
Abstract
It is now routinely possible to sequence and recover microbial genomes from environmental samples. To the degree it is feasible to assign transcriptional and translational functions to these genomes, it should be possible, in principle, to largely understand the complete molecular inputs and outputs of a microbial community. However, gene-based tools alone are presently insufficient to describe the full suite of chemical reactions and small molecules that compose a living cell. Metabolomic tools have developed quickly and now enable rapid detection and identification of small molecules within biological and environmental samples. The convergence of these technologies will soon facilitate the detection of novel enzymatic activities, novel organisms, and potentially extraterrestrial life-forms on solar system bodies. This review explores the methodological problems and scientific opportunities facing researchers who hope to apply metabolomic methods in astrobiology-related fields, and how present challenges might be overcome.
1. Introduction
Linking discrete molecular compounds to biological activity in the search for extraterrestrial life has been a priority since the time of the Viking lander (Klein et al., 1976). Mars has been the main target of this effort by virtue of its proximity to the Earth, and the perceived similarity of Mars to Earth has heightened the interest, although the following discussion can be related to other extraterrestrial environments as well. For over 40 years, attempts have been made to measure discrete organic compounds on Mars (Biemann et al., 1977; Eigenbrode et al., 2018). Future missions to Mars and the outer planets will include mass spectrometers capable of at least constraining mass distributions of small organic compounds (Sephton et al., 2018). Recently, the Curiosity rover confirmed the presence of organic compounds in Mars' shallow subsurface (Eigenbrode et al., 2018). The identities of these compounds are not yet known, and their source is likewise unclear.
Laboratory experiments and field observations have shown that small organic compounds (e.g., amino acids, carboxylic acids, and nucleotides), including molecules known to be essential for terrestrial microorganisms (Miller, 1953; Eschenmoser and Loewenthal 1992; Cleaves, 2012), can be synthesized abiotically. The ability to distinguish biotic from abiotic molecules is therefore crucial in the search for life on other worlds. Ideally, given that life seems to be a phenomenon that combinatorially generates and then explores huge chemical spaces, these searches need to be as open as possible (Cleland, 2019).
Many advances in the rapid analysis of metabolites (small organic molecules, generally <1000 Da) for medical diagnostics are now being used in the environmental sciences, enabling the correlation of environmental genetic information with biochemical data (Abram, 2015; Cao et al., 2019). However, various phenomena currently complicate intentions to link genomic and small-molecule data for astrobiological exploration. For example, the geological conditions that favor DNA survival in the environment are fairly well understood. The maximum lifetime of useful DNA sequence data does not exceed one million years (1 Ma) even under the most favorable circumstances on Earth (Allentoft et al., 2012; Hofreiter et al., 2015), although claims have been made for the recovery of much older (∼100 Ma) sequences (Inagaki et al., 2005). Thus, the potential for linking genomic information to molecular biomarkers as a function of time is only possible for the very recent paleontological record even on Earth, where life is abundant, and samples are easy to obtain.
In the context of the detection of potential extraterrestrial biochemistries (Giri et al., 2018), there are several possibilities for carbon-based life: (1) such life-forms use conventional nucleic acids as a repository of genetic information, in which case conventional sequencing methodologies may be useful for solar system exploration (Mojarro et al., 2017); (2) such life-forms use alternative nucleic acids as a repository for genetic information (Cleaves et al., 2015) in which case novel sequencing methods would be required (Carr et al., 2016); (3) such life-forms use non-nucleic acid polymers as genetic molecules (Sowerby and Petersen, 2002); or (4) such life-forms do not depend on conventional notions of molecule-based genetic inheritance (Segré et al., 2000). It should be noted that even if the first possibility is true, it may be very unlikely that alien biochemistries would use the same genetic code or coded amino acids (or indeed that nucleic acids and proteins will be universally paired biochemical features), have cognate genes or protein folds, or use similar metabolic pathways. All of these phenomena may be deeply contingent aspects of terrestrial biological evolution.
Even if conventional or nonconventional nucleic acids prove too fragile to survive in extraterrestrial environments (or to be made in the first place), many other types of biomolecules could provide information about past biology (Briggs and Summons, 2014). In the four cases mentioned above, extraterrestrial metabolism could be based on similar or dissimilar pathway transformations and/or compounds, and novel metrics for distinguishing living versus degraded versus abiogenic organic compounds would be required.
Metabolomics, the study of the intermediates and end products of metabolism, or metabolites, produced by living cells, is rapidly revolutionizing the understanding of biochemistry (Aldridge and Rhee, 2014). Environmental metabolomics, which uses the techniques of metabolomics to describe the interaction between organisms and their environment, offers promising tools for life detection in the Solar System beyond the Earth. The techniques used to identify metabolites in modern microbial communities can assist in detecting quantifiable substances whose presence are indicative of ancient or extinct life, that is, biomarkers, depending on the preservation of the original biomarkers. Furthermore, inasmuch as organic compound-based biochemistry will almost certainly involve controlled transformations of specific, relatively low-molecular-weight compounds (Hoehler et al., 2018), metabolomic techniques offer an unbiased (Johnson et al., 2018; Chan et al., 2019) way to search for alien biochemical components, independent of known biological pathways that are encoded by genes and carried out by protein-based catalysts (enzymes). Here we review some contemporary environmental metabolomic methodologies and propose how they could be tailored for astrobiological purposes.
2. The “Omics” Revolution and the New Frontier of the Metabolome
Biology is currently undergoing a rapid and expansive “omics” revolution (Kuska, 1998), which focuses on multiple classes of living systems' molecular components and integrates different types of data to obtain a more comprehensive understanding of biological systems as a whole. The list of omics disciplines is ever-increasing. Several omics disciplines encapsulate steps in the central dogma of molecular biology: DNA is transcribed to RNA, which is translated to protein (Crick, 1958). Discoveries at each level of this information-transfer can help elucidate an organism's holistic metabolic capabilities (Fig. 1).
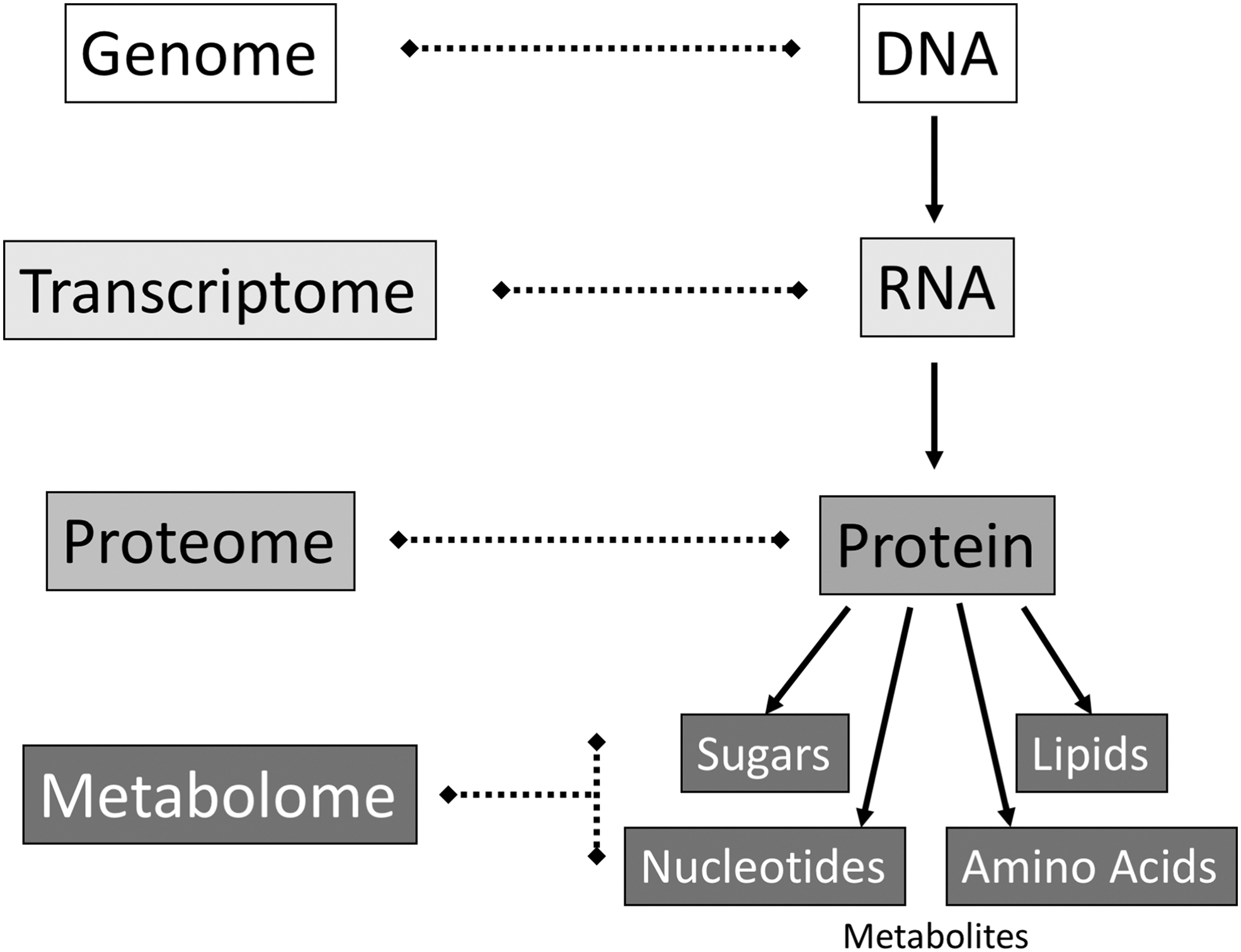
The central dogma of molecular biology and its corresponding omics disciplines.
With regard to astrobiology, these omics techniques have provided insights into the survival capabilities of microbes living in Earth environments that resemble potentially habitable environments on other worlds (Rothschild and Mancinelli, 2001). As stated above, however, extraterrestrial life may not use DNA or RNA to store genetic information, and even if it does, the lability of nucleic acids provides a narrow temporal detection window following an organism's demise. It may therefore be helpful to seek out compounds or physical phenomena that may serve as longer lasting, process-agnostic biomarkers on other worlds (Chan et al., 2019). Analyzing the suite of small metabolic products of living organisms—the metabolome—may help constrain and direct the search for such biomarkers.
Metabolomic investigations aim to characterize the complexity of biological molecules in samples with minimum preparation. This can be a daunting task considering that cells may contain a mixture of many thousands of metabolites, over a large range of abundances (Zamboni et al., 2015). Only recently have analytical technologies been specifically developed for this purpose, and many challenges remain. Mass spectrometry (MS), in particular, now enables simultaneous detection of thousands of putative metabolites from minimal amounts of sample (Bowen et al., 2011; Patti et al., 2012; Zamboni et al., 2015). MS-based metabolomics have been used to demonstrate the existence of complete pathways not detected by genome annotation (Tang et al., 2009), to demonstrate the function of theoretical pathways (Peyraud et al., 2009), to discover novel metabolic pathways (Fürch et al., 2009; Liu et al., 2016), and to describe novel biomarkers associated with disease (Sabatine et al., 2005; Wang-Sattler et al., 2012). Thus, even if we are ignorant of a particular extraterrestrial biology's “genetic” system, we may be able to infer something about its metabolic capabilities via the distributions of its detected metabolites.
3. Metabolomics and the Search for Biomarkers
Although extraterrestrial metabolisms could be wildly different from terrestrial ones, the search for biomarkers on other worlds might begin with the identification of molecules that are diagnostic of metabolic pathways in analog environments on Earth. It might be expected that there would be some overlap of at least some “switchboard” compounds such as those found in the various incarnations of the tricarboxylic acid or other carbon-fixation cycles (Braakman and Smith, 2012a, b).
Few extraterrestrial bodies in the Solar System host environments are considered “clement” on Earth, although environments with conditions overlapping those considered “extreme” on Earth may abound. Generally, environments are considered “extreme” on Earth when they are characterized by conditions (e.g., of pH, temperature, salinity, pressure) that are inhospitable to humans (Rothschild and Mancinelli, 2001). Metabolomic profiling of microbial communities in such environments can assist in understanding metabolite preservation as well as microbial community adaptation to extreme conditions and response to environmental change (Blanchowitz et al., 2019). However, characterizing organisms, genomes, and metabolomes from extreme environments is challenging due to limited genomic data (Hedlund et al., 2014) and the difficulty of interpreting fragmented sedimentological and geobiological data (Hodgson et al., 2018).
No standard environmental metabolomic protocol is ideal for applying metabolomics to all extreme environments; protocols are usually both sample and environment specific (Riekeberg and Powers, 2017). In general, extremophilic communities and their mesophilic counterparts differ with respect to the number of microbial species present and their phylogenetic diversity (Mesbah and Wiegel, 2012). Extreme settings typically contain a relatively low diversity of microbial species, all adapted to the dominant ecological stress of their environment (Stahl et al., 1985; Liu et al., 2014; Sharp et al., 2014; Poli et al., 2017). While species diversity is low, these environments can have extremely high abundances of selected species (Cowan et al., 2002; Brazelton et al., 2006; Kubo et al., 2011). The metabolome of a community of interest can be compared with that of the dominant species observed in situ (if it can be cultured in isolation), or the metabolome can be searched for intermediates or products suggestive of the metabolic pathway(s) under investigation (Zamboni et al., 2015). This allows for novel species to be associated together based on common metabolites and metabolomes, and can also aid in describing key metabolic pathways (Maifiah et al., 2017). Distinct organisms adapted to similar extreme environments may have many shared metabolites (Poli et al., 2017), which could act as unique signatures of that environment and its associated adaptive metabolisms.
A reliance upon shared metabolic products results in a conundrum for astrobiological assessment. Truly unbiased biosignature searches (i.e., independent of genes, proteins, or pathways) may either yield no shared metabolites that can be used for eventual targeted analyses, or there may be simple, highly conserved metabolites associated with central metabolism (housekeeping pathways that are common across all domains of life) that can yield many downstream compositional possibilities. For example, low-molecular-weight organic acids act as intermediates in multiple central metabolism pathways, including amino acid metabolism, nucleic acid synthesis, and carbon fixation, but these compounds can also be produced abiotically through geochemical reactions (McDermott et al., 2015). These outcomes make it difficult to distinguish between biotic versus abiotic compounds. In addition, many, if not most, primary metabolites are ions at physiological and environmental pH values, which facilitates their retention inside cells (Bar-Even et al., 2011), and many metabolites, especially the most abundant ones (e.g., amino acids, cofactors, and nucleotides), are common across a wide variety of organisms (Peregrín-Alvarez et al., 2009). Secondary metabolites, in contrast, are often more species-specific, and often more likely to diffuse into the surrounding environment (Breitling et al., 2013; Covington et al., 2016).
The molecules present in microbially inhabited environmental samples are likely to be a mix of two types: (1) compounds that are being actively metabolized and synthesized, and are therefore relevant to extant microbes and their communities, and (2) compounds that are recalcitrant to microbial reuse, and may be ancient remnants of communities. This simple distinction may allow for the identification of active biology, as an abundance of chemically labile compounds may suggest the existence of extant organisms. Active microbial communities might be expected to effectively maintain low steady-state concentrations of certain metabolites, and compounds that can accumulate and dominate geochemical analyses may do so because the microbiome cannot consume them (Kleber, 2010).
Organisms construct themselves from many types of organic compounds, which are out of thermodynamic equilibrium with the environment (Ornes, 2017). When organisms die, their components remineralize. Thermodynamics (as well as the kinetics of the associated reactions), rather than genetic capability, will ultimately constrain the ability of microbiomes to effect environmental organic transformations. Thus, one might expect the geochemical context to determine the distribution of organic compounds in environmental settings, rather than the composition of the microbial community (Louca et al., 2019). For example, long-term geological burial of organic carbon (e.g., petroleum, kerogen, coal) requires a lack of oxidants during burial (Berner, 2003). Understanding the genomic, metabolic, and geochemical processes that allow biological carbon to evade remineralization is therefore important. Specific organic molecules could be markers of the limits of remineralization under specific Eh/pH conditions. Assuming extraterrestrial organisms share some common metabolites with terrestrial ones, pulse-chase experiments, in which microbial communities are exposed to a “pulse” of stable isotope-labeled compound followed by a “chase” of the same compound in an unlabeled form, could provide a way to distinguish between active and recalcitrant compounds, provided the organisms in the sample metabolize the pulse feed rapidly and efficiently. Lability of organic compounds can be also assessed with analytical techniques discussed further on in this review, including nuclear magnetic resonance (NMR) spectroscopy (Knicker, 2004) and Fourier transform ion cyclotron resonance mass spectrometry (FTICRMS) (D'Andrilli et al., 2015).
4. An Overview of Metabolomic Methods
Metabolomic experiments generally follow one of two approaches: targeted or untargeted. In targeted metabolomic studies, a suite of metabolites are preselected for analysis and then quantified by using standards. In untargeted studies, as many metabolites as possible are measured from samples without preconceived notions of which compounds may be present (Patti et al., 2012; Baig et al., 2016). Untargeted approaches would likely be most valuable to the discovery of novel biomarkers in astrobiological applications.
All metabolomic methods seek to detect and quantify metabolite profiles within organisms or communities. No one method is ideal for all applications or compound classes, and each has its advantages and disadvantages (Aldridge and Rhee, 2014; Kido Soule et al., 2015). NMR-based methods can structurally elucidate and quantify metabolites with minimal sample preparation, but require large samples due to lower sensitivity than MS-based methods. (In addition, the size and power requirements of NMR instruments likely preclude the possibility of including NMR on a spacecraft.) In contrast, MS-based methods can detect thousands of metabolites over a large abundance range, but are limited in their ability to provide structural information for annotation (Aldridge and Rhee, 2014). In brief, NMR-based methods are ideal for structural characterization, while MS-based methods are better suited to detecting large suites of metabolites. A summary of methods is provided in Fig. 2.
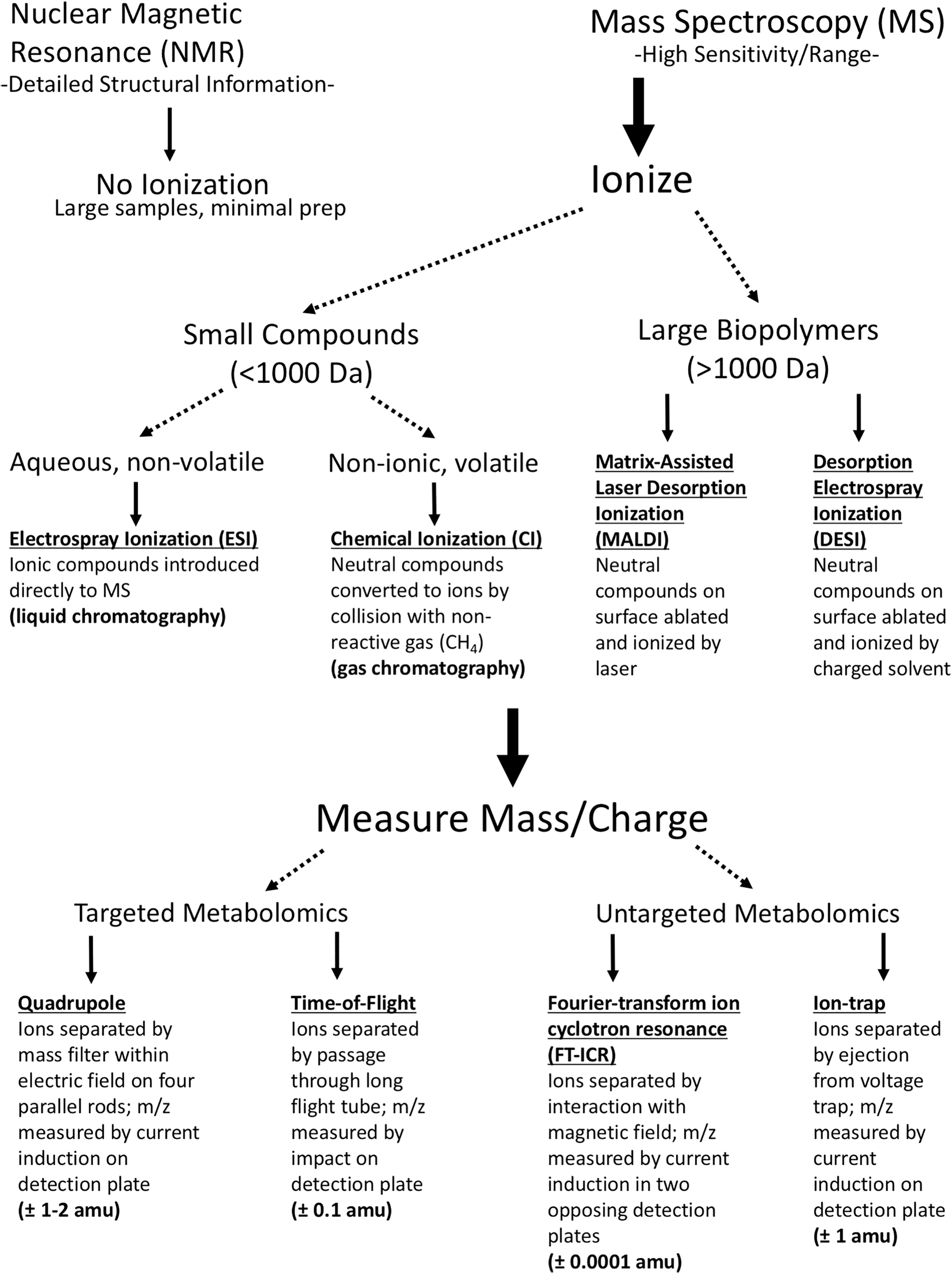
A summary of metabolomic methods.
MS-based methods require ionization of metabolites before analysis. The most common ionization methods are electrospray ionization (ESI), chemical ionization (CI), matrix-assisted laser desorption ionization (MALDI), and desorption electrospray ionization (DESI). ESI ionizes molecules that occur as ions within aqueous or polar solvent solutions, and thus is used to couple liquid chromatography with mass spectrometry (LC/MS). LC/MS allows for the detection of the greatest number of metabolites (Jonsson et al., 2005; Schrimpe-Rutledge et al., 2016), and can resolve metabolites in their native state from complex mixtures containing thousands of analytes ranging over 10,000-fold differences in abundance (Aldridge and Rhee, 2014). This approach, however, does not ionize neutral (nonionic) or volatile metabolites, such as hydrocarbons. Instead CI such as that in gas chromatography is used to introduce these molecules into a mass spectrometer. Large biopolymers such as proteins or polysaccharides can be ionized by MALDI or DESI before MS (MALDI-MS) (Edward and Kennedy, 2005).
Many metabolites are polar and water-soluble, and thus amenable to LC/MS (Aldridge and Rhee, 2014; Kido Soule et al., 2015). Detection and identification of metabolites rest on the choice of mass spectrometer used in these studies. Targeted metabolomic studies emphasize sensitive quantification over comprehensive analysis and thus often use quadrupole-based mass detection or time-of-flight mass spectrometers. These mass spectrometers are sensitive to changes in ion abundance, but have limited mass resolution for detecting molecules with small changes in molecular mass. Untargeted metabolomic studies, in contrast, often leverage high mass resolution to deconvolute complex mixtures and thus rely on Fourier transform-based mass analyses such as Orbitrap or FTICRMS. In each of these techniques, structural identification rests on tandem fragmentation spectra of selected ions, while quantification, where possible, requires authentic standards.
Annotation of metabolite “features” (ions with unique mass/charge ratios and retention times) in MS-based untargeted metabolomic data begins through comparison of observed mass with metabolite databases. Two types of such databases exist (Longnecker et al., 2015). The first type contains chemical information (e.g., formula, mass, structure, and physicochemical properties) for any compound regardless of source. Examples include PubChem (Bolton et al., 2008) and ChemSpider (Pence and Williams, 2010), which include both predicted and experimentally observed compounds, and databases such as the Human Metabolome Database (Wishart et al., 2007) and METLIN (Smith et al., 2005) that include experimental metadata for compound contextualization. The best current example of the latter type of database is MetaboLights (Steinbeck et al., 2012; Haug et al., 2013), which contains only known naturally occurring metabolites (as opposed to predicted ones).
Mass measurement alone cannot provide metabolite information beyond molecular formula (Schrimpe-Rutledge et al., 2016), masking the enormous diversity of structural isomers (Meringer et al., 2013). Combined accurate mass and retention time may still not be sufficient to unambiguously identify compounds of interest (Baig et al., 2016). Tandem MS (e.g., MS/MS), which results in unique fragmentation patterns for each compound, is therefore required to assign metabolite identity with more confidence for compounds larger than 50 Da (Hoffman et al., 2014). Importantly, each detected feature may not represent a distinct metabolite, resulting in overestimates of the number of unknown compounds present in a given sample (Dunn et al., 2013). Naturally occurring isotopologues may be present, metabolites may ionize as more than one adduct, or metabolites may fragment or form noncovalent interactions with other compounds upon entering the mass spectrometer (Zamboni et al., 2015). Accurate identification of metabolites must therefore discriminate between metabolites of different nominal masses, metabolites with the same nominal mass but different molecular formulae, and metabolites with the same monoisotopic masses but different chemical structures (e.g., enantiomers and structural isomers) (Dunn et al., 2013). A number of bioinformatic tools have recently been developed to help automate metabolite peak identification, with methods for peak picking and grouping related ion peaks, nonlinear retention time alignment, relative quantification, metabolite identification, and statistical analysis (Patti et al., 2012; Lynn et al., 2014; Schrimpe-Rutledge et al., 2016). Furthermore, the Metabolomics Standards Initiative has developed a protocol for assigning confidence to metabolite identification (Sumner et al., 2007).
Metabolomics provides opportunities for discovery, and challenges in data processing and management, similar to those experienced during the development of high-throughput DNA and RNA sequencing technologies. Like high-throughput sequence data, untargeted metabolomic data sets are massive (gigabytes per sample) and far too complex to be analyzed manually. When simple mass-to-charge ratios are searched against metabolite databases, there are often surprisingly few hits (Zamboni et al., 2015). The situation is not much better for MS/MS spectral matching: of the over 60 million molecules in the PubChem database, only 20,000 include MS/MS spectral data (Johnson and Lange, 2015). The distribution of fragment ions within MS/MS spectra depends on instrument parameters (Herman et al., 2017), limiting the general applicability of these libraries within a given instrumental configuration. Consequently, a few percent, at most, of spectral features in an untargeted metabolomics experiment can presently be annotated (da Silva et al., 2015), underscoring the large gaps in current understanding of metabolism (Kind et al., 2009; Patti et al., 2012). As more metabolite features are discovered and characterized, the number of compounds that may serve as potential biomarkers on other worlds will likely grow.
Metabolomic researchers are rising to meet analytical challenges with improved databases, bioinformatic software, and crowdsourcing platforms such as Global Natural Products Social Molecular Networking (GNPS) (Wang et al., 2016). For example, fairly exhaustive databases of potential molecular isomers can be generated, blanketing a given chemical formula space in silico (e.g., Meringer and Cleaves, 2017). It is increasingly possible to accurately simulate MS/MS fragmentation spectra (e.g., Bauer and Grimme, 2016; Ruttkies et al., 2016) although comparisons with laboratory data still suffer from the analytical dependencies described above and the increasing complexity of fragmentation patterns within large molecules. There is also still room for significant improvements in sample preparation standardization and analytical and data reduction methods (e.g.,
5. The Promise and Challenges of Spatially Resolved Metabolomics
The colocalization of metabolites, with each other and with other observables such as cell-like morphologies, is particularly important in an astrobiological context. MS imaging compiles mass spectra from individual locations within a sample and rasters across an area of interest to build a map of spectral features and their corresponding parent molecules (Dunham et al., 2017). The contextual information that can be gained from such determinations can give otherwise uninteresting molecules new meaning if present in clustered or out-of-equilibrium arrangements. MS imaging allows unique evaluation of distinct spatial aspects of a sample's chemical distribution to better infer the processes involved in compound formation and/or preservation.
The most salient trade-off in MS imaging is between spatial resolution and the size of detected molecules. Nanoscale secondary ion mass spectrometry uses a high-energy primary ion (e.g., Cs+ or O−) beam to provide high spatial resolution spot sizes (down to 50 nm), but fragmentation results in low-mass secondary ions from ∼1 to 300 amu (Fletcher and Vickerman, 2010). This is an extremely useful tool for studying the isotopic composition of the components of individual cells (Marlow et al., 2014; Kopf et al., 2015), and when coupled with knowledge of relevant reservoirs and fractionation factors, this information can point toward metabolic pathways and/or interspecies interactions (Orphan et al., 2009; Pasulka et al., 2018) by tracking relative abundances of C, H, N, O, and S stable isotopes. However, the identity of the molecules that possess these isotopic ratios (e.g., lipids, proteins, and metabolites) cannot be obtained due to the fragmentation accompanying high-energy ion beams and the inefficiency of ionization (typically only about one in 100,000 molecules is ionized by the primary ion beam; Fletcher and Vickerman, 2010).
To better analyze larger molecules more relevant to metabolomics, softer ionization methods are needed. Several variants exist, but MALDI (Caprioli et al., 1997) is most commonly used. Sample surfaces are coated with a chemical matrix that absorbs laser light at a given wavelength, and the sputtered material is sent through a mass spectrometer, determining the fragment sizes and inferred composition of the sample entrained within the matrix. MALDI approaches generally result in relatively large analytical spot sizes (e.g., 50 μm; Cornett et al., 2007), but recent advances in instrument configuration and matrix application have enabled 1.4 μm spatial resolution in a study of metabolite, lipid, and peptide distributions (Kompauer et al., 2016), approaching the scale of microbial cells.
As secondary ion mass spectrometry- and MALDI-based techniques converge in terms of spatial resolution, submicron-scale analyses of a wide range of molecular weight compounds with resolution amenable to isotopic studies may become possible. Maintaining the spatial arrangements of geobiological samples is critical for their interpretation, as distributions of key parameters (e.g., lipid or metabolite type, isotopic composition) with respect to pore space, conduits, mineral type, or texture may offer valuable information on biogenicity. With this additional context, astrobiologists will gain new perspectives on observed features, enabling nuanced, environment-specific interpretation of metabolomic data sets. While the deployment of these tools for space-based missions is infeasible currently due to size and power constraints, sample-return missions will undoubtedly benefit from their use.
6. Integrating Metabolomics with Other Omics Data
To understand the function of metabolites within cellular processes, it is necessary to develop metabolomic analysis approaches that integrate data across different omics data sets (Johnson et al., 2016)—an effort that has some overlap with the field of systems biology (Kitano, 2002). A desired outcome of this type of data integration is the characterization of unknown metabolites within the context of known genes and proteins. As stated previously, it cannot be assumed that alien life uses nucleic acids or the same genetic code as terrestrial life, or even that protein folding could be predicted from an alien gene sequence. Indeed, predicting the structure and by extension the function of terrestrial proteins remains an ongoing problem (for a complete review, see Lee et al., 2017). Metagenomic analyses must also make assumptions about homology between organisms that may or may not be true (Prakash and Taylor, 2012). As the integration of omics techniques enables the development of a deeper and more fundamental understanding of terrestrial cellular metabolism (Ritchie et al., 2015), it may also provide clues to the functions of unknown metabolites associated with metabolisms of astrobiological interest.
There is a growing gap, however, between the large (and rapidly increasing) amount of omics data and researchers with skills to process and interpret these data (Barone et al., 2017). The lack of standard laboratory and data processing procedures across databases and studies is a critical problem, as is the elucidation of the function of unannotated “hypothetical” genes that often dominate environmental omics annotation pipelines (Jiao et al., 2017).
Ideally, metagenomic data would provide information about which compounds a microbial cohort is capable of biosynthesizing, and then MS analysis would be able to corroborate that a physiologically relevant subset of such compounds is in fact present. For example, identification of sterols based on the detection of unique mass species in geological specimens has allowed estimation of divergence times in the tree of life as determined by phylogenetic comparison. Using such techniques, Brocks et al. (1999) estimated the earliest eukaryotes to have appeared at 2.7 Ga (although these data have since been questioned; French et al., 2015), and the Ediacaran fossil Dickinsonia was identified as one of the first animals based on MS-derived molecular fossil data (Bobrovskiy et al., 2018).
However, there are multiple challenges in interpreting combined environmental genomic and molecular data. Genomic data may be amplified in ways that skew interpretation of the activity and abundance of organisms (Kim and Bae, 2011; Shakya et al., 2013; Solonenko et al., 2013; Quince et al., 2017). Furthermore, it cannot be assumed that the abundance of a given gene cohort is directly proportional to the abundance of specific molecules (Chan et al., 2010). Gene annotation, including accurate identification of start and stop codons, can be challenging (Pauli et al., 2014; Mattick and Rinn, 2015; Borriss et al., 2017). The ability to annotate genomes has greatly improved as a consequence of the increase of annotation databases such as NCBI's RefSeq (O'Leary et al., 2016), UniProtKB (Bateman et al., 2017), NCBI's Clusters of Orthologous Groups (COGs) (Galperin et al., 2015), the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2016), Gene Ontology (GO) (Huerta-Cepas et al., 2017), and Protein Families (Pfam) (Finn et al., 2016). The growth of these databases has enabled vast improvements in the interpretation of environmental metagenomic, transcriptomic, and proteomic data (e.g., as reviewed in Konopka and Wilkins, 2012; Haider and Pal, 2013; Franzosa et al., 2016).
Programs such as ModelSEED (Henry et al., 2010) and Computation of Microbial Ecosystems in Time and Space (COMETS) (Harcombe et al., 2014) have been developed to reconstruct metabolic community networks from multiple genomes, and can be used to identify key metabolic “nodes” within these networks (Henry et al., 2016) and describe metabolic networks over time (Granger et al., 2016). Researchers are developing data analysis pipelines that integrate metabolomes with other omics data sets to develop more complete pictures of the metabolism of organisms or communities of interest (Noecker et al., 2016; Huan et al., 2017; Nagler et al., 2018; Witting et al., 2018). In vivo labeling of metabolite pools with 13C, 15N, and/or deuterium (2H) can be used to improve metabolite annotation accuracy (Birkenmeyer et al., 2005; Hegeman et al., 2007) and for metabolic network-wide elucidation of pathways (Kikuchi et al., 2004; Winder et al., 2011; Creek et al., 2012). The combined use of omics techniques thus allows for an expansion of our understanding of metabolism on Earth, improving our ability to search for analogous biological processes on other worlds.
7. Conclusions
Like astrobiology, metabolomics is highly interdisciplinary and often requires collaboration across different research areas to complete all stages of an experimental workflow. For those who are new to metabolomics or who are not specialized in certain informatics approaches, several initiatives have been developed, including resources through the US National Institutes of Health (NIH) Common Fund Metabolomics Program, and the Coordination of Standards in Metabolomics (COSMOS) (Salek et al., 2015; Johnson et al., 2016). Both vendor-provided and open-access MS analysis software offer user-friendly options for beginners, as well as advanced tools for experts (Johnson et al., 2016).
Metabolomics has much to offer to the advancement of astrobiology, particularly in cases in which noncanonical modes of information storage and metabolism may be present. Applications of metabolomic-derived tools will undoubtedly increase as data collection, storage, and analysis techniques improve. The integration of metabolomics with other omics data will also provide insight into the functions of newly discovered metabolites and their connectivity in metabolic networks. These advances will help astrobiologists search for biochemical signs of life on other worlds.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was partially supported by the ELSI Origins Network (EON), which is supported by a grant from the John Templeton Foundation. The opinions expressed in this publication are those of the authors and do not necessarily reflect the views of the John Templeton Foundation. This work was partially supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research on Innovative Areas “Hadean Bioscience,” grant number JP26106003, and also partially supported by Project “icyMARS,” funded by the European Research Council, ERC Starting Grant No. 307496. A.A-B thanks the contribution from the Project “MarsFirstWater,” funded by the European Research Council, ERC Consolidator Grant No. 818602 and the HFSP Project UVEnergy RGY0066/2018.
Abbreviations Used
Associate Editor: Lewis Dartnell