe irradiation experiments) of each sample were prepared in thin-walled 0.2 mL PCR tubes (Eppendorf). As in the X-ray experiments, typically 4 mg (6.4 μmol) of [FeCl(oep)] or an intimate mixture of 4 mg of [FeCl(oep)] and 100 mg of the NaCl/NaHCO3 mixture (see above) was used. Transport controls were sent with the other samples by airplane to Japan and returned without being irradiated. In addition, as a laboratory control, identical samples were stored at room temperature in the dark.
Samples were irradiated at the HIMAC facility at room temperature in air. The orientation of the PCR tubes to the particle beam was random. After irradiation, the samples were kept at 4°C in the dark for no longer than 30 days before extraction and analysis. The extraction of the contents of the PCR tubes was performed in the same manner as described for the X-irradiated samples (see above).
The results of the irradiation experiments were statistically analyzed by using SAS version 9.4 (SAS Institute, Inc.). The statistical significance of differences between the mean values for different doses was tested by the Tukey test. The significance level was p ≤ 0.05.
2.8. Quantification of H2(oep), [{Fe(oep)}2(μ-O)], and [FeCl(oep)]
The method described here was applied to all dichloromethane extracts from all experiments. In the first step of the analytical procedure, each sample was checked for the presence of the free base H2(oep) and the complex [{Fe(oep)}2(μ-O)]. Both compounds were quantified by IR spectroscopy. For the quantification of H2(oep), an aliquot of the dichloromethane extract of the sample was transferred to a tightly sealable NaCl IR cell.
The area of the N–H stretching band at 3317 cm−1 was measured, and the H2(oep) content was calculated by using a calibration curve (Supplementary Fig. S7). In some experiments, the μ-oxido dimer [{Fe(oep)}2(μ-O)] formed, which was also quantified from the dichloromethane extracts. The [{Fe(oep)}2(μ-O)] content was calculated from the area of the Fe–O–Fe stretching band at 875 cm−1; the corresponding calibration curve is shown in Supplementary Fig. S8.
In the second step of the analytical procedure, [FeCl(oep)] was quantified. Here, IR spectroscopy could not be used for direct determination, because there is no suitable band without overlap with bands of H2(oep) or [{Fe(oep)}2(μ-O)]. Therefore, an indirect method was developed. After determining the content of H2(oep) and [{Fe(oep)}2(μ-O)] in the sample (see above), the iron complexes, including [FeCl(oep)], were transferred to the free-base porphyrin H2(oep) by demetallation with concentrated sulfuric acid (Buchler, 1975; Sanders et al., 2000). H2(oep) was then quantified as described earlier. Subsequently, the molar amount of [FeCl(oep)] (n
[FeCl(oep)]) was calculated by considering the amounts of [{Fe(oep)}2(μ-O)] and H2(oep) before demetallation, according to the equation:
Demetallation was performed as follows. Exactly 500 μL of the dichloromethane extract, which should not contain more than 1 μmol of [FeCl(oep)], was transferred to a 5 mL V-vial. If the concentration of the extract was too low, up to 4000 μL was used instead of 500 μL. The dichloromethane was carefully removed in a stream of nitrogen gas. To the dry residue, 50 μL (0.90 mmol) of 96% sulfuric acid was added. The vial was closed, vigorously shaken on a vortex mixer, and finally kept on a roller mixer for 15 min for complete demetallation.
During this procedure, the dark reddish-brown residue dissolved and a pink solution formed. Then, 1800 μL (1.80 mmol) of a 1 mol/L sodium hydrogen carbonate solution was added dropwise while cooling the vial in an ice/water bath. Evolution of carbon dioxide occurred, and concomitantly a dark brown precipitate formed. After gas evolution had stopped, 1000 μL of dichloromethane was added and the precipitate was dissolved by shaking the closed vial.
A 600 μL portion of the dichloromethane phase was transferred to a smaller vial, and the solvent (including residual water that might have interfered with the subsequent IR measurement) was carefully removed in vacuo. The completely dry residue was dissolved in 300 μL of dichloromethane, and a portion of the solution was transferred to an NaCl IR cell, which was tightly sealed. The amount of H2(oep) present was then determined by IR spectroscopy, as described earlier.
In addition, UV-visible spectroscopy was used to quantify [FeCl(oep)], but only if the IR analysis revealed minimal decomposition (<5%). In cases of stronger decomposition, the UV-visible method was not reliable, because decomposition products could distort the analytical results. In the absence of interfering substances, however, quantification by UV-visible spectroscopy was even superior to the IR method with respect to accuracy and reliability (see below).
For the quantification of [FeCl(oep)] by UV-visible spectroscopy, an aliquot of the dichloromethane extract (typically 100 or 200 μL) was diluted with dichloromethane to a volume of 5 mL. A portion of the solution was transferred to a gas-tight 10 mm quartz glass cuvette to measure the absorbance of the Soret band at 380 nm. The [FeCl(oep)] content of the sample was calculated by using a calibration curve (Supplementary Fig. S9).
Control experiments were performed to ascertain the accuracy and reliability of the quantification methods used. First, it should be mentioned that all calibration curves were linear, and the fits were very good (R 2 > 0.999, Supplementary Figs. S7–S9). Recovery rates of known amounts were determined after performing the different types of extraction with dichloromethane. For example, after an extraction procedure identical to that used in the experiments with acids and bases (see above), the recovery rate of H2(oep) was on average 99.8% and ranged from 98% to 102% (IR method) (Supplementary Fig. S10). Thus, the error was estimated as ±2%.
After the same extraction procedure, the recovery rate of [{Fe(oep)}2(μ-O)] was on average 100.4% and ranged from 99% to 102% (IR method) (Supplementary Fig. S11). Again, the error could be (conservatively) estimated as ±2%.
For [FeCl(oep)], too, the IR method had an average recovery close to 100% (99.2%). The range, however, was slightly wider than for the other two compounds, namely 96–102%, corresponding to an estimated maximum error of ±3% (Supplementary Fig. S12). The larger scatter of the data is probably caused by the complex sample preparation procedure involving, for example, a demetallation step (see above). An additional factor is that the amount of [FeCl(oep)] was calculated from three values, with each having an experimental error (see equation above).
Better results for [FeCl(oep)] were obtained with the UV-visible method. The main reason for this is probably the simpler sample preparation procedure (no demetallation is necessary) compared with the IR method. The recovery rate was on average 99.8% and ranged from 99% to 101% (Supplementary Fig. S13). Thus, the estimated error was only ±1%. Unfortunately, the UV-visible method could only be applied when decomposition was minimal (see above). There were no significant differences between samples with and without salt matrix and between the different extraction procedures.
The results of the control experiments—particularly the recovery rates and estimated errors associated with the various experimental and analytical methods—were used in calculating and interpreting the analytical results of the experiments described in Sections 2.3–2.7.
3. Results
3.1. Stability to aqueous acids, bases, and oxidizing agents
[FeCl(oep)] was exposed to pH values ranging from 0 to 13.5 at three different temperatures for 24 h. Hydrochloric acid or sodium hydroxide was used to adjust the pH. At 1°C and 25°C, [FeCl(oep)] was stable between pH 0.1 and 12.9 (Supplementary Table S1; the 25°C data are plotted in Fig. 2). In highly alkaline solution (pH 13.5), the amount of [FeCl(oep)] decreased to 91% ± 2% (1°C) and 79% ± 1% (25°C), respectively, of the initial amount.
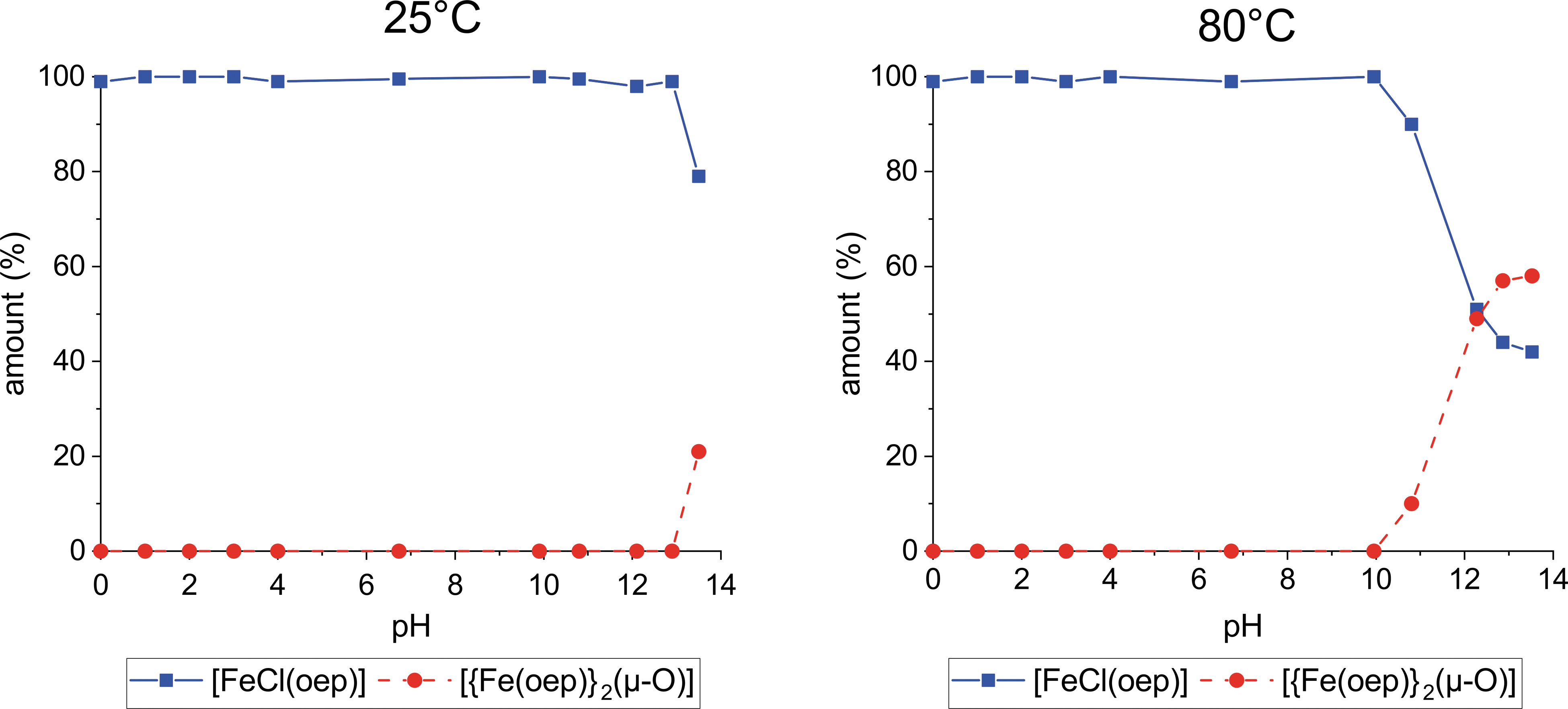
Stability of [FeCl(oep)] in the pH range 0–13.5 at 25°C and 80°C. Hydrochloric acid and sodium hydroxide were used to adjust the pH. Squares represent the amount of recovered [FeCl(oep)] relative to the initial amount. Circles represent the relative amount of [FeCl(oep)] that was transformed into the dimer [{Fe(oep)}2(μ-O)]. Each point is the average of two independent 24 h experiments. The pH stability curve determined at 1°C is very similar to the 25°C curve and, therefore, not shown.
The UV-visible spectrum indicated the formation of the dimer [{Fe(oep)}2(μ-O)] (Fig. 3) whose presence was confirmed by IR spectroscopy, which revealed the characteristic Fe–O–Fe stretching band at 875 cm−1. Quantification of [{Fe(oep)}2(μ-O)] showed that the missing [FeCl(oep)] had been completely transformed into the dimer (Fig. 2 and Supplementary Table S2). Thus, the iron porphyrin core remained intact.
UV-visible spectra of
At 80°C, [FeCl(oep)] was stable between pH 0 and 10. The formation of [{Fe(oep)}2(μ-O)] started in the moderately basic pH region. At pH 10.8, 10% ± 1% of the [FeCl(oep)] was transformed into the dimer (Fig. 2 and Supplementary Table S2). At the highest pH (13.5), more than half of the initial amount of [FeCl(oep)] (58% ± 2%) was transformed. Comparable results were obtained when the experiments were repeated with sulfuric acid instead of hydrochloric acid and sodium carbonate instead of sodium hydroxide (Supplementary Tables S1 and S2). In particular, no demetallation occurred in any of these experiments. This observation is consistent with [FeCl(oep)] being in stability class II (Section 1).
In contrast to hydrochloric and sulfuric acid, nitric acid at a concentration of 1 mol/L decomposed the iron porphyrin core of [FeCl(oep)]. Decomposition was observed at 25°C and 80°C, whereas at 1°C [FeCl(oep)] was stable to nitric acid over the entire concentration range studied (Fig. 4a and Supplementary Tables S1 and S3). At 25°C and 80°C, [FeCl(oep)] was stable to nitric acid concentrations up to 0.1 mol/L. After treatment with 1 mol/L nitric acid, 85% ± 1% (25°C) and 81% ± 1% (80°C) of the initial [FeCl(oep)] was still present.
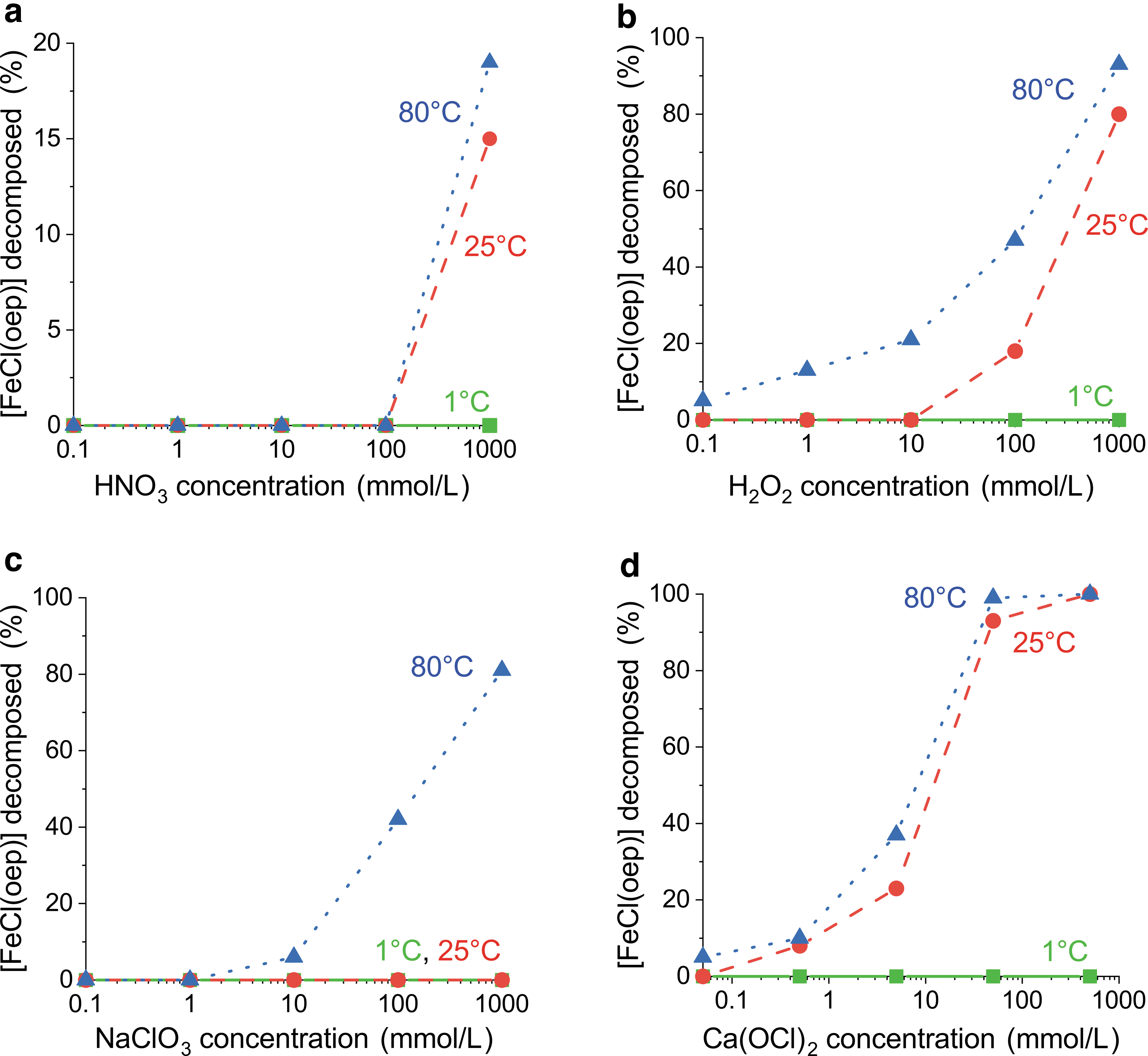
Stability of [FeCl(oep)] to potential oxidants as a function of oxidant concentration at 1°C, 25°C, and 80°C.
The formation of [{Fe(oep)}2(μ-O)] was not observed (Supplementary Table S2). Therefore, it is likely that nitric acid attacks [FeCl(oep)] oxidatively. Nitric acid is well known to act as an oxidant at higher concentrations.
The next potential oxidant used was hydrogen peroxide. At 1°C, [FeCl(oep)] was not decomposed by H2O2 even at the highest concentration of 1 mol/L (Fig. 4b and Supplementary Table S3). At 25°C, the decomposition was 8% ± 2% at an H2O2 concentration of 0.1 mol/L and reached 80% ± 2% at a concentration of 1 mol/L. Finally, at 80°C, 5% ± 1% decomposition was observed even at the lowest H2O2 concentration used (0.1 mmol/L), and at the highest concentration the decomposition of [FeCl(oep)] was almost complete (93% ± 4%).
By contrast, [FeCl(oep)] showed excellent stability to magnesium perchlorate solutions and remained unchanged under all conditions studied (Supplementary Table S3). This result was not unexpected, as perchlorate is known to be kinetically inert. The stability to sodium chlorate solutions was also relatively high. At 1°C and 25°C, [FeCl(oep)] was stable over the entire NaClO3 concentration range of 0.1 mmol/L to 1 mol/L (Fig. 4c and Supplementary Table S3).
At 80°C, slight (6% ± 1%) decomposition was observed in 10 mmol/L NaClO3; in the most concentrated NaClO3 solution, the degree of decomposition reached 81% ± 2%. Finally, we studied calcium hypochlorite Ca(OCl)2, which is commonly used as a disinfectant and bleaching agent. Again, [FeCl(oep)] proved stable at 1°C, without any signs of decomposition up to the maximum ClO− concentration of 1 mol/L (Fig. 4d and Supplementary Table S3).
At 25°C, decomposition (8% ± 1%) was first observed at a ClO− concentration of 1 mmol/L; after treatment with 1 mol/L ClO−, [FeCl(oep)] was no longer detectable. At 80°C, slight (5% ± 2%) decomposition was measurable even at the lowest ClO− concentration (0.1 mmol/L); a concentration of 0.1 mol/L was sufficient to oxidize all [FeCl(oep)] within 24 h. This showed that hypochlorite was the most potent oxidant for [FeCl(oep)] used in this study (Fig. 4). In none of the experiments with oxidants was the formation of [{Fe(oep)}2(μ-O)] observed, not even with high-pH hypochlorite solutions (Supplementary Table S2). The free porphyrin could also not be detected.
3.2. Thermal stability
The experiments with acids, bases, and oxidants showed that [FeCl(oep)] can withstand 80°C for 24 h in the presence of aqueous solutions (see above). In the following experiments, the limits of thermal stability of solid [FeCl(oep)] were explored in detail. First, we performed TG analyses up to 900°C. As the thermal behavior of solids can depend on the atmosphere present, three different astrobiologically relevant atmospheres were used, namely: synthetic air, nitrogen, and carbon dioxide.
A first small (∼1.7%) mass loss was observed between 100°C and 170°C, regardless of which atmosphere was used (Fig. 5). It was likely caused by adhering water or organic solvent. In air, [FeCl(oep)] was stable up to 276°C. Above this temperature, a large and rapid mass loss occurred because of a reaction with atmospheric oxygen. Differential thermal analysis showed a few strong exothermic peaks. At 432°C, the combustion process abruptly stopped, and no further thermal processes were observed. By the end of the experiment, the residual mass was 13.1% of the initial mass. The residue had a reddish color.
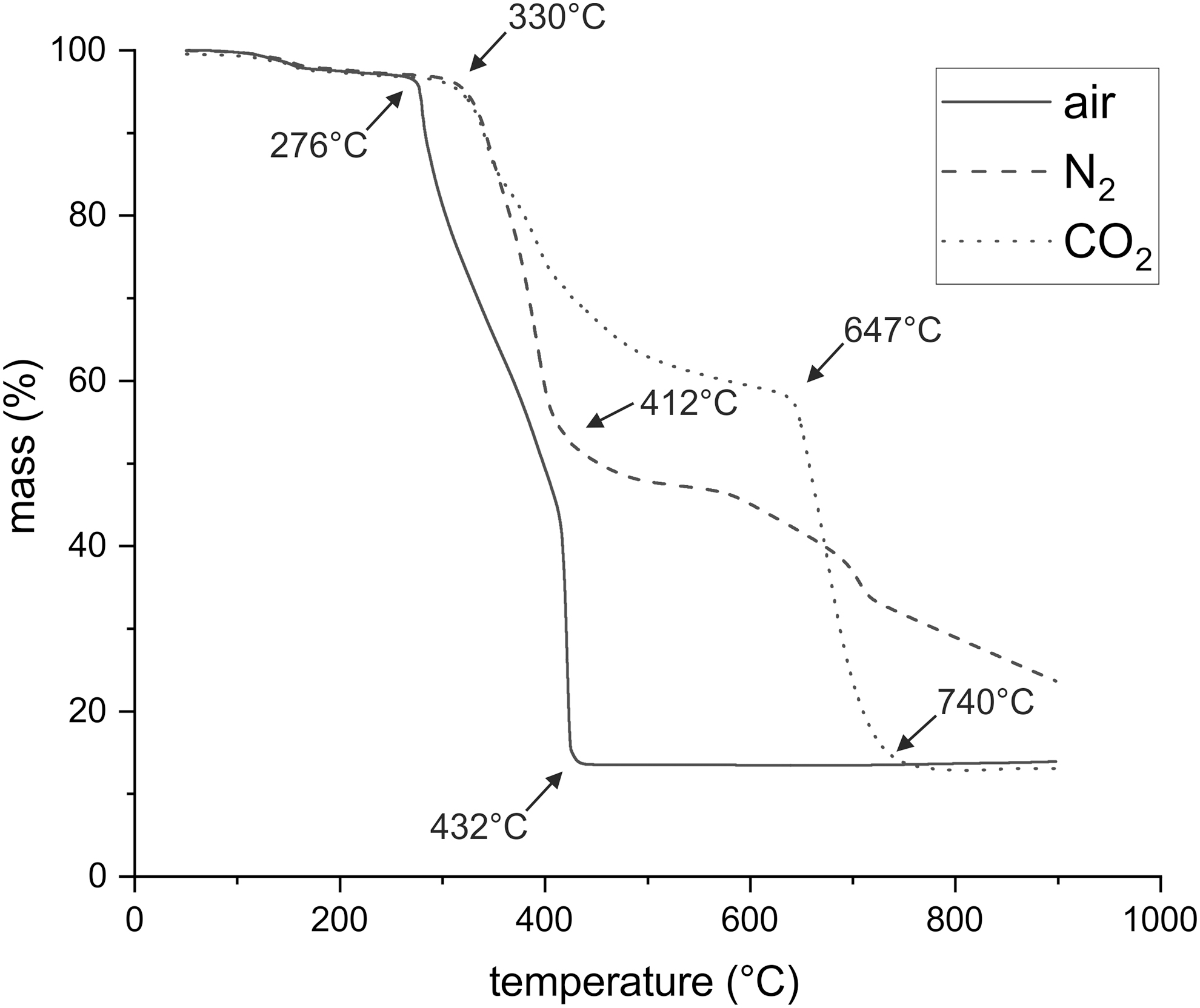
Thermogravimetric analysis of [FeCl(oep)] in air, nitrogen, and carbon dioxide at a heating rate of 5°C/min.
In an atmosphere of pure nitrogen, [FeCl(oep)] was stable up to 330°C, which is rather remarkable for a complex organic molecule (Fig. 5). Between 330°C and 412°C, a precipitous mass loss of ∼42% occurred, followed by a slower one that was not finished at 900°C. The residual mass was 23.6%. The residue was glass-like, brittle, and almost black. Differential thermal analysis was not particularly informative, showing only that several thermal processes overlapped.
The TG curve recorded in a carbon dioxide atmosphere was identical to that in nitrogen up to 350°C (Fig. 5). Thus, here also, [FeCl(oep)] was stable up to 330°C. Starting from 350°C, the first mass loss became less steep than that under nitrogen gas and ended almost in a plateau. Between 647°C and 740°C, a second, steep mass loss was measured. The residual mass (12.6%) was similar to that in the air experiment, and the residue was also reddish in color.
The clear differences between the TG curves in carbon dioxide and nitrogen showed that above 350°C carbon dioxide did not behave as an inert gas. We hypothesized that the thermal decomposition products of [FeCl(oep)] took up oxygen from carbon dioxide. This would explain the initially smaller mass loss compared with the nitrogen experiment. Then, in the second mass loss step, the decomposition products rapidly burned in the carbon dioxide atmosphere. If this was right, carbon monoxide would form at temperatures above 350°C. This was checked by using colorimetric carbon monoxide gas detection tubes. The detection limit was 2.5 ppm, which was above the CO content of the carbon dioxide used.
At the start of the experiment at 50°C and at 200–220°C, where the TG curves in carbon dioxide and nitrogen were still identical, no CO was detected (Table 1 and Supplementary Fig. S14). At 400–420°C and 500–520°C, a CO content of 110 and 120 ppm, respectively, was measured. This supports the earlier assumption that in this temperature region oxygen uptake from carbon dioxide slowed the mass loss rate. Subsequently, the CO level decreased to 30 ppm at 600–620°C, before it again increased sharply to 25,000 ppm at 700–720°C.
Carbon Monoxide Content in the Gas from the Outlet of the Thermogravimetric Analysis Instrument During Thermogravimetric Measurement of [FeCl(oep)] in a Carbon Dioxide Atmosphere
Before the start of the experiment.
This second phase of CO formation is associated with the steep mass loss above 647°C, which is believed to result from a combustion process with carbon dioxide as the oxidizing agent. Although no further mass loss occurred above 740°C, CO was still detectable at higher temperatures (20 ppm was measured at 880–900°C). This observation suggests that it takes some time for the CO to leave the furnace of the TGA instrument. CO formation, therefore, appeared delayed (Supplementary Fig. S14). Consequently, the CO content data in Table 1 should be regarded as approximate.
The TGA experiments were repeated on a larger scale in a tube furnace to obtain sufficient material of the 900°C residues for IR, X-ray diffraction (XRD), and elemental analyses. Great care was taken to maintain the same conditions (e.g., the atmospheric composition, gas flow rate, and heating rate) as used in the TG analyses. The XRD pattern of the reddish residue that remained after heating in air showed hematite (Fe2O3) as the only crystalline phase (Supplementary Fig. S15A).
The residue had a high iron content of 68.2%, whereas the carbon, hydrogen, nitrogen, and chlorine contents were below the detection limits (Supplementary Table S4A). The iron content was very close to the theoretical value for Fe2O3 (69.9%). Thus, the residue was clearly identified as almost pure hematite. This conclusion was further supported by the mass of the residue measured in the TGA experiment. Within the experimental error, the observed value of 13.1% of the initial mass was identical to the theoretical value of 12.8% expected if hematite was the only solid product.
The 900°C residue that was obtained in a carbon dioxide atmosphere was also reddish. Again, only hematite could be detected by XRD (Supplementary Fig. S15B). The iron content was 68.6%; carbon, hydrogen, nitrogen, and chlorine were not detectable (Supplementary Table S4B). In the TGA experiment, the residual mass was 12.6% of the initial mass of [FeCl(oep)]. Taken together, these results show that here, too, the residue consisted almost entirely of hematite.
The residue obtained by heating [FeCl(oep)] to 900°C in a nitrogen atmosphere was almost black and glass-like in appearance (Supplementary Fig. S15C). It consisted mainly of carbon (83.00%) and iron (14.3%) with small amounts of nitrogen (0.51%) and hydrogen (0.19%) but no chlorine (Supplementary Table S4C). This composition differs strongly from those of the residues obtained in the air and carbon dioxide atmosphere.
The XRD showed the presence of four crystalline phases, namely: α-iron, graphite, the iron carbide cohenite (Fe3C), and hematite (Supplementary Fig. S15C). Since oxygen sources were absent, one must assume that the hematite was not a product of the experiment, but rather formed from α-iron before and during analysis, when the sample came into contact with air. A dark reddish-brown sublimate formed on cooler parts of the apparatus in nitrogen and carbon dioxide atmospheres, but not in air (see Supplementary Fig. S16A for an example). The sublimate was identified as [FeCl(oep)] by IR spectroscopy.
The results just described prompted us to perform another experiment in which an [FeCl(oep)] sample was kept at 500°C in a nitrogen atmosphere for 24 h. In this way, [FeCl(oep)] was exposed for an extended period of time to a temperature in the near-plateau region of the TG curve that followed the first decomposition step (Fig. 5). In this experiment too, a significant amount of [FeCl(oep)] sublimed and condensed in pure form outside the heating zone (Supplementary Fig. S16A). This is an important result, because it shows that [FeCl(oep)] can sublime at atmospheric pressure and thereby partly escapes thermal decomposition. For technical reasons, it was not possible to determine the amount of sublimate.
The residue of the 500°C experiment was reddish on the surface but black underneath (Supplementary Fig. S16B). Therefore, it was thoroughly homogenized with a mortar and pestle before analysis. The XRD showed the residue to contain hematite (Supplementary Fig. S16C). In addition, there were two reflections, one considerably broadened, that were indicative of carbonaceous material. They were reminiscent of certain types of charcoal (Mochidzuki et al., 2003), a material structurally related to graphite but with a low degree of ordering (e.g., Lin et al., 2013).
This observation was supported by the rising baseline, which indicated larger amounts of amorphous material, and the high carbon content. The residue contained 63.09% carbon, 15.8% iron, 10.4% nitrogen, and some hydrogen and chlorine (Supplementary Table S5). The oxygen content was 8.43% (calculated by difference).
The reddish surface color, which was likely due to the formation of hematite (Fe2O3), was visible immediately after opening the thermolysis tube. At that moment, the sample was still protected by a stream of nitrogen and could not react with atmospheric oxygen. Therefore, we conclude that oxygenation, at least in part, occurred during the experiment. The residual oxygen concentration in the nitrogen gas used was low (3 vol ppm, see Section 2). However, as shown earlier (Fox et al., 2019), the amount of oxygen introduced by the gas stream may become relevant in experiments of long duration, such as the one described here.
In a final series of experiments on thermal stability, loss of [FeCl(oep)] by sublimation was prevented by sealing the samples in glass ampoules. For safety reasons, the ampoules were evacuated instead of being filled with nitrogen. The samples were kept at constant temperatures for 24 h. In these experiments, [FeCl(oep)] proved to be essentially stable up to 250°C. Only slight decomposition (5.8%) occurred at this temperature (Fig. 6).
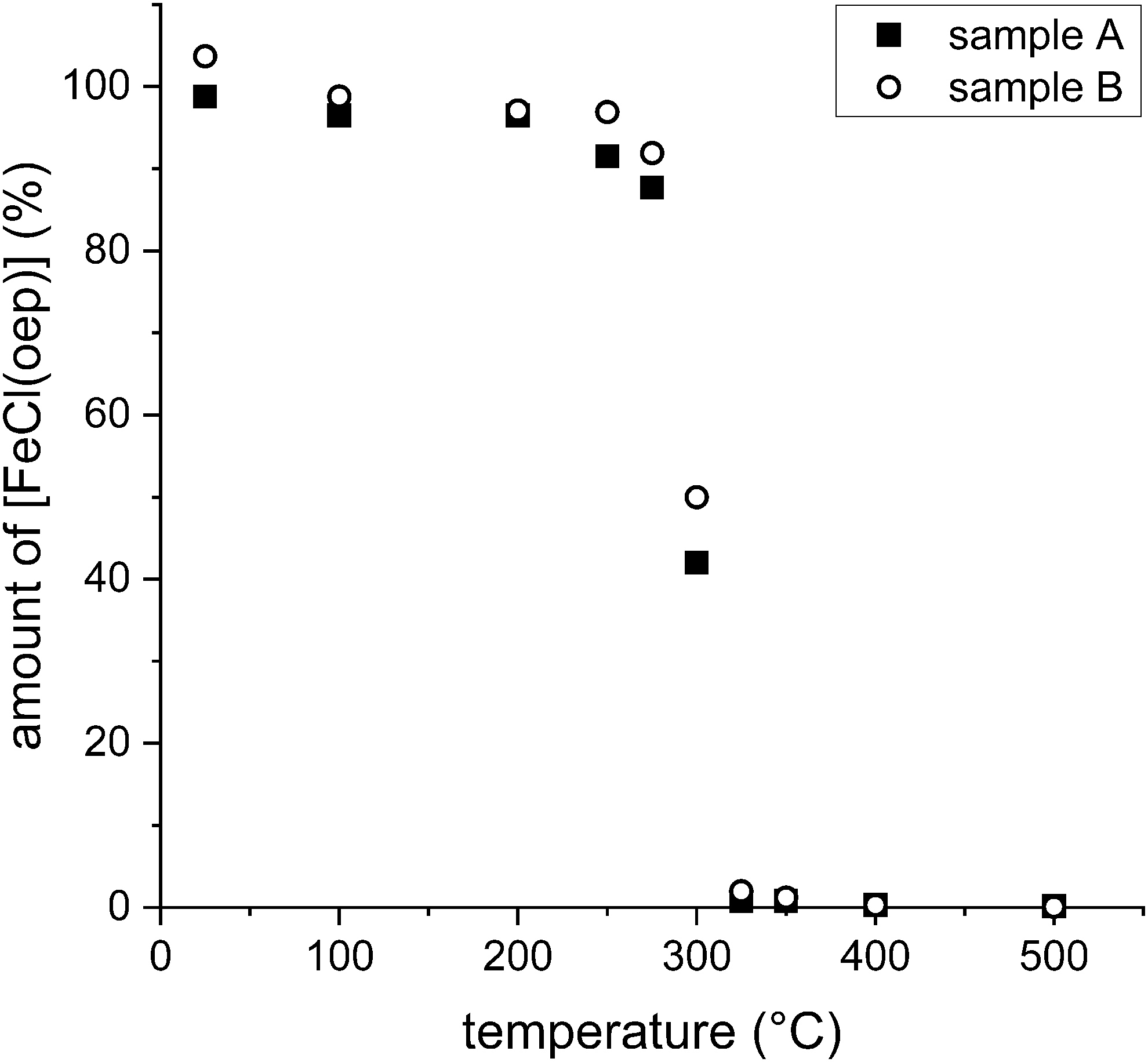
Thermal stability of [FeCl(oep)] in evacuated and sealed ampoules at various temperatures over a 24 h period. The amount of [FeCl(oep)] found at the end of the experiment is expressed as percent of the initial amount present. All experiments were performed in duplicate (samples A and B).
At 275°C, decomposition reached 10.2%, which is still a relatively low value. At 300°C, however, approximately half of the initial amount (54%) was decomposed. Here, in contrast to the experiments at lower temperatures, the inner wall of the ampoule was covered with a thin brown layer and the UV-visible spectrum showed reduced intensity of the Soret band at 380 nm (Supplementary Fig. S17). At 325°C, decomposition was nearly complete (Fig. 6) and the Soret band had almost disappeared. At this and higher temperatures, the residue was black. Finally, at the highest temperature (500°C), more than 99.9% of the [FeCl(oep)] was decomposed. The ampoule was completely black, and the Soret band was no longer observable. Further analytical studies, such as XRD or elemental analyses, were not possible, because of insufficient material.
The residue of the 500°C experiment showed no sign of red color and remained black after opening the ampoule. This supports the earlier conclusion that the hematite formation observed in the tube furnace experiment at 500°C was caused by residual oxygen in the nitrogen gas.
3.3. Stability to UV-visible radiation
The UV-visible irradiation experiments were carried out under nitrogen with a lamp emitting from 230 nm into the IR region (Supplementary Fig. S5). Small quartz glass fragments were added to assist mixing during spinning of the sample tubes. It has been reported that quartz grains can react with methane in tumbling experiments (Knak Jensen et al., 2014; Jakobsen et al., 2016). Therefore, it may be that under similar conditions quartz glass also reacts with organic compounds.
To test this possibility for [FeCl(oep)], two spinning experiments were conducted under standard conditions but without irradiation. They lasted for 570 h, which was the maximum duration of the irradiation experiments. However, no evidence was found for a reaction between [FeCl(oep)] and quartz glass.
After 95 h of UV-visible irradiation (corresponding to a radiant exposure of 13.7 MJ/m2), 4.5% of the initial amount of [FeCl(oep)] was decomposed (Fig. 7). This percentage increased to 7.5% after 190 h. After extraction of the contents of the 190-h tubes, a thin light brown film was found on the walls of the tubes (Supplementary Fig. S18A). This film was insoluble in dichloromethane, acetone, toluene, and hexane, but it was soluble in concentrated sulfuric acid. The amount was not sufficient for most analytical methods, except UV-visible spectroscopy.
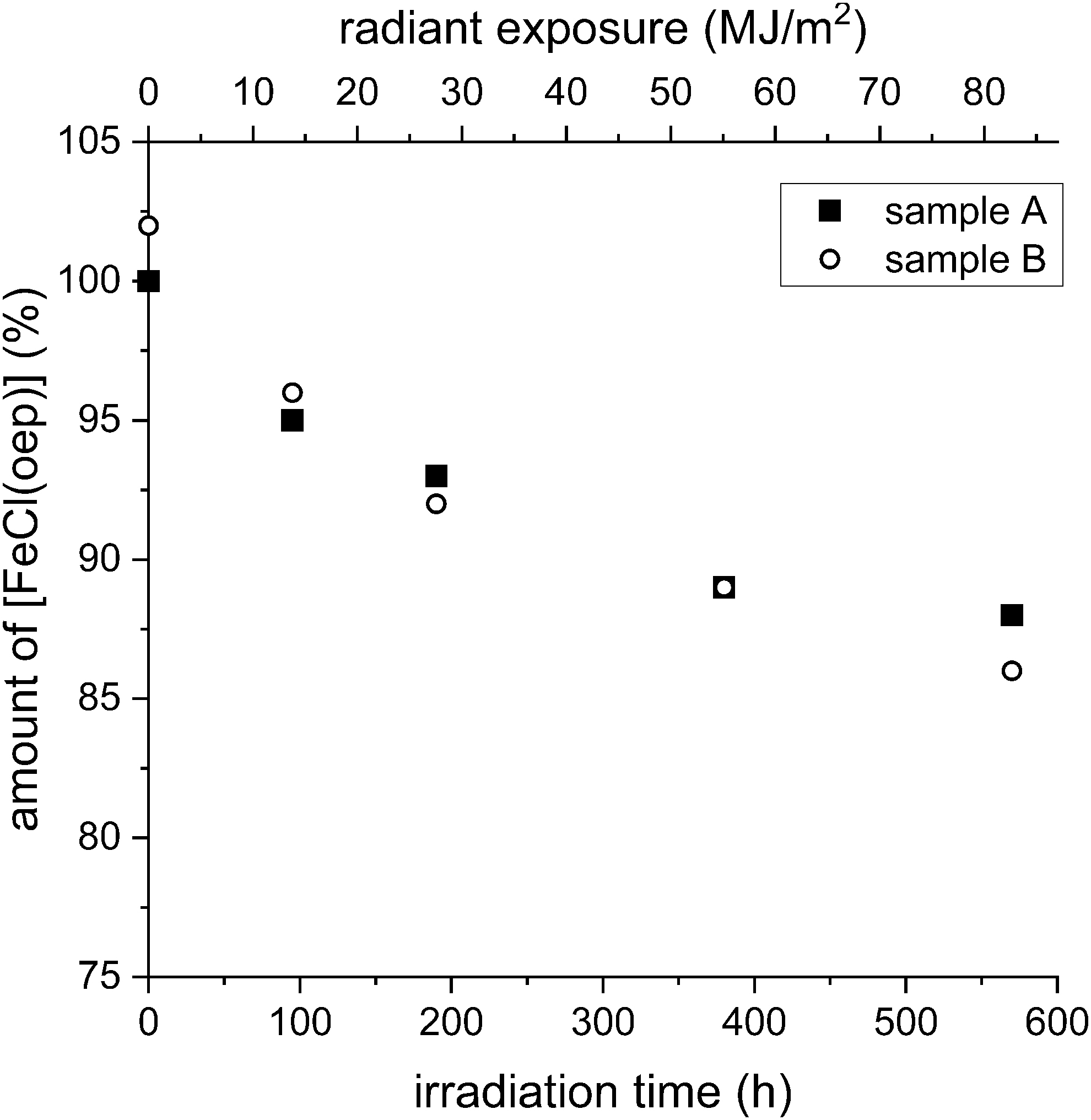
Stability of [FeCl(oep)] to UV-visible radiation. The amount of [FeCl(oep)] found at the end of the experiment is expressed as percent of the initial amount present. The radiant exposure values refer to the 230–800 nm range. All experiments were performed in duplicate (samples A and B).
The UV-visible absorption spectrum of the solid film showed a shoulder at ∼420 nm and increasing absorbance from 700 to 250 nm but was otherwise featureless (Supplementary Fig. S18B). After 380 h of irradiation, the brown film was now clearly visible. Quantitative analysis of the extractable fraction showed that 11% of the initial amount of [FeCl(oep)] was decomposed. The percentage decomposition at the longest irradiation time (570 h, corresponding to 82.1 MJ/m2) was only slightly higher (13%) than after 380 h. The brown film on the inner tube wall was also present.
Zero-, first-, or second-order kinetics are usually assumed for photodecomposition processes, but neither of them satisfactorily describes the behavior of [FeCl(oep)] (Supplementary Fig. S19). This may be explained by a protective effect of the brown film that forms during UV-visible irradiation of [FeCl(oep)]. As can be seen in Supplementary Fig. S18B, the brown material absorbs in the relevant wavelength range. The thicker the film becomes, the better it will protect the remaining [FeCl(oep)].
We, therefore, assumed a bounded decay model based on the von Bertalanffy growth model (von Bertalanffy, 1938; Mangel, 2006). It is described by the general equation
In addition, this model allows estimation of the time course of the photodecomposition in the absence of the protective film. During the first few hours of irradiation, the brown material was present in low amounts and therefore had virtually no protective effect. Thus, assuming initial zero-order kinetics, a linear approximation can be made for small values of t (Supplementary Fig. S20). The resulting equation is
Using this equation, we estimate that the decomposition of [FeCl(oep)] would reach 100% after ∼1526 h (∼64 days) of irradiation (corresponding to a radiant exposure of 220 MJ/m2) if no protective film is formed.
3.4. Stability to X-rays
The X-irradiation experiments on [FeCl(oep)] were conducted both with and without the presence of a sodium chloride/sodium hydrogencarbonate matrix. [FeCl(oep)] and the salt mixture were used in a 4:100 mass ratio. When the salts were present, no decomposition was observed up to the highest dose used (10 kGy). By contrast, without salts [FeCl(oep)] was only stable up to ∼3 kGy. At 10 kGy, 5.5% of the initial amount was decomposed. The decomposition followed zero-order kinetics according to the equation
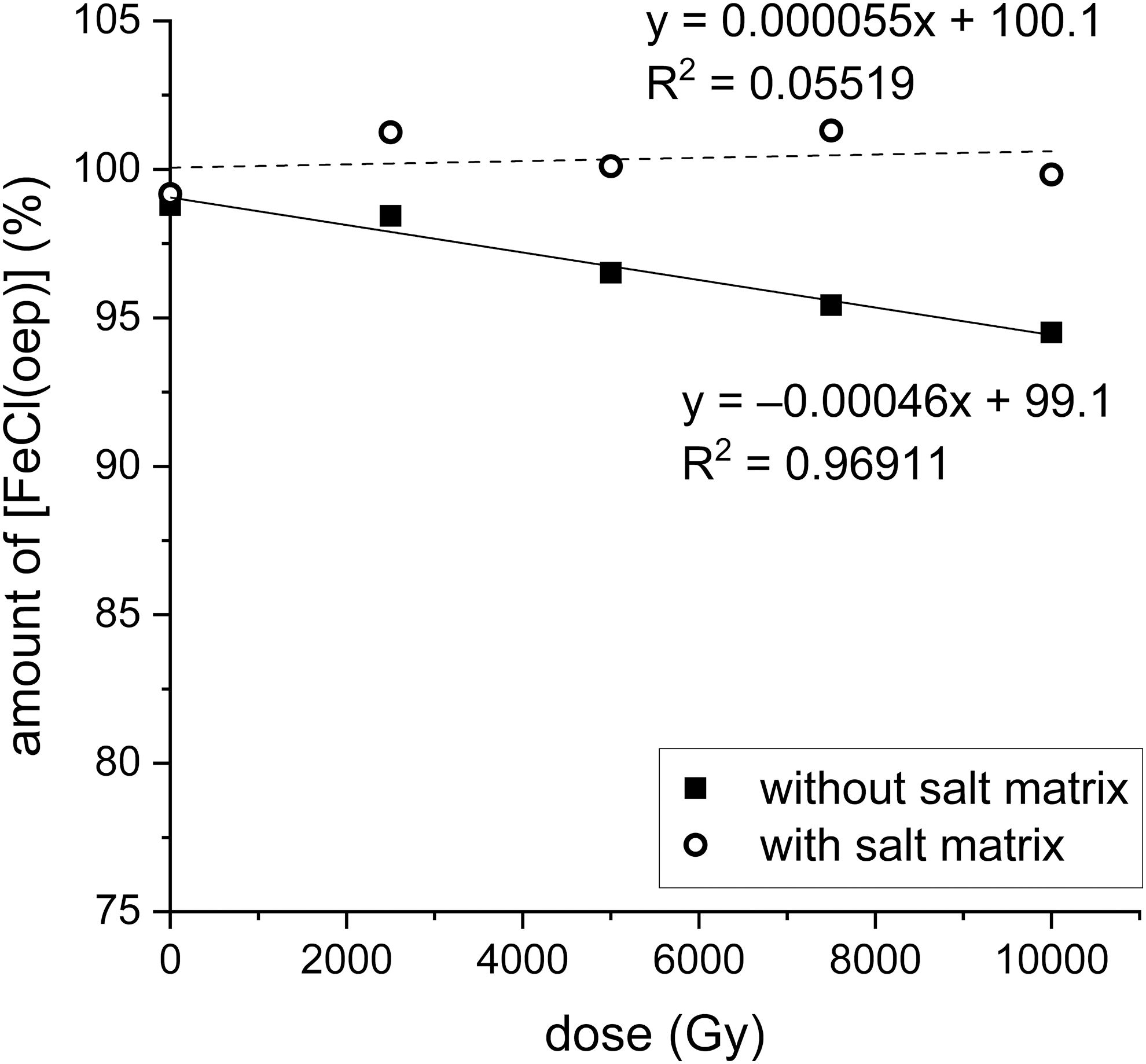
Stability of [FeCl(oep)] to X-irradiation in the presence and absence of a sodium chloride/sodium hydrogencarbonate matrix. The amount of [FeCl(oep)] found at the end of the experiment is expressed as percent of the initial amount present. All experiments were performed in duplicate, and the mean value is shown. The corresponding data can be found in Supplementary Table S6.
From this, the doses required for 50% and complete decomposition of [FeCl(oep)] can be estimated at 107 and 215 kGy, respectively. However, one must bear in mind that the data are only for the initial phase and the rate law may be different for higher doses. Similarly, it is not known how effectively the salt matrix protects [FeCl(oep)] at higher doses.
3.5. Stability to high-energy ion bombardment
In a first series of experiments, [FeCl(oep)] was irradiated with 145.2 MeV helium ions up to a dose of 1000 Gy (Fig. 9a). In none of the experiments was the amount of [FeCl(oep)] after irradiation significantly different from the controls. In a second series of experiments, [FeCl(oep)] embedded in excess sodium chloride/sodium hydrogencarbonate was used, as in the earlier experiments with X-rays. As with neat [FeCl(oep)], helium ion irradiation caused no decomposition (Fig. 9b). Thus, it was not possible to detect a potential effect of the salt mixture. Unfortunately, higher helium ion doses were not available.
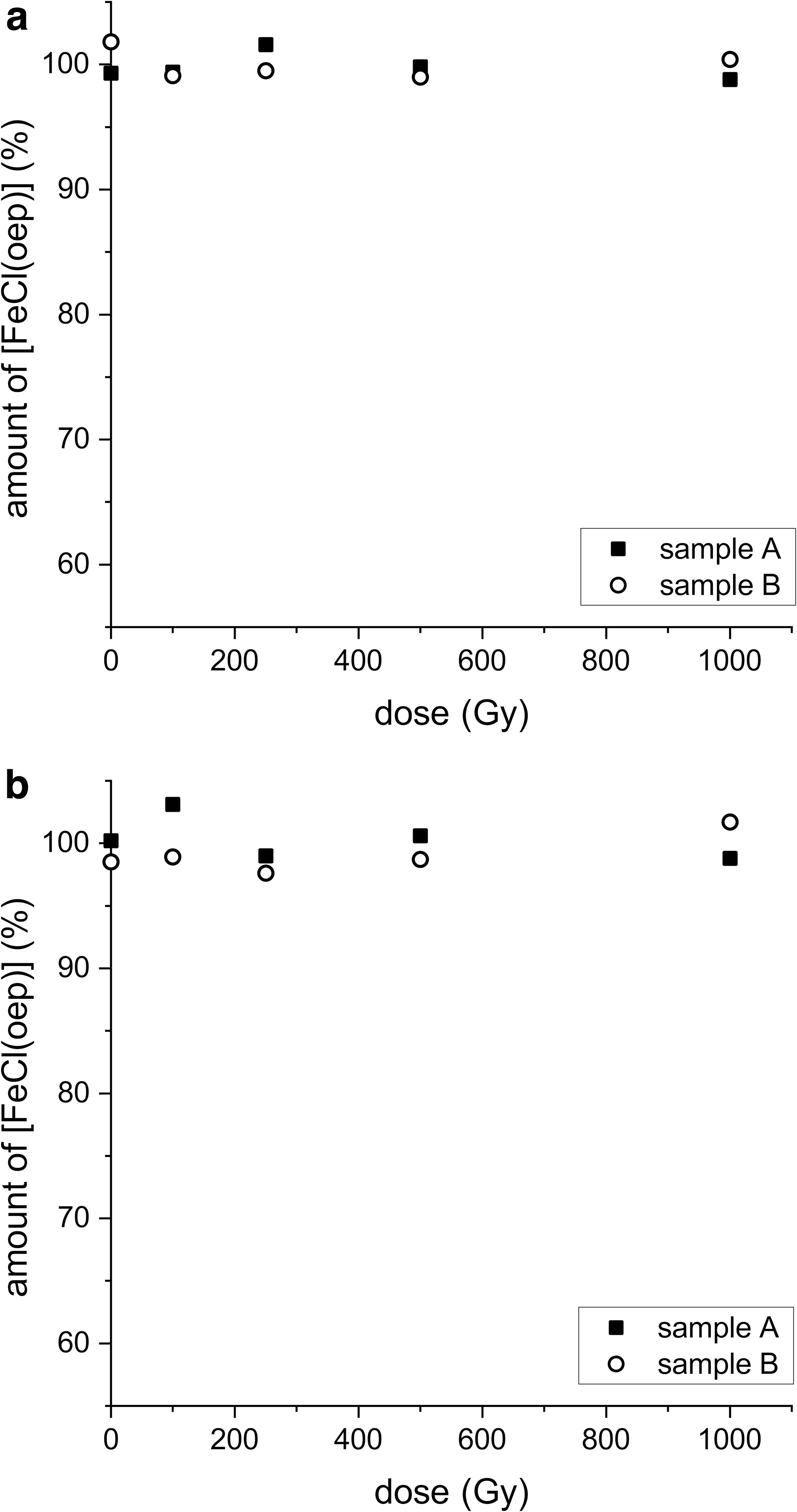
Stability of [FeCl(oep)] to helium ion irradiation in the absence
In another set of experiments, 418.3 MeV iron ions were used. Neat [FeCl(oep)] proved stable to iron ion bombardment up to a dose of 2000 Gy (Fig. 10a). Samples exposed to 3000 Gy, however, were statistically significantly different from the controls and the other samples (Supplementary Fig. S21). On average, 7.2% of the initial amount of [FeCl(oep)] was decomposed by exposure to 3000 Gy. The 3000 Gy dose corresponded to 1.98 × 1017 MeV/m2 and 4.74 × 1014 Fe ions/m2. Since higher doses were not available, an estimate of the relationship between irradiation dose and degree of decomposition was not possible.
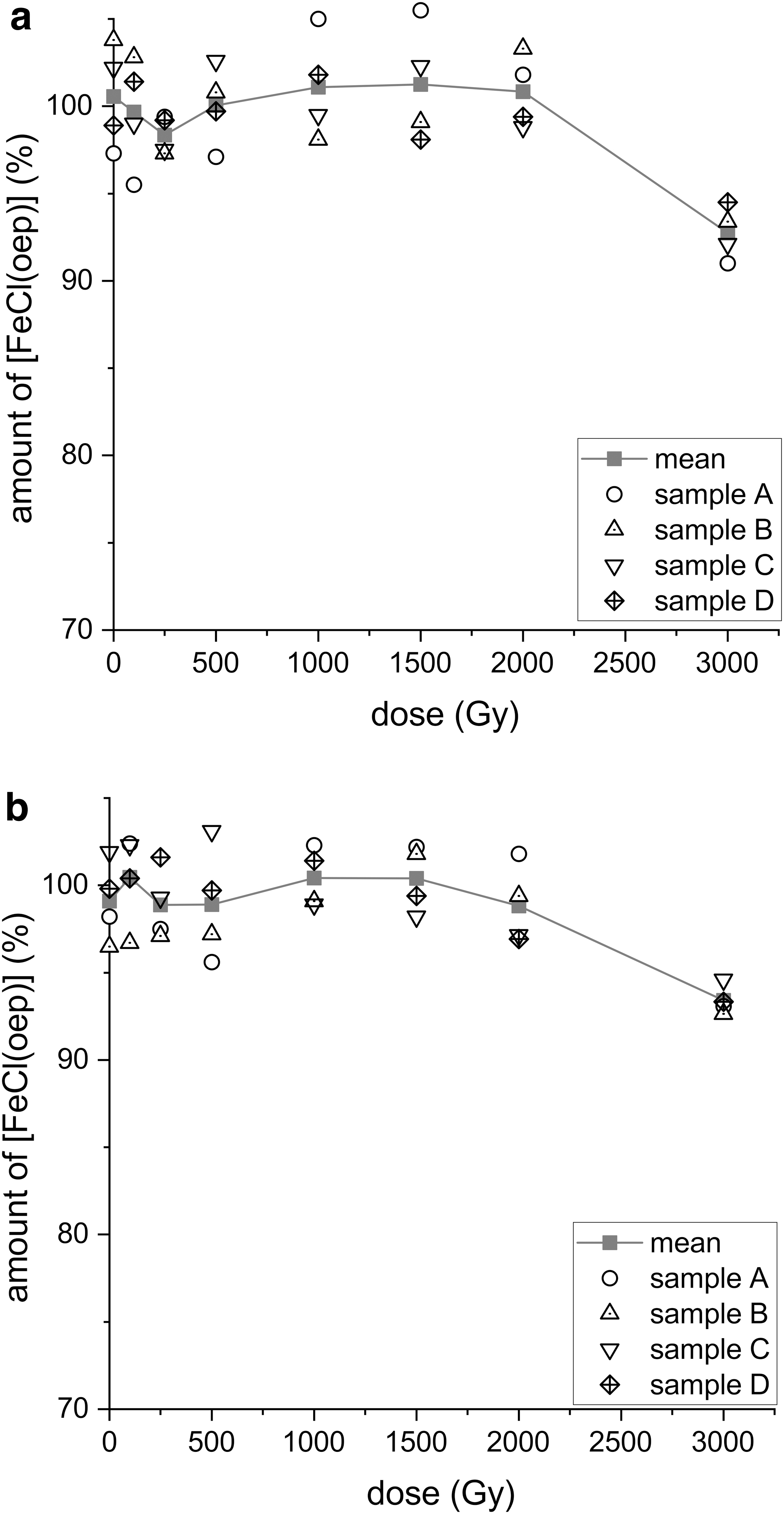
Stability of [FeCl(oep)] to iron ion radiation in the absence
When a mixture of [FeCl(oep)] and sodium chloride/sodium hydrogencarbonate was irradiated with 418.3 MeV iron ions, the results were similar to those obtained with neat [FeCl(oep)]. In the salt matrix, the iron porphyrin was stable up to 2000 Gy, whereas at a dose of 3000 Gy 6.6% decomposition was observed (Fig. 10b). Statistical analysis showed that the mean of the 3000 Gy samples was the only one that differed significantly from the control means (Supplementary Fig. S22). In addition, the significance of difference between the means of the 3000 Gy samples both with and without salt was determined by using a two-sample t test (SAS software version 9.4, significance level p ≤ 0.05). The t test revealed that there was no statistically significant difference and, thus, no evidence of an effect of the salt matrix.
4. Discussion
4.1. Stability to aqueous acids, bases, and oxidizing agents
Very acidic natural environments, sometimes having pH values below zero, are known on Earth. A brief overview and a method for measuring negative pH values are given by Nordstrom et al. (2000). Extreme acidity of natural waters is often caused by magmatic gases, which provide HCl, HF, and (after oxidation in air) H2SO4; thus, it is associated with volcanic activity. Another source of high acidity is the oxidation of pyrite, which produces sulfuric acid. In our study, we set the lower pH limit at zero and used hydrochloric, sulfuric, and nitric acid.
Nitric acid was included, because it is a constituent of volcanic plumes (Mather et al., 2004). Natural sulfuric acid occurs not only on Earth but also on Jupiter's moon Europa (Carlson et al., 1999b, 2005) and in the atmosphere of Venus (Krasnopolsky, 2015). Both the subsurface ocean of Europa and the Venus atmosphere are of astrobiological interest because they are potential habitats for extraterrestrial life (Schulze-Makuch and Irwin, 2018).
Our results showed that [FeCl(oep)] was stable to acidic solutions for at least 24 h even at pH 0 and 80°C, provided that the acid was not strongly oxidizing (Fig. 2 and Supplementary Table S1). It is further known that in 100% sulfuric acid demetallation occurs at room temperature, but the porphyrin ligand survives as porphyrinium cation (Falk, 1964; Buchler, 1975). Thus, acidic environments appear not to be generally problematic for the preservation of (iron) porphyrin biosignatures.
The upper pH limit of our experiments was 13.5. To our knowledge, the highest pH values reported for natural environments on Earth do not exceed 13. For example, pH 12.9 and 12.5 were measured in hyper-alkaline groundwater (Pedersen et al., 2004) and in pore water from serpentinite seamounts, respectively (Mottl et al., 2003). The astrobiologically interesting subsurface ocean of Saturn's moon Enceladus is believed to have a pH of ∼11–12 (Glein et al., 2015). Its alkalinity is caused by the presence of sodium carbonate.
We found that at 25°C [FeCl(oep)] was stable to a sodium carbonate solution of pH 11.4 for 24 h; at slightly higher pH, decomposition commenced (Supplementary Table S1). At 80°C, the stability to alkaline solutions (sodium carbonate or sodium hydroxide) was strongly decreased, but the decomposition consisted solely of the formation of the dimer [{Fe(oep)}2(μ-O)] (Supplementary Table S2). The dimerization of [FeCl(oep)] under alkaline conditions is known as a synthesis method for [{Fe(oep)}2(μ-O)] (Stolzenberg et al., 1981). In this reaction, the axial ligand at the iron porphyrin core is exchanged; the reaction is reversed in the presence of hydrochloric acid (Fig. 11) (Wijesekera and Dolphin, 1994).
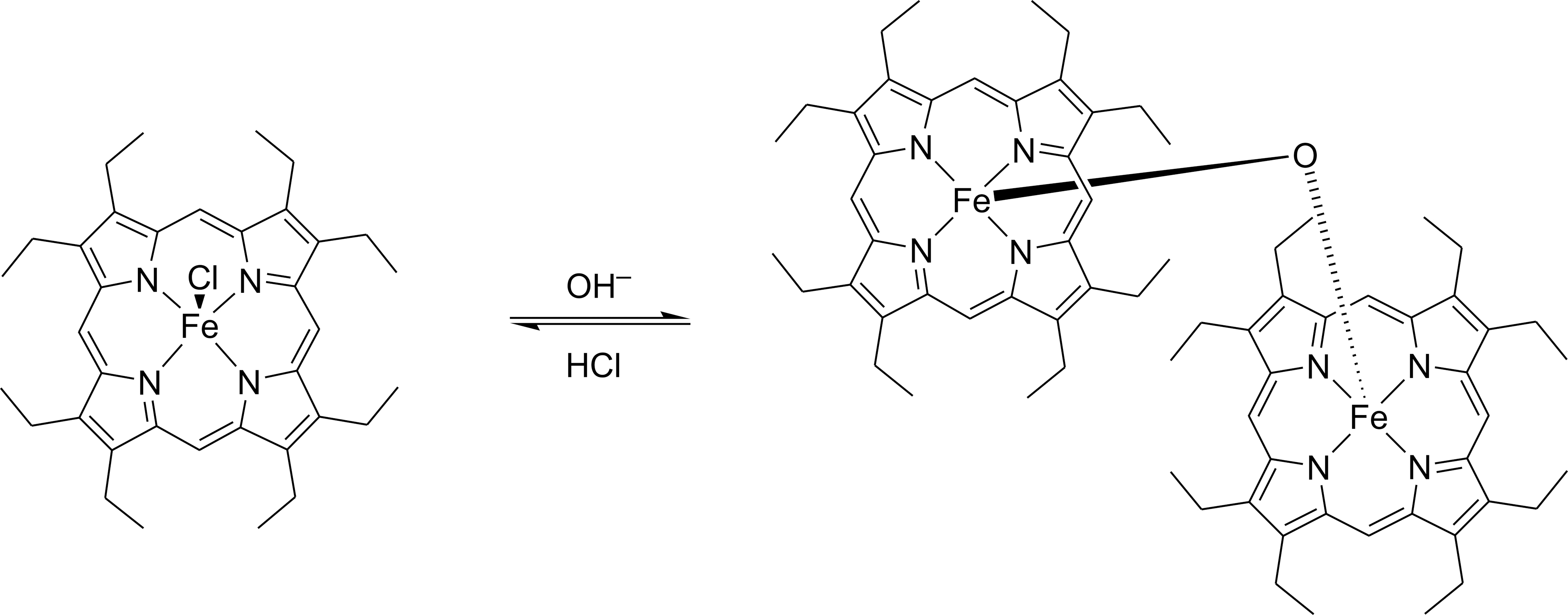
Formation of [{Fe(oep)}2(μ-O)] from [FeCl(oep)] in alkaline solution. The reaction can be reversed by hydrochloric acid.
Our pH experiments show that an iron porphyrin core can withstand a wide range of pH values (0.0–13.5), even at 80°C. Thus, we conclude that iron porphyrin biosignatures may remain intact under extreme pH conditions, at least over short time periods.
Not only can the pH of extraterrestrial environments but also the redox conditions vary. In this context, oxidizing agents, such as perchlorate, have attracted particular attention. Perchlorate salts have been detected in martian soil (Hecht et al., 2009; Kounaves et al., 2010, 2014; Glavin et al., 2013), where they may occasionally be dissolved by thin water films or small amounts of meltwater (Cull et al., 2010). Martian perchlorate chemistry is believed to be linked to the chemistry of hypochlorite and chlorate, which is why these two anions are also believed to occur on Mars (Hanley et al., 2012; Quinn et al., 2013; Carrier and Kounaves, 2015; Clark and Kounaves, 2016).
Perchlorate, and possibly chlorate, were found to be associated with the cations Mg2+, Ca2+, Na+, and K+ (Toner et al., 2014). For our study, we chose aqueous magnesium perchlorate, sodium chlorate, and calcium hypochlorite solutions.
[FeCl(oep)] withstood exposure to magnesium perchlorate solutions for 24 h up to the maximum concentration and temperature used (1 mol/L ClO4 −, 80°C). This seems particularly interesting, because perchlorate may be the most abundant potential oxidant in martian soils (Toner et al., 2014). Chlorate, which is possibly the second most abundant oxidant on Mars, destroyed [FeCl(oep)] only at 80°C. At 0°C and 25°C, [FeCl(oep)] was stable to chlorate concentrations up to 1 mol/L (Fig. 4 and Supplementary Table S3).
Thus, iron porphyrins such as [FeCl(oep)] appear to be quite resistant to perchlorate and chlorate. Hypochlorite was the most effective oxidant of the three oxychlorine species studied. A 1 mol/L solution was capable of destroying [FeCl(oep)] completely within 24 h at 25°C. For comparison, the highest surface temperature measured at the Opportunity rover landing site on Mars was 22°C (Spanovich et al., 2006). The martian daily mean temperature, however, is much lower (around −60°C near the equator) and may not have been substantially higher for most of the past 4 billion years (Shuster and Weiss, 2005).
Hypochlorite yields obtained from radiolysis of calcium perchlorate were relatively low (Quinn et al., 2013), but the sensitivity of [FeCl(oep)] to ClO− suggests that even small concentrations of ClO− may be sufficient to destroy potential (iron) porphyrin biosignatures on Mars, at least over longer periods. Therefore, radiation-protected or perchlorate-free locations appear necessary for long-term preservation of iron porphyrins. The direct effects of ionizing radiation on [FeCl(oep)] will be discussed later. Chlorine dioxide, which is also formed by radiolysis of perchlorate, was not part of this study. As Quinn et al. (2013) have already shown, this compound, in contrast to hypochlorite, does not readily destroy organic molecules.
Nitrate is another species of interest in the context of biosignature stability. Besides its association with volcanic activity on Earth (see above), it has been found in martian sediments (up to a few hundred mg/kg) (Stern et al., 2015, 2017) and in a martian meteorite (Kounaves et al., 2014). Nitrate can act as a strong oxidant in acidic solution (Wiberg, 2001). For Mars, there is evidence from mineralogy that acidic environments existed in the past (Klingelhöfer et al., 2004) and that low-pH conditions dominated chemical weathering (Hurowitz and McLennan, 2007).
With this in mind, we used nitric acid as a model for nitrate in acidic solution and investigated its effects on [FeCl(oep)] at different concentrations (pH values) and temperatures. At 1°C, the iron porphyrin was stable to 1 mol/L nitric acid (i.e., pH 0) for 24 h. At higher temperatures of 25°C and 80°C, partial oxidation was observed (Fig. 4 and Supplementary Table S3). However, at a nitric acid concentration of 0.1 mol/L (i.e., pH 1), [FeCl(oep)] was stable even at 80°C.
The latter pH and temperature conditions are extreme compared with most natural environments on Earth and Mars. From these results, we conclude that “simple” iron porphyrins will be stable to nitrates under moderate conditions of acidity and temperature for periods much longer than the duration of our experiments.
The final oxidant studied was hydrogen peroxide. H2O2 is a widespread molecule, which has been detected, for example, in the interstellar medium (Bergman et al., 2011) and on the icy moons Europa (Carlson et al., 1999a) and, tentatively, Enceladus (Newman et al., 2007). In addition, hydrogen peroxide occurs at low concentrations (ppb) in the martian atmosphere (Clancy et al., 2004; Encrenaz et al., 2004, 2019; Hartogh et al., 2010). The atmospheric hydrogen peroxide may be bound and accumulated in the martian regolith (Encrenaz et al., 2012). It is interesting to note that hydrogen peroxide is generated in a reaction between the disulfide mineral pyrite (FeS2) and oxygen-free water (Borda et al., 2001, 2003; Cohn et al., 2006).
The oxidative degradation of heme and hemin by hydrogen peroxide is well documented (Nagababu and Rifkind, 2004). In addition, it has been reported that the hemin model [FeCl(oep)] is rapidly destroyed by 5% (1.5 mol/L) alkaline hydrogen peroxide solution at 37°C (Schaefer et al., 1985; note that the authors used the formula Fe(III) OEP instead of [FeCl(oep)]). In the latter study, [FeCl(oep)] was dissolved in acetone, whereas in our experiments the solid was used.
We found that the effect of hydrogen peroxide solutions on [FeCl(oep)] is strongly temperature dependent (see Section 3)—an observation also made with the other oxidants (Fig. 4). The temperature at the martian surface, for example, can be high enough for hydrogen peroxide to destroy iron porphyrins, if present, in relatively short periods of time. Of course, the rate of decomposition would largely depend on the H2O2 concentration in the martian regolith, which is unknown. Moreover, interactions with regolith minerals may alter the reactivity of hydrogen peroxide (Quinn and Zent, 1999).
When [FeCl(oep)] was oxidized with hydrogen peroxide, the characteristic reddish-brown color disappeared and the resulting UV-visible spectra showed comparatively weak bands. No attempts were made to identify the products, but it seems likely that cleavage of the porphyrin ring and subsequent breakdown to oxidation products of mono- and dipyrroles occurred, as previously reported for slightly different reaction conditions (Schaefer et al., 1985). It is interesting that the nature of the metal center is key to the oxidation stability of metalloporphyrins.
The Fe(III) complex [FeCl(oep)] is oxidized by hydrogen peroxide under relatively mild conditions, as shown in this study. Schaefer et al. (1985) found similar results for the oep complexes of Co(II) and Mn(III)Cl, whereas the complexes of, for example, Mg(II) and Cu(II), as well as the metal-free porphyrin proved stable to hydrogen peroxide. Finally, we note that hydrogen peroxide is a more effective oxidant for [FeCl(oep)] than chlorate or nitric acid, but it is less effective than hypochlorite.
4.2. Thermal stability
Volcanic activity on rocky planets can cause local high temperatures, which on Earth cover a range of several hundred degrees. For example, the highest temperature measured by Pinkerton et al. (2002) for the surface skin of lava flows on Kilauea volcano, Hawai'i, was ∼900°C, whereas the temperature of solid crusts on lava flows ranged from 425°C to 750°C and the temperature of the inner levee walls of lava channels was between 300°C to 650°C.
The 500°C used in our static temperature experiments is, thus, comparable to the temperatures that were observed in hot volcanic environments. The upper temperature limit of the TGA experiments and their repetition in a tube furnace was 900°C.
Thermogravimetry showed that the thermal stability of [FeCl(oep)] depended on the atmosphere used. In air, the compound combusted above 276°C, leaving hematite as residue. Hematite is an abundant mineral that also forms in volcanic environments. Therefore, the hematite from thermally decomposed iron porphyrins is not particularly useful as a biosignature. In nitrogen and carbon dioxide atmospheres, decomposition of [FeCl(oep)] started at 330°C under TGA conditions. Thus, [FeCl(oep)] has a substantially lower thermal stability than its tetraphenylporphyrinate analogue [FeCl(tpp)], which is stable in air up to ∼380°C (Gokakakar and Salker, 2010).
[FeCl(oep)] is in the middle range of potential biosignatures in terms of thermal stability. For example, the decomposition onset temperatures of the nucleobases guanine and adenine are ∼420°C and ∼300°C, respectively, and the amino acid glycine shows clear decomposition above 234°C (Fox et al., 2015; Abdelaziz et al., 2019). The onset temperatures given were determined by TGA under nitrogen; note that they depend on the heating rate.
It has been shown that the thermal behavior of glycine is strongly affected by minerals and wet–dry cycles (Dalai et al., 2017; Fox et al., 2019). Therefore, it would be worthwhile to also investigate the influence of minerals on the thermal behavior of iron porphyrins; however, this is beyond the scope of this work.
As Fig. 5 shows, the TG curves of [FeCl(oep)] in nitrogen and carbon dioxide are identical up to 350°C. At higher temperatures, the two curves depart from each other because of oxygen uptake from carbon dioxide, a process in which carbon dioxide was reduced to carbon monoxide. At 647°C, rapid combustion started. The final product of the carbon dioxide experiments was hematite (Fe2O3) and thus the same as in air, whereas a completely different residue was obtained under nitrogen. The results were surprising and indicated that carbon dioxide-rich planetary atmospheres can support the combustion of organic molecules even in the absence of free oxygen.
In contrast to the experiments in carbon dioxide and air, the heating of [FeCl(oep)] to 900°C under nitrogen produced a mixture of α-iron, graphite, the iron carbide cohenite, and hematite. The latter is believed to be an artifact of air oxidation of α-iron, which formed after the experiment. The mixture had a high carbon content of 83% (Supplementary Table S4C). We conclude that carbon-rich mixtures of iron, graphite, and iron carbides could be characteristic of iron porphyrins that were thermally decomposed in non-oxidizing atmospheres. However, it is unclear whether such mixtures can be useful as biosignatures, for example in combination with stable carbon isotope measurements.
Based on the TG results, a tube furnace experiment was performed in which [FeCl(oep)] was kept at 500°C under nitrogen for 24 h. At this temperature, the TG curve showed a near plateau after a mass loss of 52% (Fig. 5). It is also interesting to note that temperatures around 500°C occur in active volcanic regions (see above). Compared with the residue obtained by raising the temperature from 50°C to 900°C over ∼3 h (at 5°C/min), the residue of the 500°C experiment had a significantly lower carbon content and higher contents of the other elements, especially nitrogen (Supplementary Tables S4C and S5).
The X-ray diffractogram was also different, showing hematite and carbonaceous material. Since [FeCl(oep)] contains no oxygen, hematite must be considered an artifact, which will not be formed in a completely oxygen-free nitrogen atmosphere (see Section 3). By contrast, the carbonaceous material must be mainly a product of anaerobic thermolysis, except a possible small oxygen content. It is partially amorphous and can be regarded as an intermediate on the route to crystalline graphite that was observed at 900°C (Supplementary Fig. S15C). Interestingly, the molar nitrogen to carbon ratio of the 500°C residue was higher than that of octaethylporphyrin (1:7.1 vs. 1:9), indicating the possible presence of pyrrole rings and/or other characteristic N-containing structures, which may be the subject of future research.
In the tube furnace experiments performed in nitrogen and carbon dioxide, some sublimed [FeCl(oep)] was recovered from cooler parts of the apparatus. It is known that porphyrins, such as H2oep and H2tpp, and metalloporphyrins can sublime, but sublimation is usually carried out under high vacuum conditions (Erdman et al., 1956; Gruhn et al., 1999). In our experiments, sublimation was observed at ambient pressure. It is, therefore, plausible that certain metalloporphyrins may sublime intact in hot, oxygen-free natural environments and thereby (partly) escape thermal decomposition.
A further series of experiments was performed in which samples of [FeCl(oep)] were sealed in evacuated glass ampoules to prevent sublimation. The samples were heated at different constant temperatures for 24 h. Decomposition started at ∼250°C, which is 80°C lower than in the TGA experiments in nitrogen and carbon dioxide. It is well known that in rising temperature (TGA) experiments thermal processes are delayed compared with isothermal conditions (i.e., they are observed at higher temperatures).
However, the observed difference in the onset temperatures seems too large to be explained solely by this effect. It appears that confinement of the sample and/or low pressure facilitate the decomposition of [FeCl(oep)]. If confinement is an important factor, this would be relevant to the thermal decomposition of metal porphyrins embedded in, for example, volcanic ash or sedimentary rocks.
4.3. Stability to UV-visible radiation
During the UV-visible experiments, air was excluded to avoid the formation of reactive oxygen species, such as singlet oxygen and ozone. Many astrobiologically interesting places are either devoid or nearly devoid of free oxygen. For example, on Mars, the mean O2 mixing ratio is ∼1.7 × 10−3 and the mean surface pressure is 6 mbar (Catling and Kasting, 2017). Thus, the partial pressure of O2 is 0.01 mbar. This corresponds to 10 vol ppm in a 1 bar atmosphere—which is considered “oxygen-free” in many (though not all) ambient pressure experiments in terrestrial laboratories. The following discussion will focus on Mars' surface, but, of course, the experimental results are relevant to UV-irradiated environments in general.
It is well known that UV radiation can alter the structure of molecular biosignatures by photoionization and bond cleavage, but little work was done on the photostability of metalloporphyrins (e.g., Sobbi et al., 1993). In the present study, [FeCl(oep)] was exposed to the unfiltered radiation of a xenon short-arc lamp (230 nm to IR). The resulting decomposition curve is shown in Fig. 7. The UV intensity in our experiments was lower than the UV intensity on Mars.
Using the model of Cockell et al. (2000) and assuming that UVB and UVC are responsible for the decomposition, we estimate that 24 h irradiation in the laboratory corresponds to ∼5 h irradiation on present-day Mars and ∼14 h on early Mars 3.5 Ga ago. The corresponding hypothetical decomposition curves for [FeCl(oep)] under martian UV conditions are shown in Supplementary Fig. S23.
The photodecomposition of solid [FeCl(oep)] followed neither zero-order, first-order, nor second-order kinetics. Instead, its time course could be explained by a bounded decay model with a boundary of ∼86% (i.e., the decomposition was limited to ∼14%). This unusual behavior was due to the formation of a brown, insoluble, UV blocking solid that protected the [FeCl(oep)] beneath from further decomposition. This kind of self-protection of [FeCl(oep)] requires that sufficient starting material is available to allow the protective layer to become thick enough before all [FeCl(oep)] is decomposed.
The decomposition product was probably a polymer, which unfortunately could not be analyzed in detail because the amounts were too small. It may also protect [FeCl(oep)] against other destructive influences. Further experiments are needed to test this possibility.
We estimate that without the formation of the protective substance, [FeCl(oep)] would have been completely destroyed within ∼64 days. The estimate is based on the assumption of zero-order kinetics (see Section 3). Zero-order kinetics are relevant for tiny crystals or thin films of [FeCl(oep)], where the product layer cannot become thick enough to exert a significant protective effect. The period of ∼64 days necessary for (hypothetical) complete photolysis in our experiment corresponds to ∼14 days under present-day martian UVB and UVC conditions (see above). Here, it is worth noting that thin layers of regolith can provide effective shielding from UV radiation (Mileikowsky et al., 2000; Cockell and Raven, 2004).
Cook et al. (2014) exposed thin films of [FeCl(tpp)] to solar radiation in low Earth orbit and to simulated solar radiation from an Xe arc lamp combined with an H2/He discharge lamp. As mentioned in Section 1, decomposition was observed in all their experiments. [FeCl(tpp)] partially survived 18,000 MJ/m2 of filtered solar radiation (>∼170 nm) in dry atmospheres but was completely destroyed by ∼17,000 MJ/m2 in a humid argon atmosphere (estimated from Fig. 5 in Cook et al., 2014).
These radiant exposure values are considerably higher than the 220 MJ/m2 estimated to be necessary for complete decomposition of [FeCl(oep)] in the absence of the protective substance. Clearly, [FeCl(oep)] is much more susceptible to UV radiation than [FeCl(tpp)]. The explanation for this difference probably lies in the fact that the meso-positions of [FeCl(oep)] are unsubstituted and, thus, more accessible. Indeed, Cook et al. (2014) showed that loss of the meso-phenyl groups of [FeCl(tpp)] preceded the destruction of the porphyrin core itself, at least in humid atmosphere.
Self-protection, as observed for [FeCl(oep)], was not reported for [FeCl(tpp)]. The reason could be that thin films were used or that [FeCl(tpp)] does not form protective decomposition products.
4.4. Stability to X-rays
In addition to UV radiation from the Sun, diffuse X-rays, gamma rays, and particle radiation (galactic and extragalactic cosmic rays, solar particle radiation) can damage molecular biosignatures. Therefore, we also studied the effects of X-rays and charged particle radiation on [FeCl(oep)]. Ionizing radiation in the form of gamma rays and X-rays reaches the solar system from outside, for example caused by gamma-ray bursts (e.g., Mészáros, 2002). On planets, this type of radiation is produced during radioactive decay, as secondary radiation, and in terrestrial thunderstorms (Dwyer et al., 2008). Martian dust storms are also believed to produce gamma and X-rays (Arabshahi et al., 2017).
Neat [FeCl(oep)] proved to be quite resistant to X-rays with an effective energy of 68 keV. Only 5.5% were decomposed even at the highest dose of 10 kGy (Fig. 8). We estimate that ∼215 kGy would be required to completely decompose the compound. For comparison, the dose rate of natural radioactivity of martian rock has been calculated to be on the order of 0.04 cGy/year or 0.4 kGy/106 years (Mileikowsky et al., 2000). There are other potential molecular biosignatures that are even more resistant. Purines and pyrimidines, for example, have been shown to be very stable under gamma irradiation (Hammer et al., 2019). In most cases, these compounds survived a dose of 992 kGy without significant decomposition. Interestingly, the decomposition of guanine was dose-rate dependent. Whether there is a dose-rate dependence in the case of [FeCl(oep)] was not studied by us.
[FeCl(oep)] was also X-irradiated in a sodium chloride/sodium hydrogencarbonate salt matrix, which was used to mimic the salt composition of Enceladus' plumes (Postberg et al., 2009). If life exists in the subsurface ocean of Enceladus, molecular biosignatures and even whole organisms may be ejected in the plumes and subsequently embedded in the salts on the Moon's surface. The decomposition of neat [FeCl(oep)] started at 5 kGy, whereas in the salt matrix no decomposition was observed up to the maximum dose of 10 kGy (Supplementary Table S6).
This clearly shows that the salts protected [FeCl(oep)] against X-rays. It would be interesting to evaluate in future studies how effective the protection is at higher X-ray doses.
Teodoro et al. (2017) have calculated the ionizing radiation dose at and a few meters below the surface of Enceladus, assuming the presence of pure water ice. They estimated that in the upper 1 m layer, complex biomolecules (e.g., proteins) would be largely decomposed within a few million years and simple organic molecules (e.g., amino acids) within less than 100 million years. These figures do not take into account the effect of salts (see above) and, in particular, the fact that fresh biomolecules would be frequently ejected. According to the calculations of Teodoro et al. (2017), 10 kGy—the dose at which [FeCl(oep)] was still stable to X-irradiation in the salt matrix—will be reached at the surface after ∼30,000 years. From these results, one can conclude that there is a realistic chance of finding intact iron porphyrins on Enceladus' surface if they are present in the plumes.
In the presence of O2 and water, ionizing radiation generates reactive chemical species such as the hydroxyl radical. These species can lead to oxidative decomposition of organic molecules (e.g., Garrison, 1987). As our X-irradiation experiments were conducted under ambient atmosphere, the stability of [FeCl(oep)] might, therefore, be greater in an oxygen- and water-free environment.
Stability enhancement by drying has recently been demonstrated for carotenoids in cells of a cyanobacterium (Baqué et al., 2020). After exposure to 113 kGy gamma irradiation, the carotenoid Raman band at 1516 cm−1 had lost only 20% of its initial intensity in dry cells, but 91% in hydrated cells. We extrapolated that [FeCl(oep)] is half decomposed by an X-ray dose of ∼107 kGy (see Section 3). It is, thus, of comparable stability to the cyanobacterial carotenoids.
4.5. Ion bombardment experiments
The nuclear component of galactic cosmic rays consists of ∼87% protons, ∼12% helium nuclei, and ∼1% heavier nuclei (Simpson, 1983; Mewaldt, 1994). The particle energies are high, ranging from ∼10 MeV to >103 GeV with a peak at ∼200–400 MeV (Badhwar, 1997). When cosmic rays reach the surface of a rocky planet, the dose maximum is sub-surface. For example, on Mars, the dose rate at the surface is 76 mGy/year, at 10 cm depth 96 mGy/year and at 2 m depth 8.7 mGy/year; the first value was measured by the Radiation Assessment Detector on the NASA Mars Science Laboratory, and the latter two were calculated (Hassler et al., 2014).
The helium and iron ions used at HIMAC had energies of 145.2 and 418.3 MeV, respectively, which were in the peak region of the energy distribution of galactic cosmic rays (Badhwar, 1997). [FeCl(oep)] survived a dose of 1000 Gy of helium ions without decomposition (Fig. 9). For iron ions, a higher maximum dose of 3000 Gy was available. At this dose, 7.2% of the [FeCl(oep)] were destroyed (Fig. 10). On the basis of these results and the data of Hassler et al. (2014), we can estimate that [FeCl(oep)] would remain largely intact 2 m below the martian surface for at least 350,000 years, the time after which a dose of 3000 Gy has been accumulated.
This estimate may give some idea of the radiation stability of iron porphyrins in the martian soil; however, it does not include the composition of the cosmic rays. Note that 2 m is the maximum depth that the drill of the ExoMars Rosalind Franklin rover can reach (Vago et al., 2018). Our experimental results do not allow us to predict the time necessary for complete decomposition of [FeCl(oep)] by cosmic radiation in the martian soil. For this, data at doses higher than 3000 Gy would be required to estimate the shape of the dose–decomposition curve.
We also studied whether a sodium chloride/sodium hydrogencarbonate matrix (the “Enceladus salt mixture,” see above) affects the stability of [FeCl(oep)] to helium and iron ion irradiation. As can be seen from Figs. 9 and 10, the salts had no effect. In particular, there was no significant difference in the degree of decomposition between salt-embedded and neat [FeCl(oep)] after exposure to 3000 Gy of iron ions (6.6% vs. 7.2%). Thus, in contrast to the experiments with X-rays, no protective effect of the salt matrix was observed.
At the surface of the Jovian satellite Europa, iron porphyrins and other possible biosignatures would be exposed to an extreme radiation environment (Paranicas et al., 2009). Assuming a water ice surface, a dose rate of ∼30–60 MGy/year, depending on hemisphere, is expected at 1 μm depth (estimated from supplementary Fig. 1 in Nordheim et al., 2018). Thus, 3000 Gy, the dose at which [FeCl(oep)] showed initial signs of decomposition, are accumulated in 1 h or less.
From this, one may infer that it would hardly be possible to find intact iron porphyrins at Europa's surface, even though the charged particles on Europa and in our experiments are different. Using the example of amino acids, Nordheim et al. (2018) showed, however, that at depths below ∼1–20 cm (depending on hemisphere and latitude) molecular biosignatures should be able to survive sufficiently long to be detectable. On Europa, the stability of organic molecules will also depend on the chemical environment resulting from irradiation, in addition to the direct radiation exposure (Carlson et al., 2009). Radiation-produced oxidants, such as H2O2, are especially important here (Carlson et al., 1999a; see Section 4.1).
One should note that the irradiation experiments described in this article were performed under fixed environmental conditions (e.g., at or near room temperature). However, it could well be that the irradiation behavior of [FeCl(oep)] depends on environmental parameters. This would not be unusual for an organic compound. For example, when the nucleobase thymine was exposed to proton radiation in water ice, it was found that the decomposition rate constant depended on the temperature and the thymine to water ratio (Materese et al., 2020).
5. Conclusions
We investigated the stability of the porphyrinato complex [FeCl(oep)], which is a good model for iron porphyrin biosignatures. Solid [FeCl(oep)] was exposed to a variety of astrobiologically relevant extreme conditions, namely: (1) aqueous acids and bases, (2) oxidizing agents, (3) heat, (4) UV-visible radiation, and (5) ionizing radiation. Under some conditions, [FeCl(oep)] proved completely stable, whereas under others the compound was either partially or completely decomposed.
Even though there is no universal stability scale that can be applied here, our results allow an assessment of the stability limits of iron porphyrins in extraterrestrial environments: Extreme pH values do not seem to pose a hazard to simple iron porphyrins such as [FeCl(oep)]. For example, no decomposition was observed with nonoxidizing acids at pH 0 and temperatures up to 80°C. Under sufficiently alkaline conditions (e.g., in the presence of sodium carbonate solution of pH 11.6), a dimerization reaction occurred, but the porphyrin biosignature remained fundamentally intact. Thus, iron porphyrin biosignatures can withstand the high pH of Enceladus' ocean and also low-pH conditions of volcanic origin on terrestrial planets, at least for short periods. At low temperature (1°C), the oxidizing agents used in this study did not react with [FeCl(oep)] within 24 h. At higher temperatures (25°C and 80°C), however, only perchlorate was unreactive, whereas hypochlorite was an effective oxidant, followed by hydrogen peroxide. Some decomposition was also noted with chlorate and nitric acid. Not unexpectedly, higher oxidant concentrations resulted in increased decomposition of [FeCl(oep)]. Our results show that on Mars the generally low surface temperatures will slow the oxidation of iron porphyrins. Nevertheless, hypochlorite and hydrogen peroxide may have high enough concentrations in the martian regolith to be detrimental to the stability of iron porphyrin biosignatures. The same could be true for hydrogen peroxide on Europa and Enceladus, where, however, surface temperatures are much lower. By contrast, direct oxidation by martian perchlorate and chlorate appears less problematic. [FeCl(oep)] was found to be moderately thermally stable for a small biosignature molecule. Our TG measurements showed that the compound was stable up to 276°C in air. Under non-oxidizing conditions, decomposition started at 330°C in TG and at ∼250°C in static temperature experiments. Carbon dioxide did not affect the onset of the decomposition compared with a nitrogen atmosphere. However, interestingly, at higher temperatures it became an oxidant, resulting in the same final decomposition product of [FeCl(oep)] as in air, namely hematite (Fe2O3). Although hematite is not specific enough, the carbon-rich mixture of iron, graphite, and iron carbides that forms at high temperatures in a pure nitrogen atmosphere could serve as an indicator of thermally decomposed iron porphyrins. However, as a major volcanic gas, carbon dioxide will often be present in significant amounts in the atmospheres of rocky planets. Finally, it is interesting that sublimation may allow certain metalloporphyrins to escape thermal decomposition, at least partly. [FeCl(oep)] proved to be sensitive to UV radiation ≥230 nm. The photodecomposition was slowed by the formation of a UV-absorbing product layer. This process can be regarded as a kind of self-protection, which may also occur with other solid molecular biosignatures. To our knowledge, this potentially widespread UV protection mechanism has not been systematically studied. A sodium chloride/sodium hydrogencarbonate mixture that mimics the salt composition of Enceladus' plumes protected [FeCl(oep)] against ionizing electromagnetic radiation (68 keV effective-energy X-rays). Our observations, together with results by Teodoro et al. (2017), suggest that plume-delivered iron porphyrins may survive long enough to be detectable at the surface of Enceladus. We also studied the effect of ionizing particle radiation on [FeCl(oep)]. The compound partially decomposed on irradiation with 418.3 MeV iron ions at a dose of 3000 Gy. Here, the salt mixture had no protective effect. We estimate that 2 m below the martian surface, [FeCl(oep)] would show initial signs of decomposition by cosmic rays after a few hundred thousand years. This suggests that on Mars, potential ancient iron-porphyrin biosignatures will only be preserved in greater depths.
This study focused solely on the effects of single factors. However, in natural environments, often two or more of these factors act together, and their combined effect may differ from the sum of the individual effects. It has been shown, for example, that NaClO3 and NaClO4 can either increase or decrease the UV decomposition rate of amino acids, depending on the amino acid and the humidity conditions (Liu and Kounaves, 2021). It is conceivable that similar (positive or negative) synergistic effects may occur for [FeCl(oep)].
Finally, our results may also be relevant in the context of prebiotic chemistry. It has been suggested that abiotically produced iron porphyrins could have played a role in the origin of life and existed, for example, on primordial volcanic islands (Section 1). It is reasonable to assume that radiation was an issue for prebiotic iron porphyrins as it was for [FeCl(oep)] in our experiments.
However, as shown, a decomposition layer can attenuate UV radiation and inorganic salts can protect against X-rays. The UV shielding by sulfur and organic haze must also be considered (Cockell, 2002). Other environmental conditions on primordial volcanic islands would have been less of a problem, namely low pH values and elevated temperatures (at least up to near 250°C). Although reasonable pathways to hydrogen peroxide on the early Earth have been identified (Borda et al., 2001, 2003; Haqq-Misra et al., 2011), under the prevailing reducing conditions strong oxidants were probably present in too low concentrations to damage iron porphyrins such as [FeCl(oep)].
Footnotes
Acknowledgments
The authors thank Claudia Görgen, Sonja Ringer, and Marius Sannemann for assistance with sample preparation and Hanna Müller for help using SAS software. Special thanks are due to Prof. Reinhard Kohlus and Theresa Anzmann (both at the University of Hohenheim, Stuttgart) for the opportunity to perform particle size measurements and their help with the measurements. The authors are also grateful to Prof. John M. Boone (University of California, Davis) for providing the spreadsheet for calculating the X-ray spectrum. H.L.P. gratefully acknowledges the receipt of an LGFG doctoral fellowship of the State of Baden-Württemberg.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
A.F. and R.M. received support by the MEXT Grant-in-Aid for Scientific Research on Innovative Areas “Living in Space” (Grant Number: 15K21745). R.M. was supported by DLR via the ISS LIFE project funding (Programm RF-FuW, Teilprogramm 475).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Figure S15
Supplementary Figure S16
Supplementary Figure S17
Supplementary Figure S18
Supplementary Figure S19
Supplementary Figure S20
Supplementary Figure S21
Supplementary Figure S22
Supplementary Figure S23
Abbreviations Used
Associate Editor: Christopher McKay
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.