
Abstract
Aims:
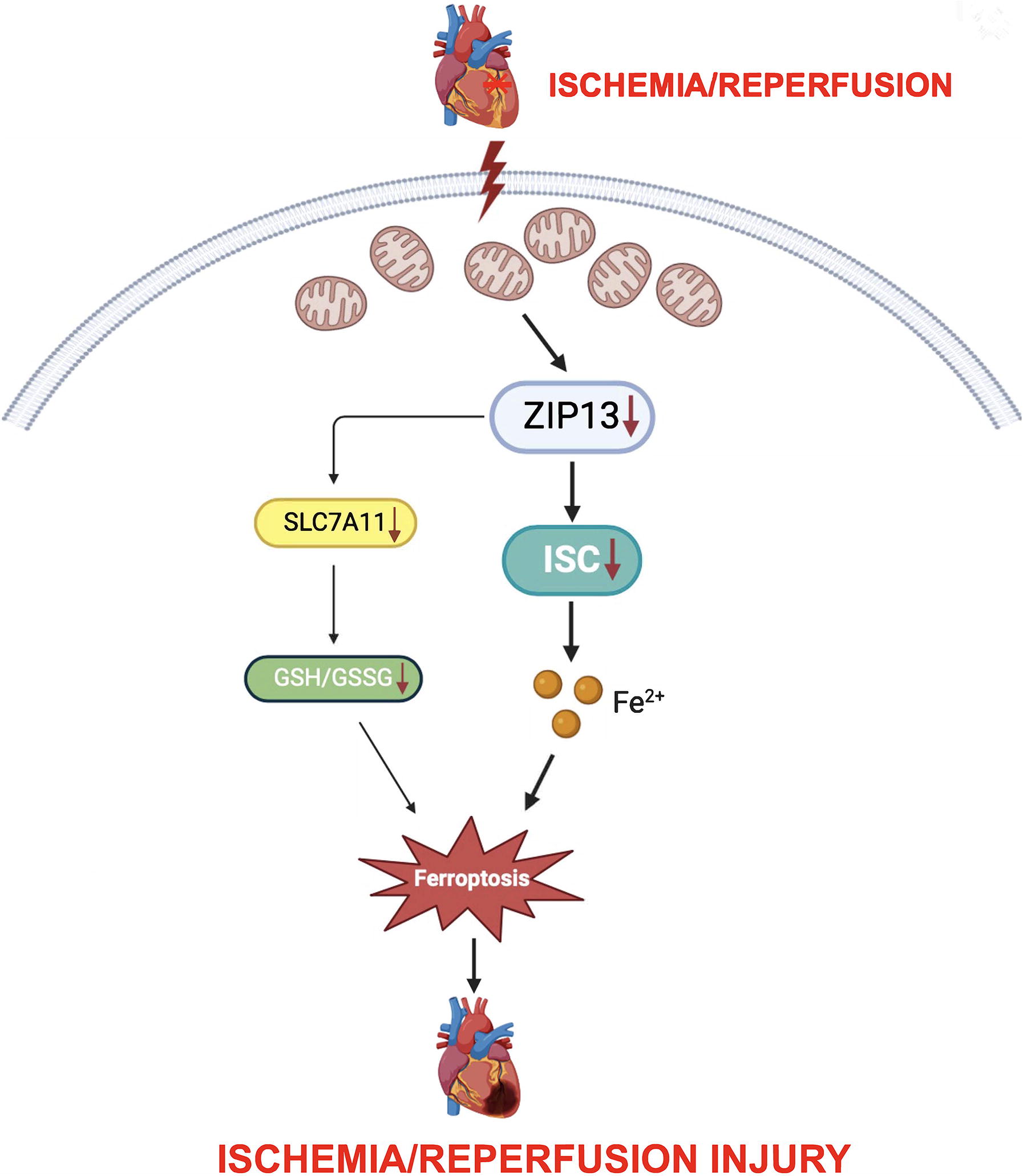
While ferroptosis is involved in the pathogenesis of myocardial ischemia/reperfusion (I/R) injury, the exact mechanism underlying the induction of ferroptosis by I/R remains elusive. Since downregulation of Zrt, Irt-like protein 13 (ZIP13) plays a role in I/R injury by targeting mitochondria, we hypothesized that ZIP13 downregulation during I/R leads to ferroptosis through a mitochondria-dependent mechanism.
Results:
ZIP13 cKO (cardiac-specific conditional knockout) induced ferroptosis and suppressed mitochondrial iron–sulfur cluster (ISC) biosynthesis. ZIP13 cKO also reduced glutathione levels as well as solute carrier family 7 member 11 (SLC7A11) expression. Moreover, cKO increased mitochondrial Fe2+ levels. Similar to the action of cKO, I/R led to ZIP13 downregulation, ferroptosis, mitochondrial Fe2+ accumulation, and suppression of ISC biosynthesis. In support, cKO of ZIP13 aggravated I/R-induced ferroptosis and mitochondrial Fe2+ accumulation. In contrast, ZIP13 overexpression prevented I/R-induced ferroptosis, mitochondrial Fe2+ accumulation, and suppression of ISC biosynthesis. Finally, ferrostatin-1, a ferroptosis inhibitor, alleviated I/R-induced ferroptosis as well as cardiac injury in cKO mice.
Innovation:
This study proposes a previously unknown mechanism by which ZIP13 downregulation contributes to ferroptosis in the setting of myocardial I/R.
Conclusions:
These findings highlight that ZIP13 downregulation at reperfusion triggers ferroptosis by suppressing the mitochondrial ISC biosynthesis followed by mitochondrial Fe2+ accumulation. Downregulation of SLC7A11 may also contribute to the action of ZIP13 downregulation. Antioxid. Redox Signal. 43, 328–344.
Introduction
As an iron-dependent form of cell death, ferroptosis is characterized by iron accumulation and lipid peroxidation (Li et al., 2020). Inhibition of the cystine/glutamate antiporter system (system Xc-)/glutathione (GSH)/glutathione peroxidase (GPX4), intracellular iron accumulation, and membrane lipid peroxidation are considered to be the mechanism underlying ferroptosis (Cao and Dixon, 2016). The system Xc− comprised the light-chain subunit solute carrier family 7 member 11 (SLC7A11) and the heavy-chain subunit solute carrier family 3 member 2 (SLC3A2), and sustains the production of GSH by exchanging extracellular cystine for intracellular glutamate. Upon being imported into cells, cystine is immediately reduced to cysteine. Cysteine is considered to be the rate-limiting precursor for GSH synthesis. As an important antioxidant enzyme, GPX4 reduces lipid hydroperoxide to nontoxic lipid alcohol in the membrane, and its expression or activity is controlled by the availability of GSH (Tang et al., 2021a). Accordingly, inhibition of SLC7A11 results in depletion of GSH to trigger ferroptosis (Dixon et al., 2012, 2014).
Innovation
Although ferroptosis is proposed to play a critical role in myocardial I/R injury, the exact mechanism by which ferroptosis is induced remains elusive. We propose a novel concept that ZIP13, a zinc transporter, contributes to the genesis of ferroptosis in the setting of I/R in the heart. We demonstrated that downregulation of ZIP13 induces ferroptosis by suppressing mitochondrial ISC biosynthesis in mouse hearts subjected to I/R. In support, overexpression of ZIP13 prevented I/R-induced ferroptosis and cardiac injury. Our findings suggest that targeting ZIP13 downregulation-induced ferroptosis may serve as a therapeutic strategy for the treatment of I/R injury (Fig. 1).
Studies have demonstrated that ferroptosis plays an important role in myocardial ischemia/reperfusion (I/R) (Fang et al., 2019; Gao et al., 2015; Li et al., 2019). Ferrostatin-1 (Fer-1), a specific inhibitor of ferroptosis, reduced cardiac cell death and blocked neutrophil recruitment following heart transplantation (Li et al., 2019). Fang et al. reported that I/R increased prostaglandin endoperoxide synthase 2 (PTGS2) expression, and Fer-1 (a ferroptosis inhibitor) or dexrazoxane (an iron chelator) reduced infarct size in mouse hearts subjected to I/R, suggesting that I/R can induce ferroptosis (Fang et al., 2019). It has been reported that inhibition of glutaminolysis alleviates cardiac I/R injury by preventing ferroptosis (Gao et al., 2015). Moreover, it was also proposed that ferroptosis occurs at reperfusion but not during ischemia in rat hearts (Tang et al., 2021b). Although these reports clearly indicate that ferroptosis plays an important role in I/R injury in the heart and that inhibition of ferroptosis may be protective against I/R injury, the mechanism by which ferroptosis is induced remains unclear.
As the main site for reactive oxygen species (ROS) generation, mitochondria play a critical role in myocardial I/R injury (Murphy and Steenbergen, 2008). Although there are controversies over the role of mitochondria in the genesis of ferroptosis, increasing evidence shows that mitochondria are crucial for the induction of ferroptosis (Lillo-Moya and Rojas-Solé, 2021). It has been reported that mitochondria play a critical role in cysteine deprivation-induced ferroptosis (Gao et al., 2019). Mitochondria are also crucial for ferroptosis induced by doxorubicin (DOX) or I/R in cardiomyocytes (Fang et al., 2019). Moreover, overexpression of mitochondrial GPX4 or chelation of mitochondrial Fe2+ prevented DOX-induced ferroptosis in cardiomyocytes, indicating the important role of mitochondria in ferroptosis (Tadokoro et al., 2020). Although the exact mechanism by which mitochondria contribute to the induction of ferroptosis is unknown, dysregulation of iron–sulfur cluster (ISC) biogenesis may play a role in the induction of ferroptosis (Lee and Roh, 2023). Most ISCs are synthesized in mitochondria and are inorganic cofactors essential in all life-forms with common roles in electron transfer, oxidative phosphorylation, radical generation, and structural support (Lill, 2009). The core machinery for the de novo biosynthesis of ISCs, located in the mitochondrial matrix, is a five-protein complex containing the cysteine desulfurase (NFS1) that is activated by frataxin (FXN), scaffold protein iron–sulfur cluster assembly enzyme (ISCU), accessory protein iron–sulfur cluster assembly factor ISD11 (ISD11), and acyl-carrier protein ACP (Lill, 2009). The inhibition of NFS1 disrupts ISC biosynthesis leading to iron starvation response and activation of ferroptosis (Alvarez et al., 2017). Likewise, inhibition of FXN can interfere with the assembly of ISC and induces ferroptosis by increasing lipid peroxidation (Du et al., 2020).
As a member of the ZIP family, Zrt, Irt-like protein 13 (ZIP13) is mainly localized in the Golgi apparatus (Takagishi et al., 2017). Recent studies demonstrated that ZIP13 is also highly expressed in the heart (Jeong et al., 2012), and downregulation of ZIP13 contributes to myocardial I/R injury by activating calcium/calmodulin-stimulated protein kinase II (CaMKII) (Wang et al., 2021). Moreover, a latest study reported that DOX downregulated ZIP13 expression in mouse cardiac cell line (HL-1) cells or mouse hearts in vivo (Hara et al., 2022). Since DOX (Fang et al., 2019) and I/R can trigger ferroptosis in the heart, it is tenable to speculate that downregulation of ZIP13 may contribute to I/R injury by mediating ferroptosis. In addition, given that downregulation of ZIP13 leads to mitochondrial dysfunction and impairment of mitochondrial respiration (Wang et al., 2021), it is worth testing if ZIP13 regulates ferroptosis by targeting mitochondria especially the mitochondrial ISC biosynthesis.
In this study, we tested if downregulation of ZIP13 induces ferroptosis in the setting of I/R in the heart and investigated the underlying mechanism. We demonstrated that ZIP13 downregulation upon reperfusion is responsible for the induction of ferroptosis thereby contributing to the genesis of myocardial I/R injury. Inhibition of ISC biosynthesis and SLC7A11 expression accounts for ferroptosis caused by ZIP13 downregulation.
Results
ZIP13 deficiency induces ferroptosis in mouse hearts and cardiac cells
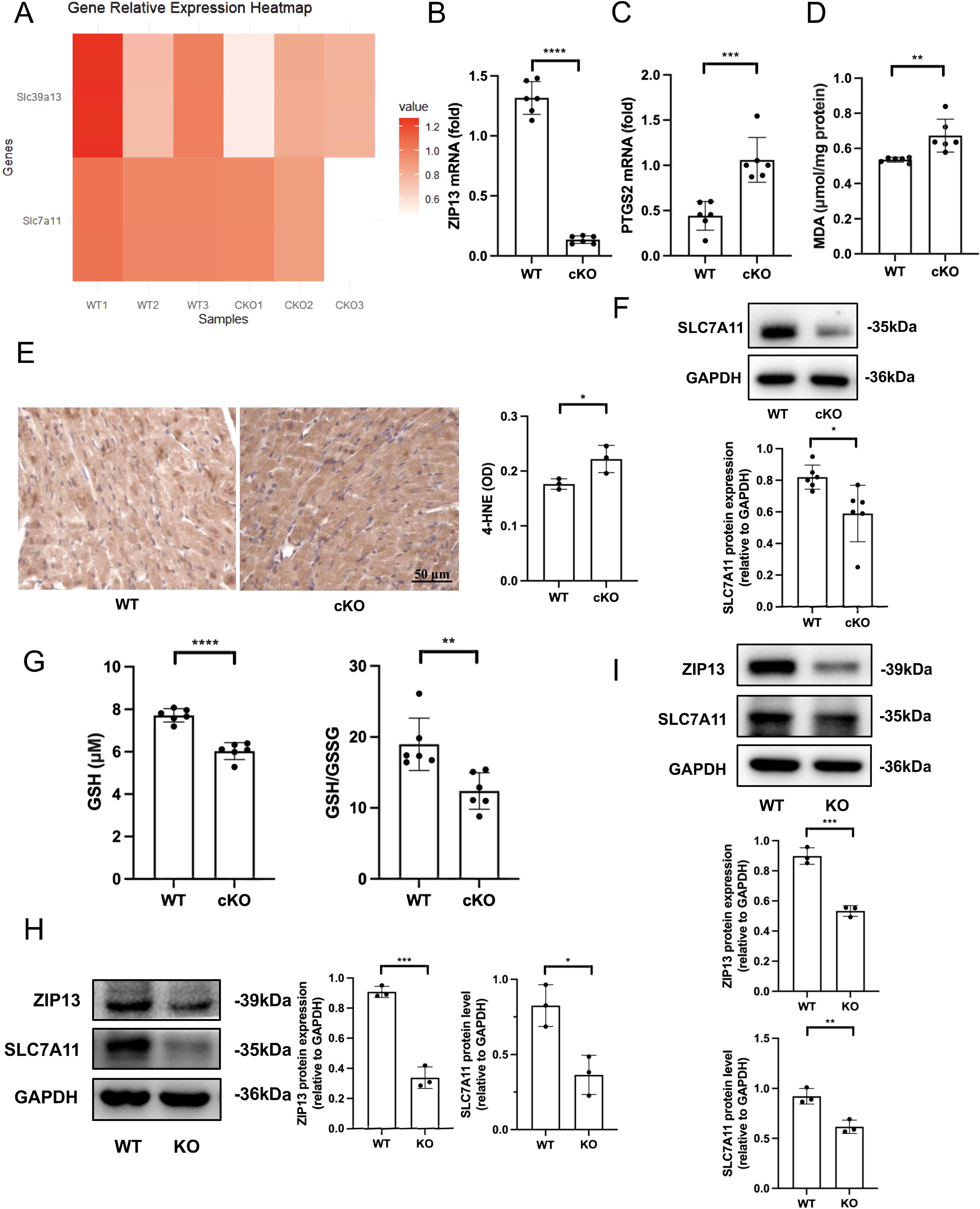
In a previous study, we have demonstrated that ZIP13 downregulation contributes to the pathogenesis of I/R injury in mouse hearts by activating CaMKII (Wang et al., 2021). In addition, ferroptosis plays a critical role in myocardial I/R injury and ZIP13 has been proposed to be an iron exporter (Xiao et al., 2014). Accordingly, we tested if ZIP13 contributes to I/R injury by regulating ferroptosis. We first examined the role of ZIP13 by performing RNA sequencing and found that cKO of ZIP13 (cardiac-specific ZIP13 gene knockout) results in downregulation of SLC7A11 in mouse hearts (Fig. 2A), indicating that ZIP13 may regulate ferroptosis. ZIP13 cKO (Fig. 2B) increased PTGS2 mRNA (Fig. 2C), malondialdehyde (MDA) (Fig. 2D), and 4-hydroxynonenal (4-HNE) (Fig. 2E) levels in mouse hearts. Further experiments revealed that ZIP13 cKO reduced SLC7A11 expression (Fig. 2F) as well as GSH and GSH/oxidized glutathione (GSSG) levels (Fig. 2G) in mouse hearts. ZIP13 cKO also reduced GPX4 mRNA expression as well as GPX4 activity (Supplementary Fig. S1). Moreover, ZIP13 KO also downregulated SLC7A11 protein expression in HL-1 (Fig. 2H) and AC16 human cardiomyocyte cell line (AC16) (Fig. 2I) cells. These data collectively suggest that deficiency of ZIP13 can induce ferroptosis in mouse hearts or cardiac cells.
ZIP13 is localized in mitochondria and ZIP13 deficiency results in mitochondrial Fe2+ accumulation
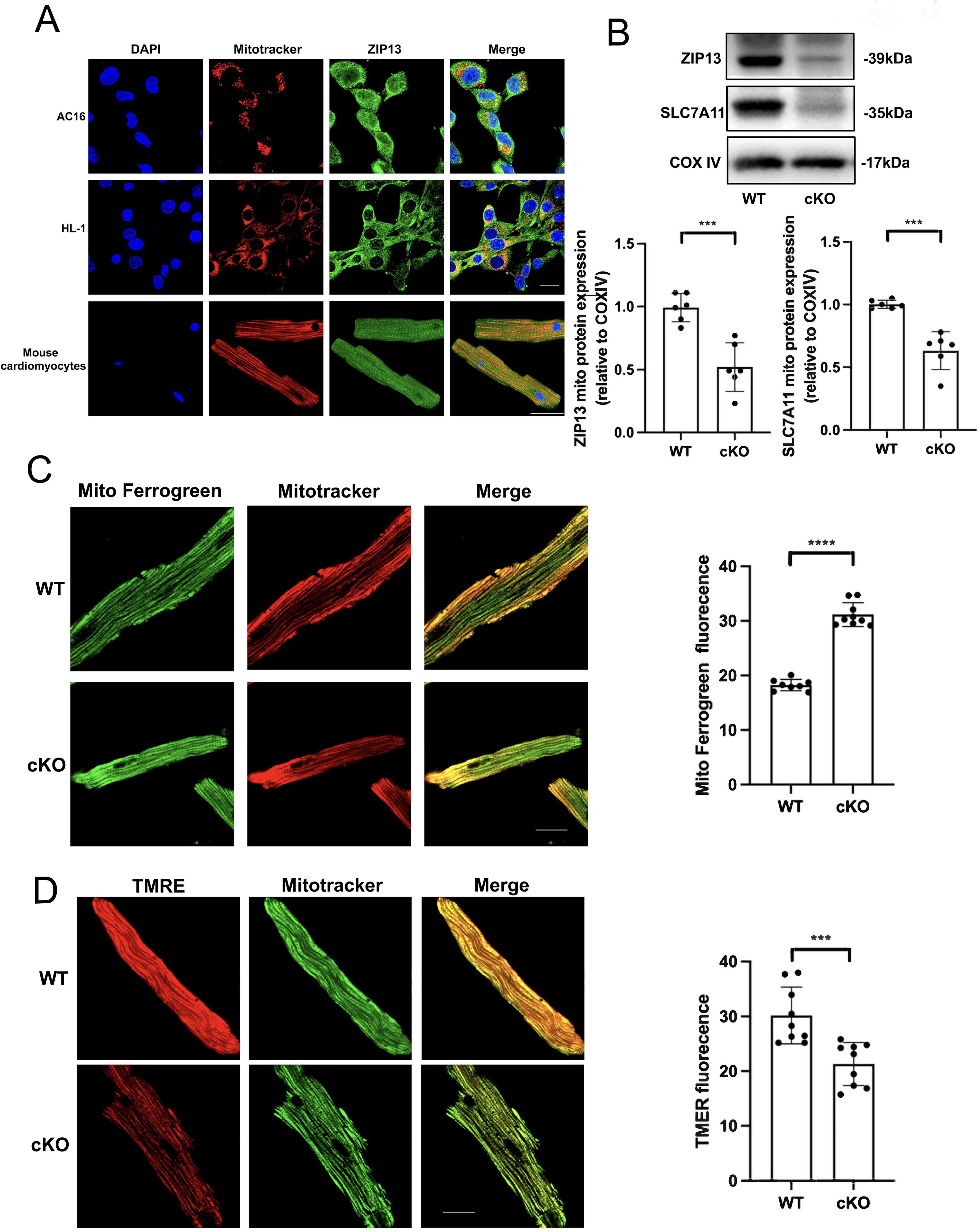
Our recent study demonstrated that ZIP13 contributes to myocardial I/R injury by targeting mitochondria (Wang et al., 2021). Therefore, we tested if ZIP13 is localized in mitochondria in cardiac cells and mouse hearts. Confocal imaging studies showed that ZIP13 is colocalized with MitoTracker in AC16, HL-1, or isolated adult mouse cardiomyocytes (Fig. 3A). Western blotting also revealed that ZIP13 is detectable within mitochondria and the cytosol isolated from mouse hearts and that cKO markedly reduced ZIP13 protein expression as well as SLC7A11 in mitochondria (Fig. 3B). Importantly, ZIP13 cKO enhanced the selective mitochondrial iron probe Mito-FerroGreen fluorescence (Fig. 3C), implying that ZIP13 deficiency leads to an accumulation of Fe2+ within mitochondria. Moreover, ZIP13 cKO caused mitochondrial depolarization (Fig. 3D). These data suggest that ZIP13 may control ferroptosis by targeting mitochondria.
ZIP13 deficiency leads to damage of the ISC biosynthesis and dysfunction of the mitochondrial electron transport chain in mouse hearts
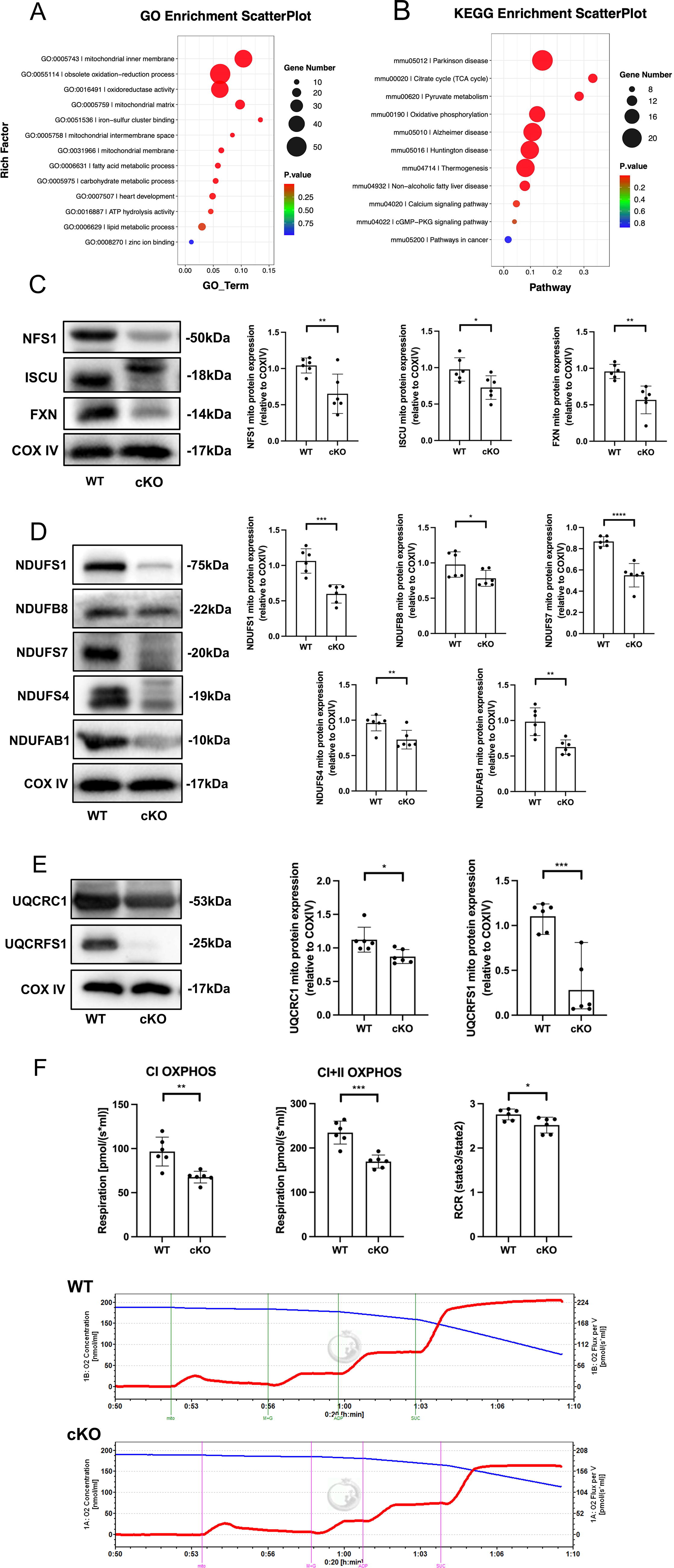
To further investigate the role of mitochondria in ZIP13 regulation of ferroptosis, we performed the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses using our RNA sequencing data obtained from wild-type (WT) and ZIP13 cKO mouse hearts and found that the differentially expressed genes between WT and ZIP13 cKO mouse hearts were enriched in mitochondria-related pathways, including the ISC biosynthesis (Fig. 4A, B). In addition, it has been reported that the inhibition of ISC biosynthesis induces the iron-starvation response and leads to ferroptosis in cancer cells (Alvarez et al., 2017). Thus, we investigated the effects of ZIP13 deficiency on expression levels of the proteins involved in the ISC biosynthesis. Figure 4C shows that cKO of ZIP13 downregulated NFS1, ISCU, and FXN protein expression in mitochondria isolated from mouse hearts, indicating that deficiency of ZIP13 can inhibit ISC biosynthesis thereby inducing ferroptosis. Since ISCs serve as important components of the electron transport chain (ETC), we next tested if cKO of ZIP13 can also alter protein expressions of complex I subunits, including NDUFS1, NDUFB8, NDUFS7, NDUSF4, and NDUFAB1. ZIP13 cKO reduced expression of all these subunits (Fig. 4D). Similarly, cKO of ZIP13 also downregulated protein levels of the complex III subunits UQCRC1 and UQCRFS1 (Fig. 4E). Moreover, ZIP13 cKO suppressed complex I and II oxidative phosphorylation, and respiratory control rate (RCR) (Fig. 4F). All these data suggest that ZIP13 cKO leads to inhibition of ISC biosynthesis, which may account for ZIP13 deficiency-induced ferroptosis.
Overexpression of FXN prevents both ferroptosis and damages of ISC biosynthesis induced by ZIP13 cKO
To corroborate the role of ISC biosynthesis in ferroptosis caused by ZIP13 cKO, we tested if overexpression of FXN can rescue ferroptosis and ISC biosynthesis inhibition caused by ZIP13 cKO. FXN overexpression (Fig. 5A) prevented decreases of complexes I (Fig. 5B) and III (Fig. 5C) in ZIP13 cKO mouse hearts, indicating that the maintenance of ISC biosynthesis machinery can rescue damages of ISC biosynthesis in the setting of ZIP13 cKO. Further experiments showed that overexpression of FXN prevented elevations of PTGS2 (Fig. 5D) and MDA (Fig. 5E) levels in ZIP13 cKO mouse hearts, indicating that ISC biosynthesis plays a critical role in ZIP13 regulation of ferroptosis.
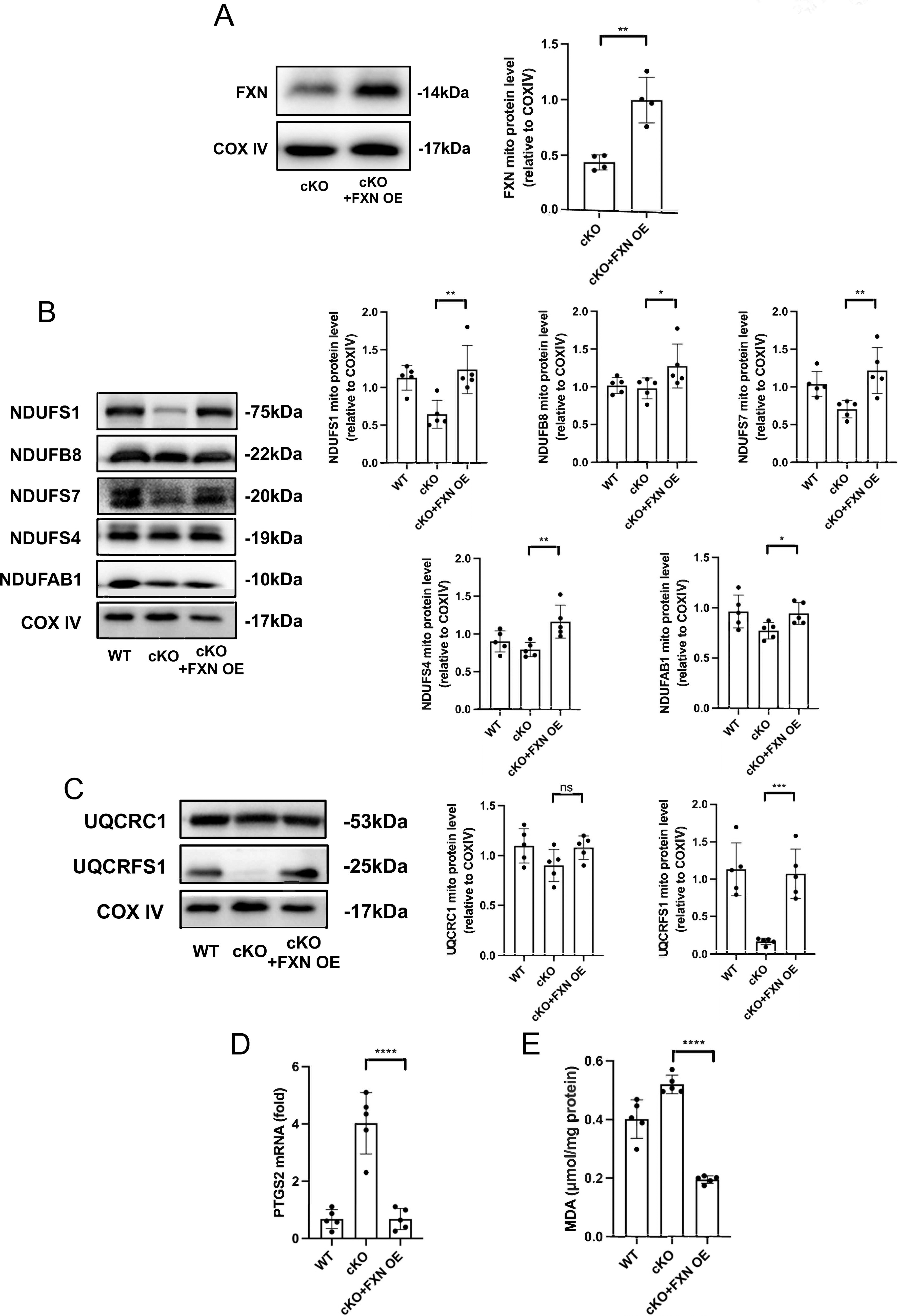
I/R downregulates ZIP13 and SLC7A11 and leads to damages of the ISC biosynthesis as well as ETC
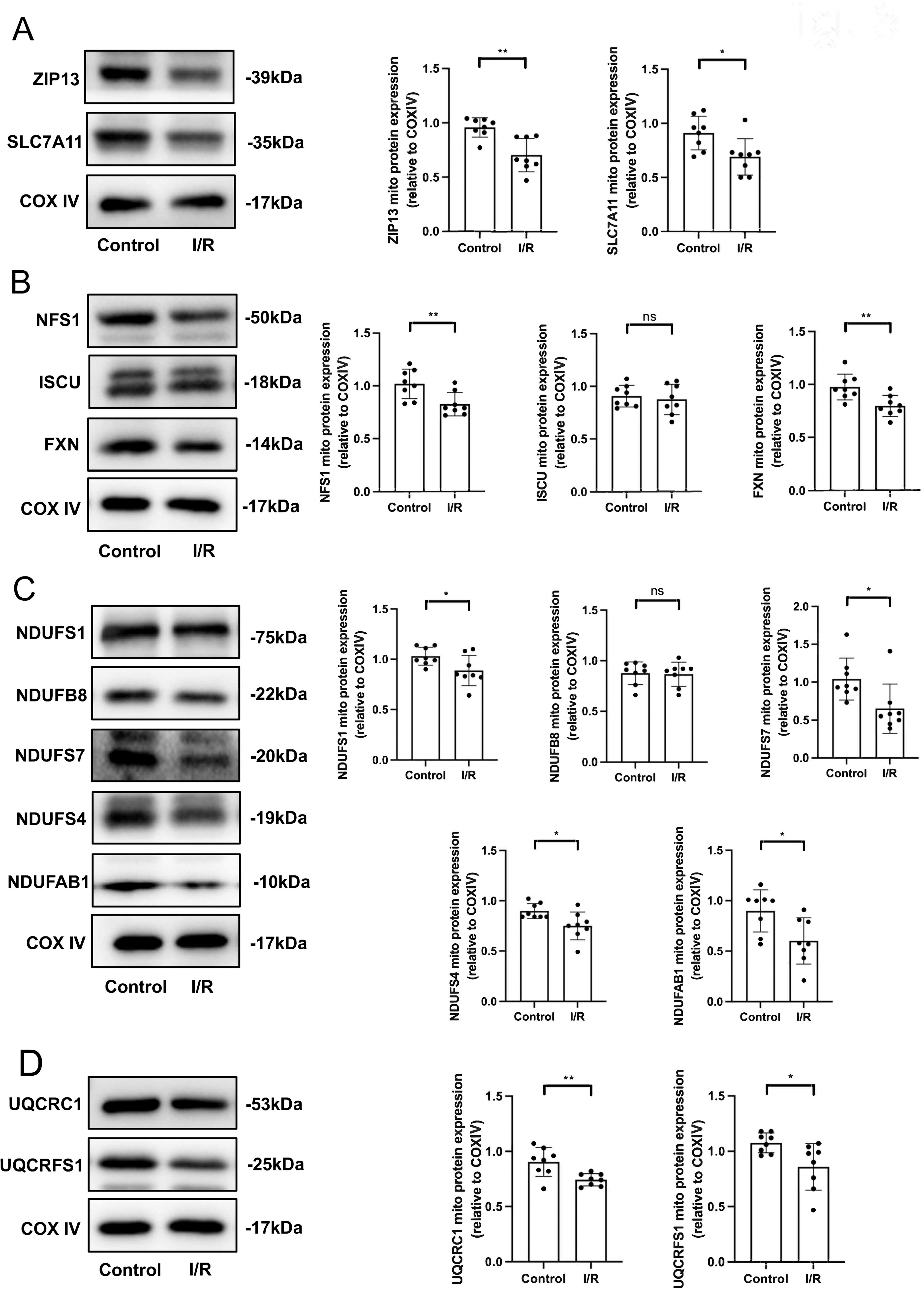
Since ZIP13 downregulation plays a role in myocardial I/R injury and deficiency of ZIP13 leads to ferroptosis, we next investigated if downregulation of ZIP13 accounts for I/R injury by triggering ferroptosis. ZIP13 and SLC7A11 were downregulated by I/R in mouse hearts in vivo (Fig. 6A). Analogous to the action of ZIP13 cKO, I/R reduced NFS1 and FXN protein expression (Fig. 6B). I/R also inhibited protein expression of the complex I subunits except for NDUFB8 (Fig. 6C). Further studies revealed that the protein expression of the complex III subunits, including UQCRC1 and UQCRFS1, was suppressed by I/R (Fig. 6D). These data suggest that I/R induces ferroptosis presumably by inhibiting ISC biosynthesis through the downregulation of ZIP13.
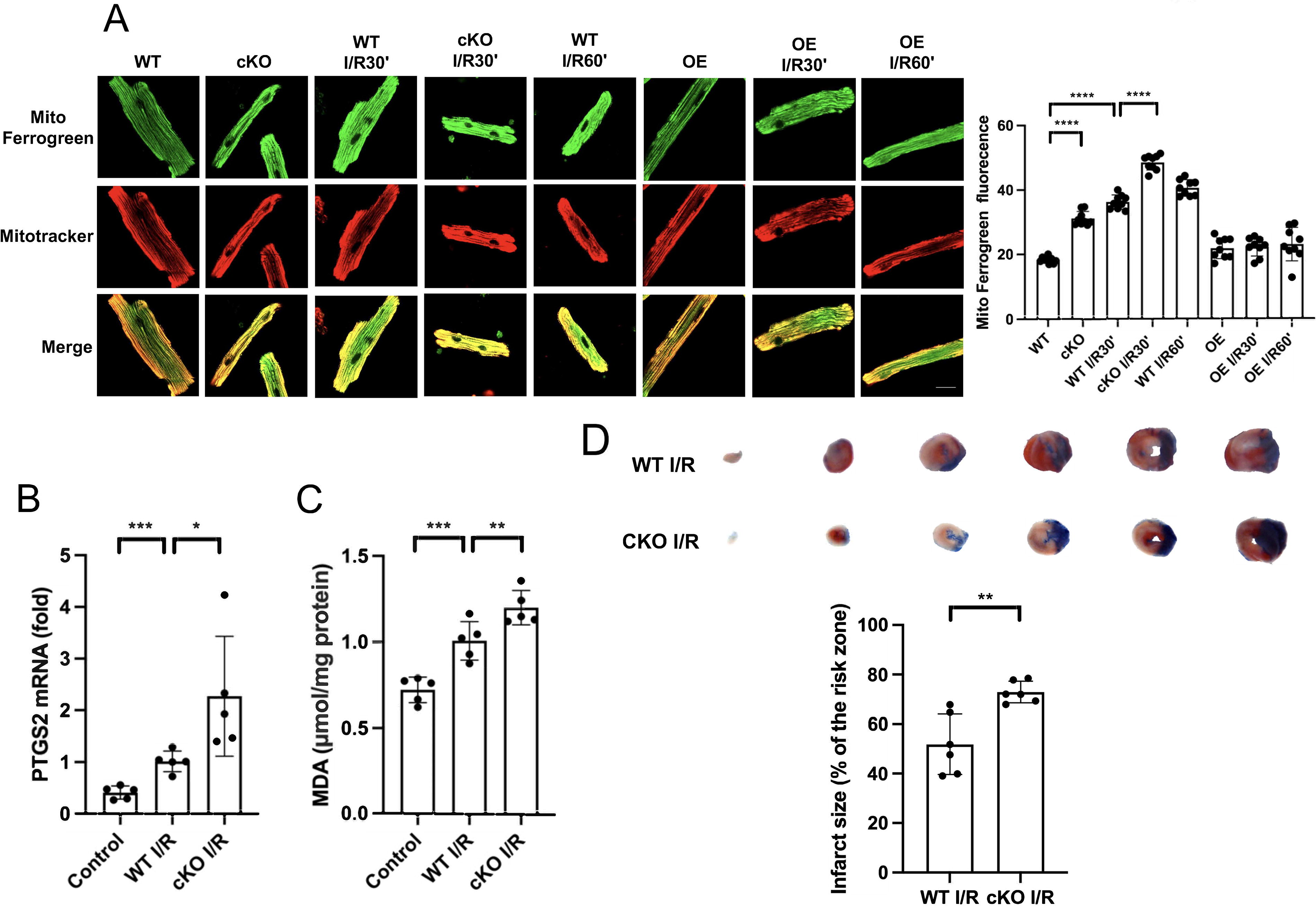
ZIP13 deficiency leads to mitochondrial Fe2+ accumulation and ferroptosis in the setting of I/R
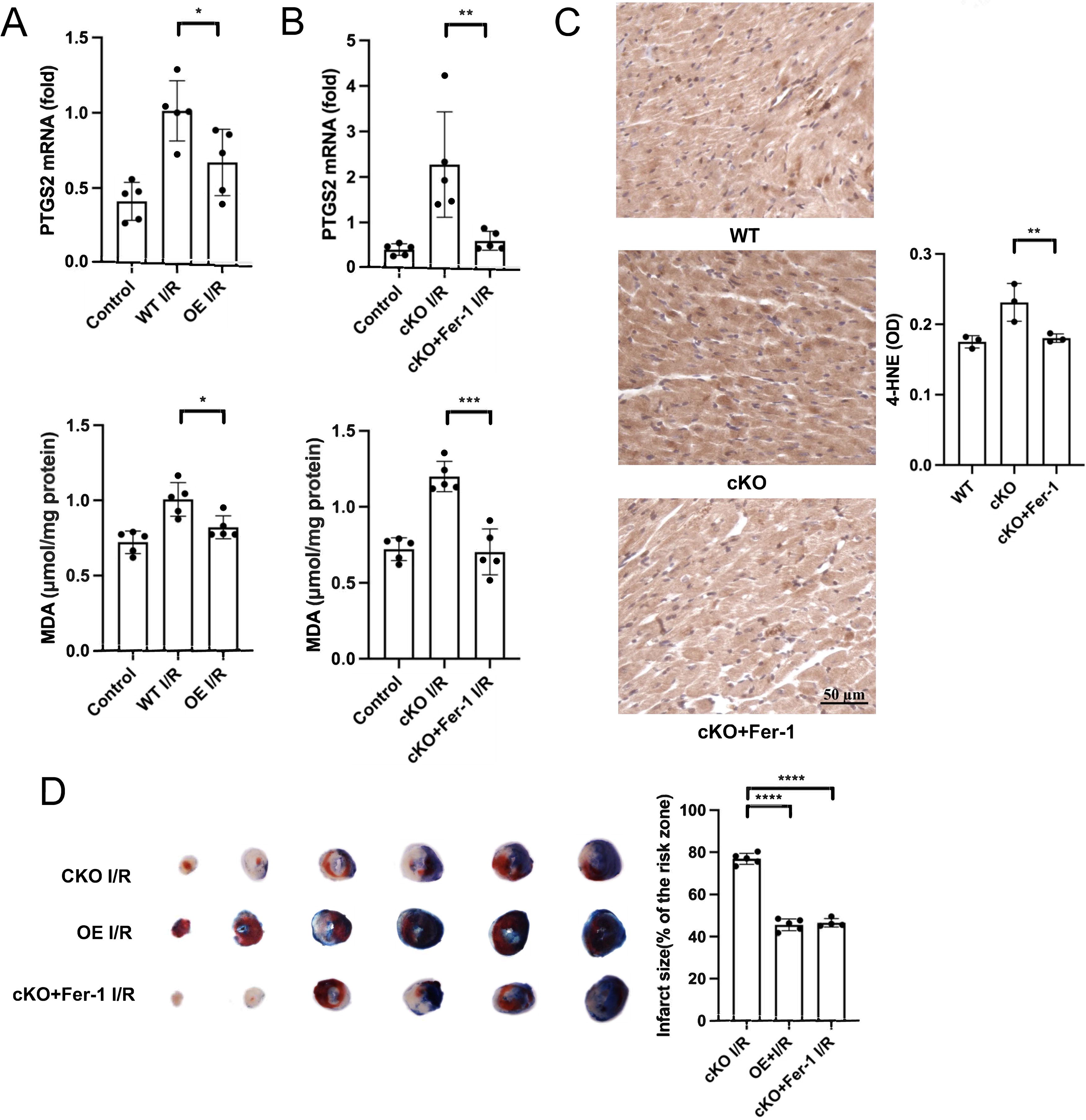
To test if I/R results in mitochondrial Fe2+ accumulation via ZIP13, we measured mitochondrial Fe2+ with confocal microscopy in isolated mouse cardiomyocytes loaded with Mito-FerroGreen in the setting of H/R. Figure 7A shows that I/R markedly increased Mito-FerroGreen fluorescence intensity and this was enhanced by cKO of ZIP13. I/R also increased PTGS2 mRNA and MDA levels in mouse hearts, which was intensified by cKO (Fig. 7B, C). In support, I/R-induced myocardial infarction was exacerbated by cKO (Fig. 7D). These data point that deficiency of ZIP13 induces myocardial ferroptosis not only under normoxic conditions but also in the setting of I/R.
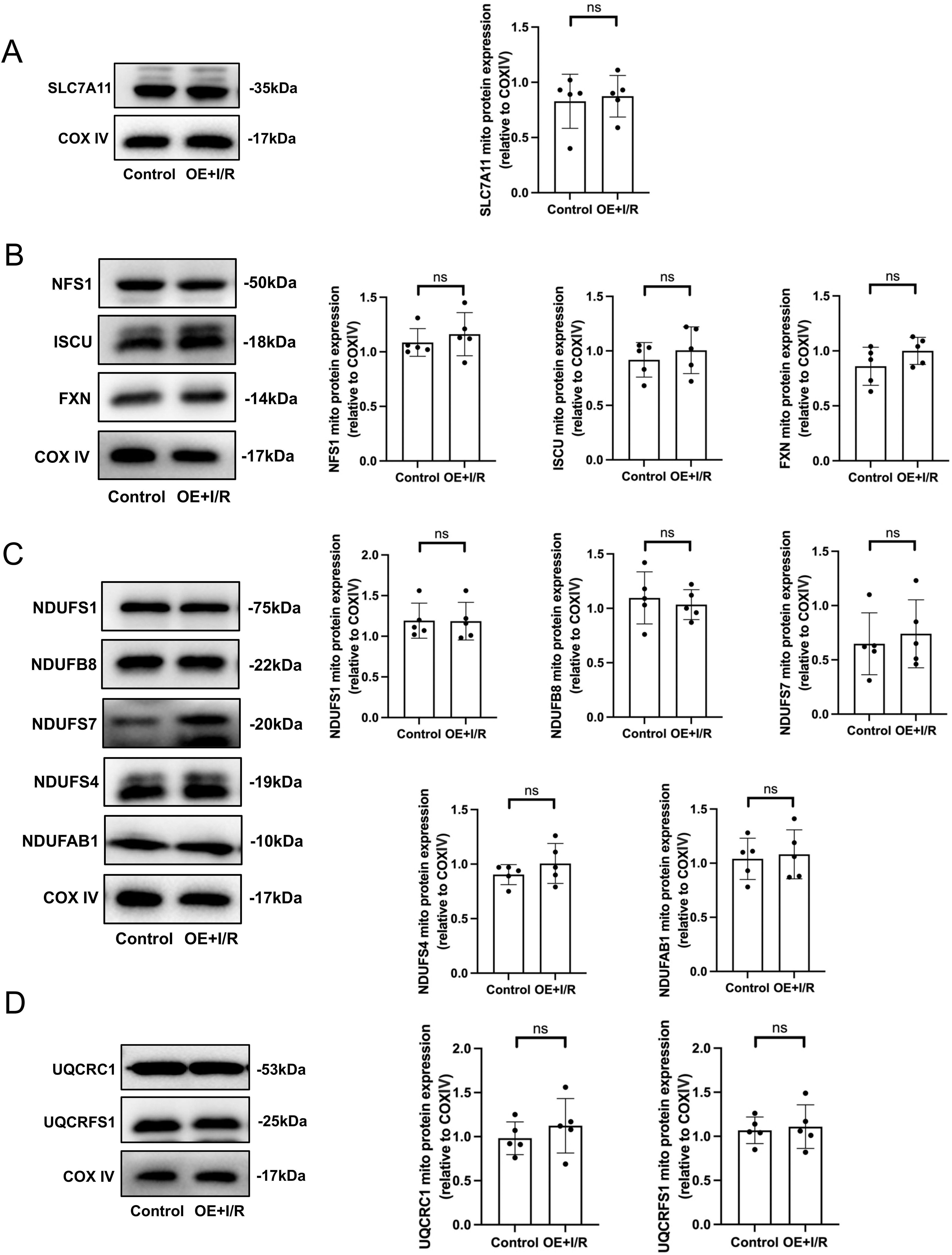
Overexpression of ZIP13 prevents I/R-induced damage of the ISC biosynthesis and dysfunction of ETC
To verify the exact role of ZIP13 in the inhibitory effect of I/R on SLC7A11 and ISC biosynthesis, we examined the effects of ZIP13 overexpression on ISC biosynthesis in the setting of I/R in mitochondria isolated from mouse hearts. Overexpression of ZIP13 prevented I/R-induced inhibition of SLC7A11 protein expression (Fig. 8A). Similarly, I/R failed to suppress the mitochondrial expression of the ISC biosynthetic machinery proteins, including NFS1, ISCU, and FXN, in the heart overexpressed with ZIP13 (Fig. 8B), compared with the control hearts. In support, I/R also did not change the mitochondrial expression of the complex I (Fig. 8C) and III (Fig. 8D) subunit proteins in the mouse hearts overexpressed with ZIP13. These data together suggest that ZIP13 downregulation is responsible for I/R-induced ferroptosis presumably by inhibiting the ISC biosynthesis.
Overexpression of ZIP13 ameliorates I/R-induced ferroptosis and Fer-1 can prevent cKO-induced exacerbation of myocardial injury
To further corroborate the role of ZIP13 in I/R-induced ferroptosis, we first tested the effect of ZIP13 overexpression on ferroptosis in mouse hearts. Figure 7A shows that increased mitochondrial Fe2+ accumulation in mouse cardiomyocytes by simulated I/R alone or I/R+ZIP13 cKO was reversed by ZIP13 overexpression, pointing that ZIP13 downregulation accounts for H/R-induced mitochondrial Fe2+ accumulation. In support, overexpression of ZIP13 inhibited the increases of PTGS2 mRNA expression and MDA levels in mouse hearts subjected to I/R (Fig. 9A). To further authenticate our hypothesis that ZIP13 downregulation contributes to ferroptosis in the setting of I/R, we examined the effects of the ferroptosis inhibitor Fer-1 on PTGS2 mRNA expression and MDA level in ZIP13 cKO hearts subjected to I/R. Figure 9B shows that I/R-induced increases in PTGS2 mRNA expression and MDA level were reversed by Fer-1, indicating a role of ZIP13 in the induction of ferroptosis in the setting of I/R. Similarly, cKO-induced increase in 4-HNE was reversed by Fer-1 (Fig. 9C). Furthermore, I/R-induced myocardial infarction was decreased in the ZIP13 cKO hearts treated with Fer-1, which was comparable with that of the hearts overexpressed with ZIP13 (Fig. 9D).
Discussion
In this study, by adopting cKO mice or KO cell lines, we established a role of ZIP13 in ferroptosis induced by I/R, identifying the inhibition of ISC biosynthesis as the major mechanism by which ZIP13 downregulation upon I/R contributes to the genesis of ferroptosis.
As a member of the SLC39A family, ZIP13 has been proposed to be localized in intracellular compartments (Fukada et al., 2008). A recent study by Fukada et al. demonstrated that ZIP13 mRNA expression in mouse hearts was downregulated by the cardiotoxic agent DOX and KO of ZIP13 resulted in irregular arrhythmia, indicating that ZIP13 is indispensable for the maintenance of normal cardiac functions (Hara et al., 2022). Similarly, we have also identified that ZIP13 protein expression is downregulated in mouse hearts upon reperfusion and ZIP13 downregulation contributes to the pathogenesis of I/R injury (Wang et al., 2021). Given that ferroptosis plays a role in cardiac I/R injury (Gao et al., 2015) and ZIP13 expression was downregulated by DOX (Hara et al., 2022), and established induction of ferroptosis (Fang et al., 2019), ZIP13 downregulation may play a role in the induction of ferroptosis in the setting of I/R in the heart. Our study revealed that KO or cKO of ZIP13 induced ferroptosis in physiological conditions and that the ferroptosis inhibitor Fer-1 prevented ferroptosis induced by I/R in ZIP13 cKO hearts. In addition, ZIP13 cKO exacerbated myocardial infarction in mouse heart subjected to I/R, an effect that was inhibited by Fer-1. Moreover, ZIP13 overexpression ameliorated I/R-induced ferroptosis. These results collectively suggest that downregulation of ZIP13 occurred during I/R plays a critical role in the induction of ferroptosis. Alternatively, ZIP13 downregulation contributes to the pathogenesis of myocardial I/R injury by inducing ferroptosis.
The mechanism underlying the induction of ferroptosis includes the accumulation of iron and ROS, GSH depletion, inhibition of system Xc−, and lipid peroxidation (Tang et al., 2021a). In this study, we found that I/R resulted in mitochondrial accumulation of Fe2+, which was enhanced by cKO of ZIP13. In contrast, I/R-induced mitochondrial Fe2+ accumulation was reversed by overexpression of ZIP13. These findings indicate that ZIP13 downregulation may induce ferroptosis by targeting mitochondria. Although mitochondria are critical for ROS generation in the setting of I/R and are involved in iron metabolism, it remains unclear if mitochondria are involved in ferroptosis. It has been reported that HT-1080 cells depleted of mitochondria were sensitive to ferroptosis inducers and ferrostatins (Gaschler et al., 2018). Likewise, in cells lacking mitochondrial DNA, ferroptosis still could be induced (Dixon et al., 2012). In addition, a recent study demonstrated that mitochondria are required for ferroptosis caused by cysteine deprivation but not for GPX4 inhibition-induced ferroptosis (Gao et al., 2019). However, there is growing evidence showing that mitochondria are involved in ferroptosis. It is well known that ferroptotic cells exhibit morphological abnormalities of mitochondria (Dixon et al., 2012; Tang et al., 2021a). Preventing mitochondrial lipid peroxidation with the mitochondria-targeted nitroxide could inhibit ferroptosis (Krainz et al., 2016). Recently, Fang et al. demonstrated that mitochondria play a critical role in either DOX- and I/R-induced ferroptosis in cardiomyocytes (Fang et al., 2019). Moreover, DOX induces ferroptosis by increasing mitochondrial Fe2+ accumulation and lipid peroxidation, and chelating mitochondrial Fe2+ can prevent DOX-induced ferroptosis in cardiomyocytes (Tadokoro et al., 2023).
Mitochondria are the main organelle for the intracellular iron utilization and regulation. Once being transported into mitochondria, iron is utilized for the biosynthesis of ISCs and heme. ISCs are essential components that consist of iron and sulfur and participate in various biological processes (Ward and Cloonan, 2019). The de novo biosynthesis of ISCs occurs on the inner mitochondrial membrane (Braymer and Lill, 2017) and a five-protein complex, including NFS1, FXN, ISCU, ISD11, and ACP, is needed for the process (Lill, 2009). The inhibition of NFS1 disrupts the ISC biosynthesis leading to iron starvation response and activation of ferroptosis (Alvarez et al., 2017). The iron starvation response triggered by NFS1 suppression can promote iron influx and release by increasing the transferrin receptor while repressing ferritin to promote cell death by ferroptosis. Likewise, FXN inhibition can interfere with the ISC assembly and induces ferroptosis by increasing lipid peroxidation (Du et al., 2020). In this study, cKO of ZIP13 inhibited expression of NFS1, FXN, and ISCU as well as the ISC-containing mitochondrial ETC subunit proteins. Moreover, overexpression of FXN prevented ZIP13 cKO-induced downregulation of ISC biosynthesis as well as ferroptosis. Interestingly, similar to the action of ZIP13 cKO, I/R also downregulated ZIP13 expression as well as the expression of NFS1, FXN, ISCU, and ETC proteins. Moreover, this effect of I/R was reversed by overexpression of ZIP13. These results indicate that ZIP13 downregulation leads to ferroptosis by suppressing ISC biosynthesis in the setting of I/R. Moreover, because the iron starvation response initiated by NFS1 suppression can increase intracellular free iron (Alvarez et al., 2017), the suppression of ISC biosynthesis by ZIP13 downregulation may account for mitochondrial Fe2+ accumulation in this study.
Although NFS1 suppression can trigger a robust iron starvation response, its alone does not cause ferroptosis because NFS1 suppression cannot decrease GSH levels or increase ROS (Alvarez et al., 2017), indicating that suppression of the ISC biosynthesis alone may not reach a threshold for the induction of ferroptosis. Inactivation or dysfunction of the system Xc−/GSH/GPX4 antioxidant system leads to ferroptosis (Chen et al., 2021). As a component of the system Xc−, SLC7A11 is involved in the production of GSH (Tang et al., 2021a). As an electron donor, GSH serves as a cofactor of the normal function of GPX4, an antioxidant enzyme that quenches phospholipid hydroperoxide. Inhibition of SLC7A11 by erastin or sorafenib triggers ferroptosis by depleting GSH (Dixon et al., 2012, 2014). In this study, cKO of ZIP13 reduced SLC7A11 expression and GSH levels, pointing that ZIP13 downregulation results in suppression of the antioxidant system. SLC7A11 expression and activity are regulated by factors such as TP53, NFE2L2, and BAP1. (Chen et al., 2017; Jiang et al., 2015; Tang et al., 2021a; Zhang et al., 2018). Although the mechanism by which ZIP13 cKO downregulates SLC7A11 and GSH remains unknown, SLC7A11 suppression followed by the reduction of GSH may cooperate with inhibition of the ISC biosynthesis to promote ferroptosis in this study.
In the present study, expression of the ETC subunit proteins was reduced due to the suppression of ISC, leading to the inhibition of mitochondrial respiration. While little is known about the relationship between the mitochondrial respiratory dysfunction and the induction of ferroptosis, the inhibition of mitochondrial respiration may contribute in a certain extent to myocardial injury caused by I/R. More studies are required to assess the role of the respiratory inhibition by ZIP13 downregulation in cardiac I/R injury.
In summary, we have demonstrated that ZIP13 downregulation upon I/R induces ferroptosis, which may contribute to the pathogenesis of myocardial I/R injury. Suppression of the ISC biosynthesis and the system Xc−/GSH/GPX4-dependent antioxidant system may underlie the mechanism by which ZIP13 downregulation leads to ferroptosis. The observation that both ZIP13 cKO and I/R lead to the inhibition of ISC biosynthesis and mitochondrial Fe2+ accumulation suggests that mitochondria play a critical role in the genesis of ferroptosis in the setting of I/R. An effective prevention of ZIP13 downregulation may be applicable in the treatment of patients with myocardial infarction or other diseases caused by ferroptosis.
Materials and Methods
Animals
C57BL/6 mice (male, 8–10 weeks) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). All the treatments and analyses were performed in a blind manner. All the experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (eighth edition) and all the procedures were approved by the Tianjin Medical University Animal Care and Use Committee.
Chemicals
Fer-1 was obtained from Selleck China. The antibodies for Ndufb8 (#ab110242), Fxn (#ab219414), ZIP13 (#ab106586), and 4-HNE (#ab46545) were purchased from Abcam. Antibodies for Tubulin (#11224–1-AP), Uqcrfs1 (#18443–1-AP), Ndufs1 (#12444–1-AP), Ndufs7 (#15728–1-AP), Nfs1 (#15370–1-AP), and ISCU (#14812–1-AP) were obtained from ProteinTech. Slc7a11 (#A13685), Ndufs4 (#A8691), Uqcrc1 (#A3339), and Ndufab1 (#A21114) antibodies were purchased from ABclonal. COXIV (#4850S) and GAPDH (#2118S) were purchased from Cell Signaling Technology.
Electronic laboratory notebook
Electronic laboratory notebook was not used.
Generation of the cardiac-specific ZIP13 conditional knockout mice
We constructed ZIP13 conditional knockout (cKO) mice with the CRISPR-Cas9 system. For details, readers can refer to our previous publication (Wang et al., 2021).
Cardiac I/R injury and infarct size measurement
C57BL/6 mice (male, 8–10 weeks old) were anesthetized with pentobarbital (80 mg/kg, i.p.). Following sedation, mice were intubated and ventilated. The ventilation frequency was set at 110 strokes/min (tidal volume, 150 μL/stroke). A left thoracotomy was performed around the third intercostal space, and the left anterior descending (LAD) coronary artery was reversibly ligated using sterile 7–0 silk suture with a slipknot. Proper ligation was confirmed by visual observation of the left ventricle wall turning pale. After 30 min of ischemia, the heart was reperfused for 24 h without ventilation. Fer-1 (1 mg/kg) was administered (i.p.) 7 days, 24 h, and 2 h before surgery. Sham-operated animals underwent the same procedure without ligation of the LAD coronary artery. The animals were re-anesthetized after a 24-h reperfusion period and the LAD was ligated again. Saline was administered before Evans blue dye (1%, Sigma) was injected through the abdominal aorta. After being excised, the heart was frozen at −20°C for 24 h and cut transversely into slices (6 slices/heart). There slices were incubated for 30 min with 1% 2,3,5-triphenyltetra-zolium chloride (Sigma) solution to visualize the unstained infarcted region. The infarct size and the area at risk were assessed by ImageJ.
Gene transfer
Recombinant adeno-associated virus 9 (AAV9) vector was used for gene transfer. The vectors carrying mouse ZIP13 with a CTNT promoter (AAV9-CTNT-Zip13-zsgreen) were constructed by Hanbio Biotechnology (Shanghai, China). AAV9- CTNT- zsgreen was used as a negative control. Polymerase chain reaction (PCR) was performed using the designed primer, and the target fragments were recovered using the agarose gel. The vector and target fragments were connected through homologous recombination, and single clones were selected for colony verification. The sequencing of the selected colonies was performed to extract the plasmid of the correctly sequenced clone samples. Finally, adeno-associated virus packaging was carried out using the plasmid. AAV9–CTNT–Zip13 and AAV9- CTNT [1.8 × 1012 vector genomes per mouse] were injected into mice (0.1 mL) via the tail vein. Studies were done 4 weeks after the injection.
Isolation of mouse cardiomyocytes
Mice were anesthetized with 2.5% avertin (0.01 mL/g, i.p.). Hearts were harvested rapidly and were perfused with a Langendorff apparatus. Hearts were perfused for 5 min with the buffer (120 mM NaCl, 5.4 mM KCl, 1.2 mM NaH2PO4, 5.6 mM glucose, 20 mM NaHCO3, 1.2 mM MgSO4, 5 mM taurine, 10 mM 2,3-buta-nedione monoxime). When there was a 10 mL perfusion buffer remaining in the reservoir, 5 mL of the collagenase buffer was added to the reservoir. The remaining buffer was discarded 2 min later and hearts were perfused with the buffer containing 100 μM Ca2+ for 20 min. Hearts were cut into pieces and the pieces were aspirated using a transfer pipette. The supernatants were removed into a 15 mL tube and were centrifuged (500 g) for 1 min. Then cell pellets were resuspended in the perfusion buffer 8 min three times. Ca2+ was added gradually to cells. Cells with a viability >90% were plated on a dish with M199 (Gibco) and 20% fetal bovine serum (FBS) (Gibco) and placed in an incubator humidified with 5% CO2−95% air for 2 h before experiments.
Cell culture
Immortalized human cardiomyocytes, the AC16 cell line was purchased from BeNa Culture Collection (Xingyang City, China). HL-1 cell line was purchased from Thermo Fisher. Cells were cultured in DMEM (Gibco) with 10% FBS (Gibco) at 37°C in an incubator humidified with 5% CO2−95% air.
Knockout of ZIP13 in HL-1 and AC16 cells
The CRISPR-Cas9 technique was adopted to generate ZIP13 KO HL-1 and AC16 cell lines (ZIP13 KO). Using the CRISPR Design Tool (https://zlab.bio/guide-design-resources), two pairs of single- guide RNAs (sgRNAs A and B) were designed. CRISPR-V2 plasmid was cut by BsmBI (NEB, USA) and was dephosphorylated by CIAP (TaKaRa, Japan). They were connected with CRISPR-V2 plasmid when sgRNAs were phosphorylated. The correct plasmids were used for transfection after being sequenced. Cells were screened with puromycin (Solarbio, Beijing, China) with a working concentration of 0.5 μg/mL for 5 days and then were continued to be cultured with puromycin with a working concentration of 0.25 μg/mL. When cells were full, the monoclonal cells were screened.
Mitochondrial isolation
Mitochondria were isolated from the left ventricle of mouse hearts. The mitochondria isolation kit was purchased from Beyotime, Shanghai, China.
Measurements of GSH, GSSG, and MDA content
For GSH and GSSG measurement, 20 mg of cardiac tissue was homogenized, and GSH and GSSG levels were measured with a commercially available kit according to the manufacturer’s instructions (ABclonal). Cardiac tissue (20 mg) was lysed with the extracting solution and MDA levels were measured with a commercially available kit according to the manufacturer’s instructions (Beyotime).
GPX4 activity assay
GPX4 activity was measured using a cellular glutathione peroxidase assay kit (Beyotime, Cat No. S0056). Cardiac tissue lysates were collected and measured according to the manufacturer’s instructions. A microplate reader was applied to measure the absorbance at 340 nm.
Confocal microscopy
A confocal microscope (Olympus FV1200) was used for cell imaging. To measure mitochondrial membrane potential in isolated cardiomyocytes, TMRE (10 nM) was loaded at 37°C for 30 min followed by two washes and the fluorescence was excited at 549 nm and collected at 574 nm. To measure mitochondrial Fe2+, isolated cardiomyocytes were loaded with Mito-FerroGreen (5 μM) for 30 min followed by two washes with PBS. The Mito-FerroGreen fluorescence was excited at 488 nm and collected at 525 nm.
Immunofluorescence staining
HL-1 or AC16 cells were preincubated with 200 nM MitoTracker Red CMXRos (Invitrogen) for 30 min at 37°C. Then, samples were fixed with 4% paraformaldehyde at 37°C for 10 min. Subsequently cells were permeabilized in 0.1% Triton-X 100/PBS and blocked with normal goat serum (1:10) in PBS for 50 min at 37°C. The samples were then incubated with the primary antibody against ZIP13 (1:100, Biorbyt) in PBS overnight at 4°C. Samples were imaged with a laser scanning confocal microscope (Olympus FV1200). Fluorescence images were analyzed with ImageJ to obtain integrated density/area.
Immunohistochemistry
Paraffin-embedded cardiac tissues were cut into 7-μm sections. After dewaxing and hydration, sections were retrieved in citrate antigen retrieval solution. Sections were stained using a commercially available kit according to the manufacturer’s instructions (BOSTER). After dehydration and vitrification, the sections were observed under a microscope. Images were analyzed with ImageJ to obtain integrated density/area.
H/R in mouse cardiomyocytes
Cells were cultured with a hypoxic solution (140 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM HEPES, 10 mM 2-deoxy-D-glucose, 10 mM sodium hydrosulfite) for 30 min. Cells were then reperfused by replacing the solution with the M199 containing 20% FBS medium for 30 min or 60 min at 37°C in an incubator (5% CO2−95% air).
Mitochondrial respiratory function and oxidative phosphorylation
Oxygraph-2k (Oroboros Instruments, Innsbruck, Austria) was used to measure the respiratory capacity of mitochondria. The respiration rate was determined with the substrate-uncoupler-inhibitor titration protocol. Data were recorded with DatLab software 5.2 (Oroboros Instruments). The respiratory leak of complex I was determined after titration with 10 mM glutamate and 2 mM malate in the absence of ADP (state 2). The complex I oxidative phosphorylation capacity was detected in the presence of 2.5 mM ADP (state 3). Succinate (10 mM) was added to induce maximal OXPHOS capacity. The respiratory control ratio (RCR) was calculated as the ratio of the respiratory rate in state 3 to that in state 2.
Western blotting
Equal amounts of protein lysates were loaded for sodium dodecyl sulfate-gel electrophoresis and were transferred to a polyvinylidene membrane. After blocking with 5% milk for 90 min at room temperature, the membrane was incubated with antibodies overnight at 4°C. The membrane was incubated with a secondary antibody and was visualized by enhanced chemiluminescence. Images were captured with Biospectrum Imaging System (UVP, Upland, CA).
Quantitative real-time PCR
Total RNA was isolated from tissues using TRIzol (Invitrogen), and RNA concentration and purity were measured using a Scientific NanoDrop (Thermo). RNA was reverse-transcribed using the GOcriptTM Reverse Transcription system Kit (Promega) in accordance with the manufacturer’s instructions. Quantitative PCR was performed using a CFX96 Real-Time System (Bio-Rad) and SYBR Green Supermix (Bio-Rad) in accordance with the manufacturer’s instructions. The fold difference in gene expression was calculated using the 2−△△Ct method and is presented relative to GAPDH mRNA. All reactions were performed in triplicate, and specificity was monitored using melting curve analysis (Table 1).
Primers for PCR Analysis
RNA-sequencing (RNA-seq)
To study the mechanism of ZIP13-induced ferroptosis, WT and ZIP13 cKO were analyzed by RNA-seq on the Illumina NovaSeqTM 6000 platform (Lc-bio Technology CO., Ltd.). Bioinformatic analysis of RNA-seq was performed at https://www.omicstudio.cn/tool.
Experimental protocol
All hearts were subjected to 30 min of ischemia followed by 24 h of reperfusion. Adult mouse cardiomyocytes were subjected to 30 min of hypoxia followed by 30 min/60 min of reoxygenation. Fer-1 (1 mg/kg) was administered (i.p.) 7 days, 24 h, and 2 h before the onset of ischemia. All samples for Western blotting or mitochondrial isolation were collected at the end of experiments.
Statistical analyses
Data are presented as mean ± standard deviation (SD). The number of samples is specified in figure legends. Statistical analyses were performed using SPSS17.0. Data were tested for normality distribution with Shapiro–Wilk test. Statistical significance was determined using the Student’s t test or one-way ANOVA followed by Tukey’s test. p Values
Footnotes
Authors’ Contributions
R.Z. and J.W. performed the experiments. Q.Y. and Y.Y. conceptualized and reviewed the article. X.C. designed the experiments and wrote the article. Z.X. supervised the project and wrote the article. All the authors approved the final version of the article.
Author Disclosure Statement
The authors have no conflicting interests to disclose in relation to this work.
Funding Information
This study was supported by the National Natural Science Foundation of China (NSFC) (grants 82270294 and 81970255 to Z.X.).
Supplementary Material
Supplementary Figure S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.