
Abstract
Significance:
Type 2 diabetes as a world-wide epidemic is characterized by the insulin resistance concomitant to a gradual impairment of β-cell mass and function (prominently declining insulin secretion) with dysregulated fatty acids (FAs) and lipids, all involved in multiple pathological development.
Recent Advances:
Recently, redox signaling was recognized to be essential for insulin secretion stimulated with glucose (GSIS), branched-chain keto-acids, and FAs. FA-stimulated insulin secretion (FASIS) is a normal physiological event upon postprandial incoming chylomicrons. This contrasts with the frequent lipotoxicity observed in rodents.
Critical Issues
: Overfeeding causes FASIS to overlap with GSIS providing repeating hyperinsulinemia, initiates prediabetic states by lipotoxic effects and low-grade inflammation. In contrast the protective effects of lipid droplets in human β-cells counteract excessive lipids. Insulin by FASIS allows FATP1 recruitment into adipocyte plasma membranes when postprandial chylomicrons come late at already low glycemia.
Future Directions:
Impaired states of pancreatic β-cells and peripheral organs at prediabetes and type 2 diabetes should be revealed, including the inter-organ crosstalk by extracellular vesicles. Details of FA/lipid molecular physiology are yet to be uncovered, such as complex phenomena of FA uptake into cells, postabsorptive inactivity of G-protein-coupled receptor 40, carnitine carrier substrate specificity, the role of carnitine-O-acetyltransferase in β-cells, and lipid droplet interactions with mitochondria. Antioxid. Redox Signal. 42, 566–622.
Introduction
Islets of Langerhans or pancreatic islets (PIs) secrete insulin, glucagon, pancreatic polypeptide (PPY), and somatostatin, and hence control metabolic homeostasis. This homeostasis must withstand fluctuations in food availability, energy consumption variations, and circadian rhythms. Physiologically, including regulations from central nervous system (CNS), this system is fine-tuned and controls secretion of hormones in responses to physiological stimulations. A central physiological role is ascribed to insulin secretion of pancreatic β-cells (Bessesen, 2001). The compounds stimulating insulin secretion are called secretagogues, and among them, glucose is the ultimate one (Campbell and Newgard, 2021). Therefore, glucose-stimulated insulin secretion (GSIS) is a major molecular mechanism, the details of which are yet to be better understood and refined.
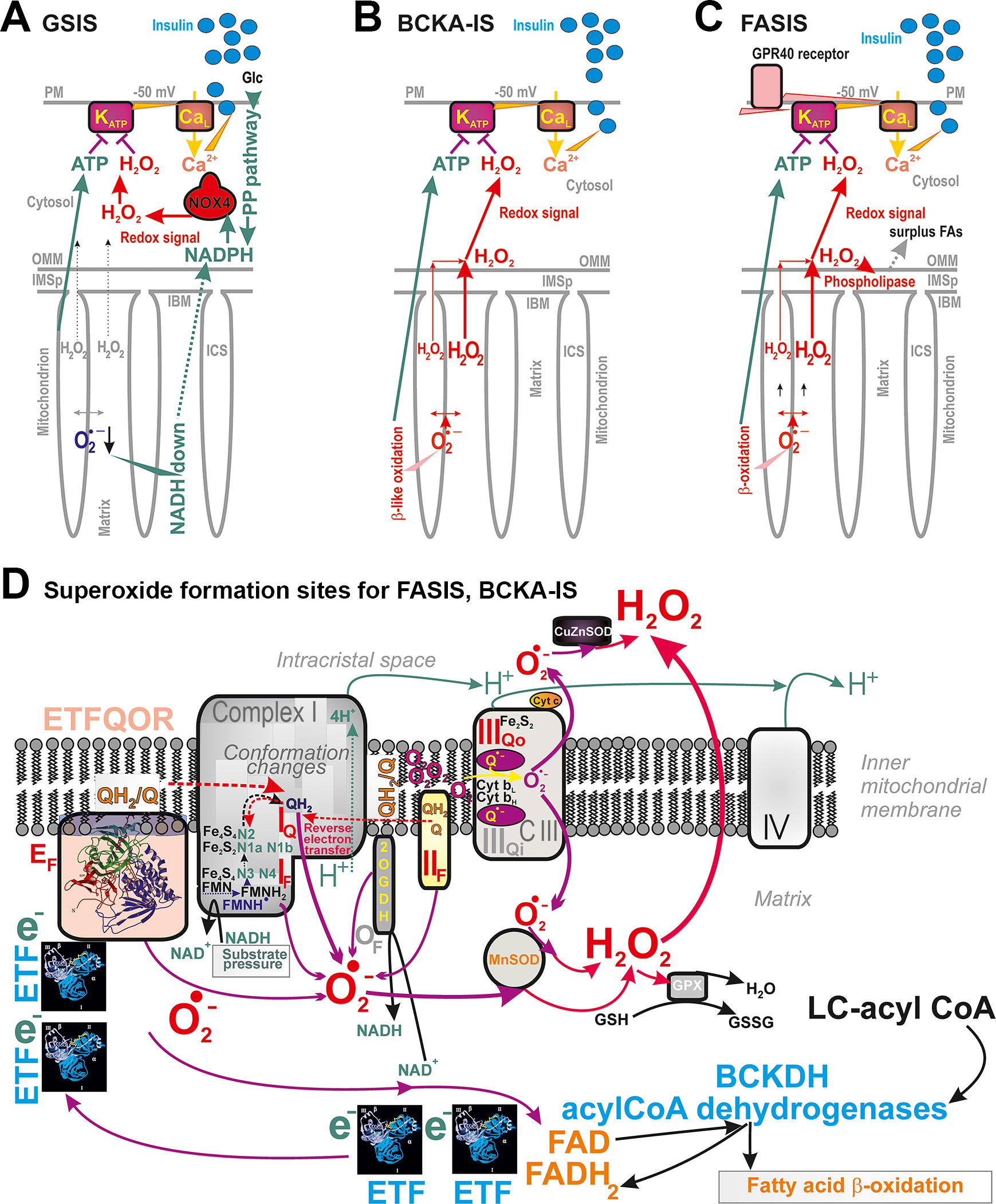
As described below, the participation of redox signaling in GSIS, originating from the NADPH-oxidase 4 (NOX4) reaction directly producing H2O2, belongs to recent discoveries, enriching a scheme for the detailed mechanism of GSIS (Fig. 1A). Redox signal (H2O2) was semi-quantified as it reached even the extracellular space of isolated PIs (Jabůrek et al., 2024). It has been demonstrated that the NOX4-derived H2O2 is essential for the required synergy to close ATP-sensitive K+ channels (KATP) (Plecitá et al., 2020). KATP could not be closed in NOX4-defficient cells. This finding also brought back a question: whether the increase of peri-plasma membrane ATP/ADP ratio is the key triggering mechanism or whether metabolic shuttles of various schemes are also essential (Merrins et al., 2022; Prentki et al., 2013), leaving mitochondrial ATP synthesis as a secondary supportive condition. For other secretagogues such as fatty acids (FAs) and branched-chain ketoacids (BCKAs), the source of the required redox signal is mitochondrial superoxide/H2O2 formation, substituting NOX4 (Fig. 1B, C). Representative molecular mechanisms are described in this review, as well as the related major sites of mitochondrial superoxide formation (Fig. 1D).
Stein et al., (1997) have recognized palmitic and stearic acids as potent insulin secretagogues. Palmitate increased Ca2+ currents and readability of releasable granule pool in pancreatic β-cells (Olofsson et al., 2004). However, a concept of FA-stimulated insulin secretion (FASIS) has not been introduced. Instead, free FAs were considered as amplifying species for GSISs (Carpinelli et al., 2002; Gehrmann et al., 2010; Graciano et al., 2011; Prentki et al., 2013). There were at least three reasons for such a point of view. First, it was the discovery of pathways for G-protein-coupled receptor 40 (GPR40/FFAR1, i.e., the one of metabotropic receptors) in pancreatic β-cells (Itoh et al., 2003). Features of the downstream GPR40 pathways were revealed, easing both KATP-dependent triggering of plasma membrane events (Cen et al., 2016; Feng et al., 2006; Graciano et al., 2013; Hauge et al., 2015; Hauke et al., 2018; Husted et al., 2017; Kristinsson et al., 2015; Latour et al., 2007; Sabrautzki et al., 2017; Tunaru et al., 2018; Yamada et al., 2016), as well as insulin granule vesicle (IGV) exocytosis, both increasing insulin secretion. The sole GPR40 pathway thus promotes IGV exocytosis (Olofsson et al., 2004). All long-chain FAs (LCFAs) (C12–C22), both saturated and unsaturated, were recognized to physiologically activate the GPR40 (Ghislain and Poitout, 2021).
The second reason paid attention to a concept of glycerolipid/free FA (GL/FFA) cycle, as it has also been recognized that GPR40 knockouts did not have completely abolished the so-called FA amplification of insulin secretion (Briscoe et al., 2003). Details of GL/FFA cycle will be described below. The third reason for the FA amplification concept was based on several observations claiming that FA-dependent triggering of insulin secretion requires sufficient glucose concentration. Nevertheless, the opposite experimental observations were reported where a low glucose concentration was present, at such concentrations which otherwise do not stimulate insulin release alone, but free FAs were still inducing insulin secretion alone as true secretagogues (Cen et al., 2016; Fernandez and Valdeolmillos, 1998; Ježek et al., 2015, Ježek et al., 2021, Ježek et al., 2022; Nyrén et al., 2012; Plecitá et al., 2020). Moreover, the requirement of redox signaling for the KATP closure was recently demonstrated in the case of free FA-dependent triggering of insulin secretion (Jabůrek et al., 2024). Therefore, the concept of FASIS must be renewed and further investigated.
The mechanisms of FASIS at low glucose concentrations are described below. However, the focus of this review will be given to the physiological meaning of FASIS. Why is such a mechanism developed, acting at low glucose for the purpose of facilitating glucose uptake to peripheral tissues by the additional insulin secretion? Could such FASIS cause hypoglycemia? Why would this mechanism lower the glycemia approaching to fasting values even more? This mechanism seems illogical. However, the key aspects lie in the timing. In humans, triglyceride (TG)-containing chylomicrons come after 3 h postprandially, when fat is included in the meal. Assessment of the insulin in a similar situation in mice with oral administration of liposomal TGs demonstrated that the insulin, peaking in three phases, is still present in the blood after 6 h (Jabůrek et al., 2024). It reached much higher levels than its initial levels prior to the meal simulation. Glycemia was much lower after 6 h than at its 10-min peak. This phenomenon was interpreted as resulting from the cleavage of TGs of lipoproteins, including chylomicrons, in PI capillaries by vascular extracellular lipoprotein lipase (LPL) (Nyrén et al., 2012). LPL could liberate free FAs for FASIS mechanism in vivo, ongoing at lower glucose concentration.
Moreover, FA transport protein 1 (FATP1/SLC27A1) was reported to be recruited by insulin to plasma membranes of white adipose tissue (WAT), that is, one of the peripheral tissues responding to insulin (Jain et al., 2009; Stahl et al., 2002; Wu et al., 2006a). Such recruitment is similar to the recruitment of the glucose transporter GLUT4 upon insulin activation of insulin receptor. The insulin-related upregulation of FATP1 was found also in placenta (Ruiz-Palacios et al., 2017). By regulating their own metabolism, FAs, via stimulating insulin secretion, represent a novel, yet uncovered detail of physiological regulation.
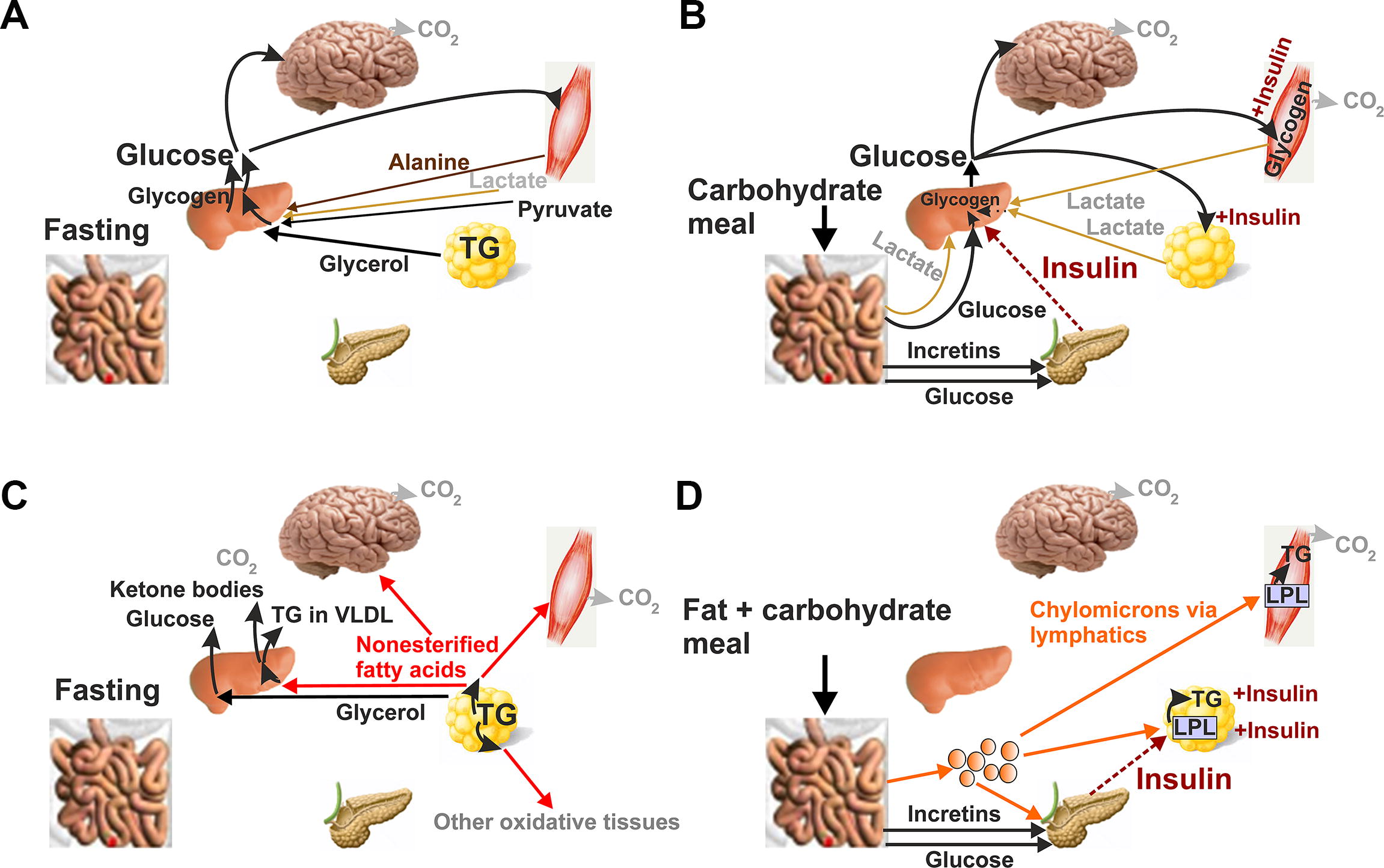
The key point of these considerations lies in the fact that glucose, TGs, nonesterified FAs (NEFAs) carried out by albumin, and several other metabolites in the plasma are being regulated, but at the same time they are the key regulators (Fig. 2). They act at the distinct timing in the pancreatic β-cells together with incretins (such as glucagon-like peptide 1 [GLP-1] or gastric inhibitory peptide [GIP]) and neurotransmitters of sympathetic nerve terminals executing the systemic control by brain (Bruning et al., 2000). Moreover, paracrine hormonal regulation by α-cells, δ-cells (Rorsman and Huising, 2018) and pancreatic polypeptide cells (PP-cells), and interactions with resident macrophages are superimposed. Such a hierarchy could be impaired in prediabetes and is disrupted or dysregulated in diabetes. The concept of FASIS with the exaggerated FA load is therefore viewed also as an initiating early event, adding on lipotoxicity effects in vivo. Investigations should be focused on these early events, when a slightly excessive FA load results in the earlier onset of FASIS, but also in prolongation of insulin secretion due to incretin action and delayed intestinal passage. Such mild starting hyperinsulinemia is a part of transformation of healthy physiological FASIS into (gluco)lipotoxic effects. This would be the key to understanding of type 2 diabetes etiology.
I devoted this review to supportive observations for the above outlined FASIS concept in vivo and its consequences. A similar concept should be considered for products of protein digestion and specifically for branched-chain amino acids. The exaggerated FASIS, in concert with pro-inflammatory responses of the immune system in β-cells as well in WAT, is then viewed as the main initiating mechanism of lipotoxicity leading to prediabetes and progressing to type 2 diabetes. Therefore, in this review, I shall discuss the physiological aspects of FASIS and its pathological consequences when the FA/lipid load is exaggerated.
Physiological Regulation of Blood Glucose
Human postprandial physiological regulations
Consequences of human PI structure
About 70% of spheroid PIs in humans have 50–250 µm dimensions being positioned along the main and interlobular duct of pancreas (Hellman, 1959). The remaining smaller islets are scattered through the acinar lobules. Their overall number in humans ranges between 5 × 105–5 × 106 and their total mass accounts only for 1%–2% of the pancreas. Islets consist of α-cells, secreting glucagon, β-cells secreting insulin, δ-cells secreting somatostatin, ε-cells secreting ghrelin, and PP-cells secreting PPY (Collombat et al., 2010; Muraro et al., 2016). Macrophages, T-lymphocytes, and other immune cells are also found in the pancreas (Hendley et al., 2021). Isolated human islets, representing the population of bigger ones, possess α-, β-, and δ-cells dispersed throughout the islet (Brissova et al., 2005). This contrasts with the isolated murine islets, in which α- and δ-cells reside at the periphery of the β-cell core. Murine islets were reported to contain 61%–88% of β-cells, 9%–31% of α-cells, and 1%–13% of δ-cells, whereas isolated human islets were much more heterogenous, with estimations of 28%–75% of β-cells, 10%–65% of α-cells, and 1%–22% of δ-cells (Brissova et al., 2005). The hormone release by each cell type can be viewed as an integrated input, while numerous paracrine relationships exist (Noguchi and Huising, Noguchi and Huising, 2019). Table 1 summarizes the main structural and consequent functional differences between human and rodent PIs.
Main Structural and Consequent Functional Differences between Human and Rodent Pancreatic Islets
IGV, insulin granule vesicle; TEM, transmission electron microscopy; TG, triglycerides; NEFA, nonesterified fatty acid.
Physiological level of GSIS in humans
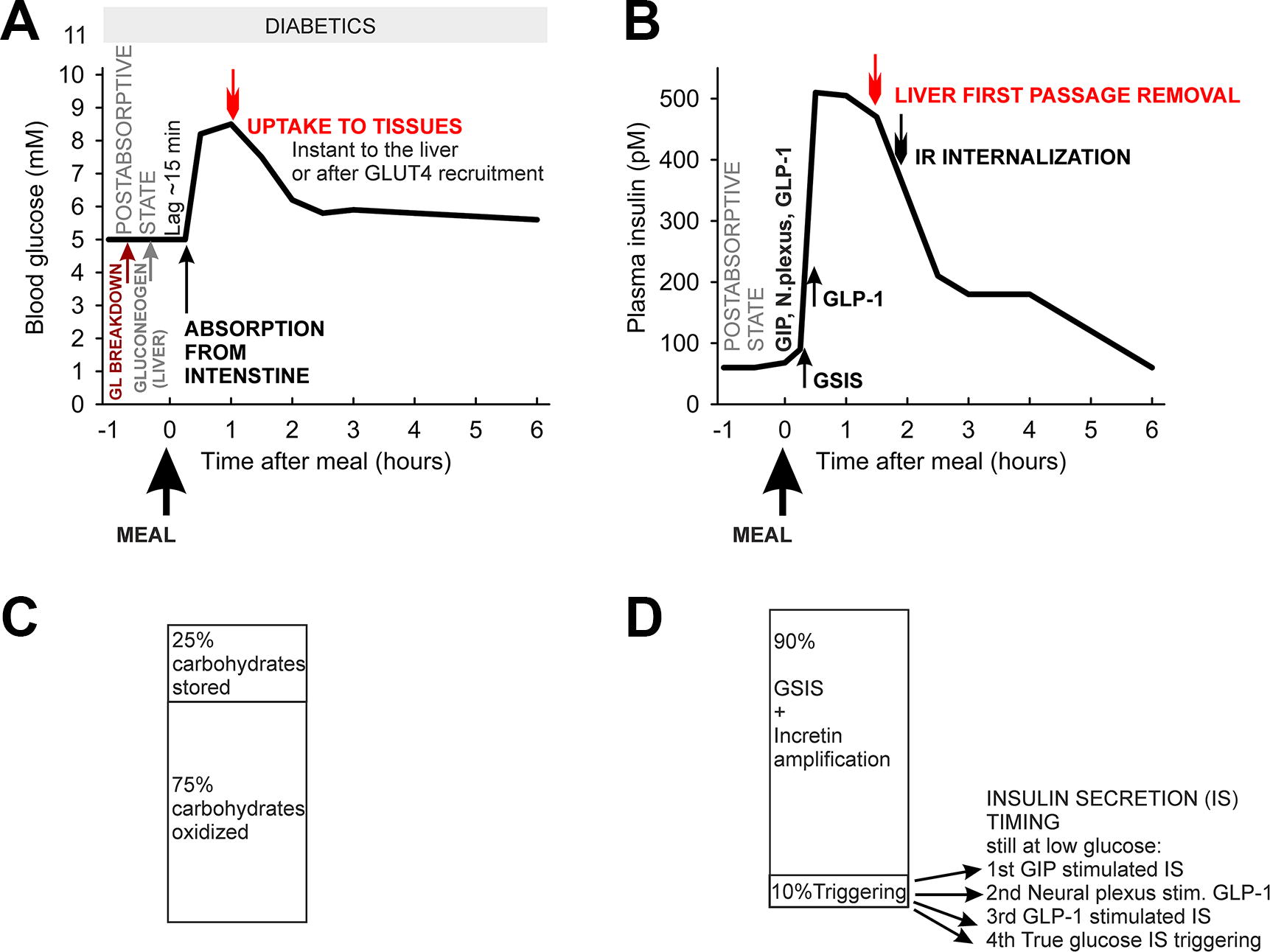
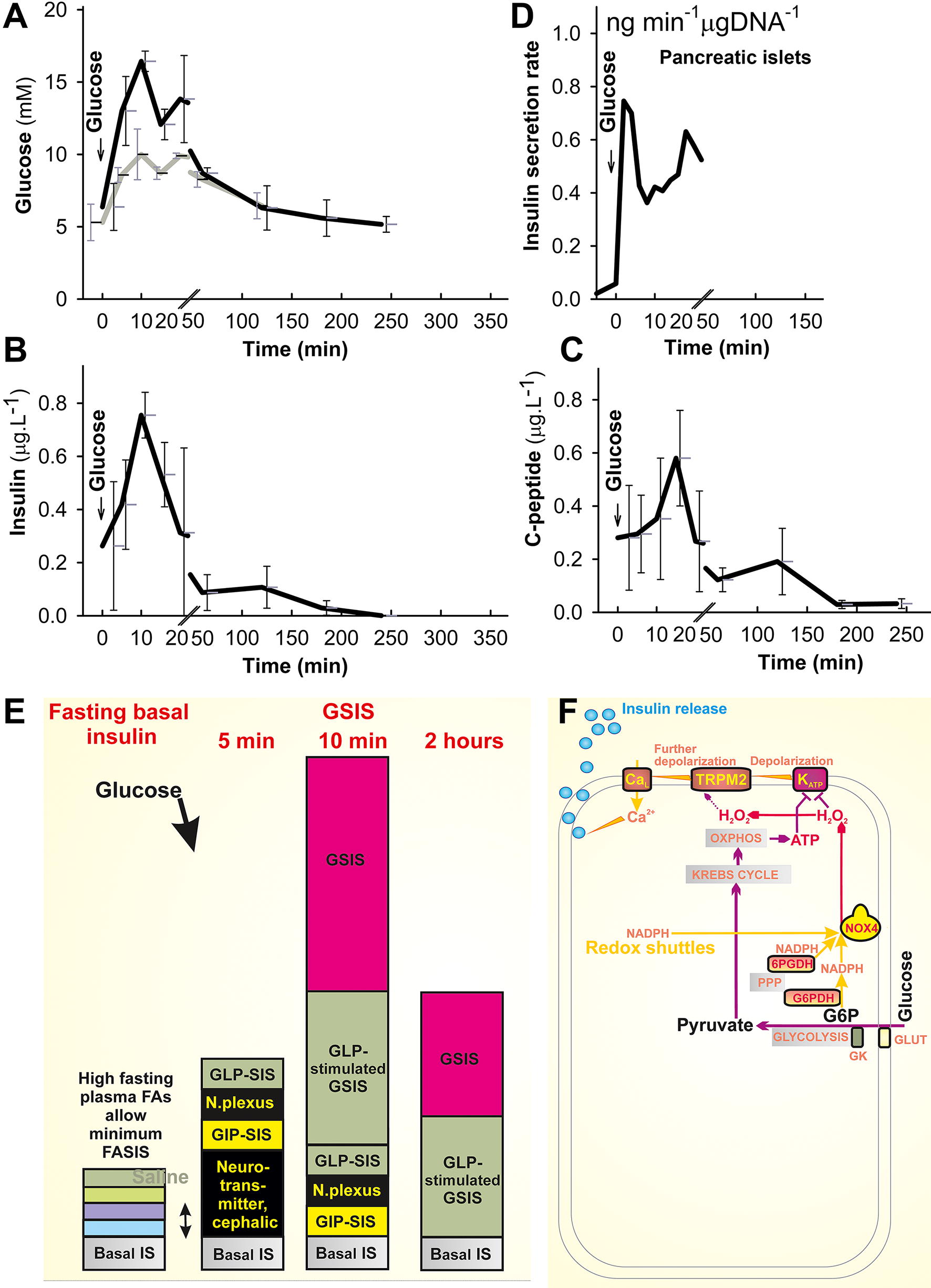
First, we describe physiological phenomena of GSIS in humans, with exception of a detailed incretin involvement, which will be discussed in the next chapter together with rodent physiology. In healthy humans, blood glucose (glycemia) of ∼5 mM is postprandially elevated up to 8–9 mM (Fig. 2B; Fig. 3). Constantly elevated blood glucose above 11 mM may have adverse effects and such glycemia reflects diabetes mellitus. The major glucose source for the blood in fed state is the absorption from the intestine.
From blood, glucose uptake to the tissues occurs by specialized GLUT transporters. GLUT1 (SLC2A1) is the primary glucose transporter in human pancreatic β-cells, having a K m of 6 mM, which is lower than K m of 11 mM for GLUT2 (SLC2A2) of rodent β-cells (Heimberg et al., 1995; McCulloch et al., 2011). In humans this enables to initiate insulin secretion at 3 mM glucose, whereas in mice it enables insulin secretion at >6 mM glucose. All GLUT1 and GLUT2 transporters, present also in the liver, kidney, and intestine, allow glucose to be equilibrated across the cell membrane. Consequently, cytosolic glucose equals its plasma concentrations. In contrast, the skeletal muscle, WAT, and heart express predominantly GLUT4, which is recruited to the plasma membrane by insulin. GLUT4 thus allows insulin-dependent glucose uptake to numerous tissues, including macrophages (Dror et al., 2017) and tumor cells.
Despite brain insulin receptor signaling playing a minor role in glucose regulation, it is important for mitochondrial function and food intake (Milstein and Ferris, 2021). Note that different organs exhibit different glucose processing rates, due to a distinct pattern of four rate-limiting enzymes of glycolysis, product inhibition of hexokinases (not existing for β-cell glucokinase, see further), and distinct GLUT isoforms. These rates could slow down if some of these key proteins are mutated.
To describe the physiological course of GSIS, it is helpful to introduce the term postabsorptive state, defining a situation when all of the last meal has been absorbed from the intestinal tract but without any excessive starvation. In humans, this happens after an overnight fast. Typical plasma insulin is then ∼60 pM (Frayn, 2010). In this postabsorptive state, glucose entering the blood comes only from the liver, partly originating from the breakdown of glycogen and partly due to gluconeogenesis from lactate (a minor one from pyruvate) (Fig. 2A). Skeletal muscle glycogen could contribute only indirectly upon anaerobic glycolysis after glucose conversion to lactate, which is then used for gluconeogenesis in the liver. About half of glucose is utilized by the brain. Among other circulating metabolites, alanine comes from skeletal muscle and glycerol from adipose tissue (Grabner et al., 2021).
The predominantly carbohydrate meal, for example, 130 g with 33 g fat, causes an increased glucose concentration in the blood after about 15 min in humans, while glycemia peaks at 30–60 min (Frayn et al., 1993) (Fig. 3). Of course, the exact timing depends on the size of the portion, fiber amount, and simple versus complex carbohydrate content. During up to 30 min, insulin levels peak at 500 pM from the resting 60 pM. If no other phenomena occur, insulin is lowered to about 30% of maximum after 3 h and reaches the initial 60 pM level after 6 h (Fig. 3). Glycemia peaks from the initial 5.5 mM up to 8.5 mM after 60 min and falls to 6 mM after 3 h. A similar course is observed for lactate, as there is a partial switch to anaerobic glycolysis in muscle and adipose tissue. Typically, 5 h after this standard meal, 25% of ingested carbohydrates will be stored (12.5% in the liver) and 75% oxidized (Frayn, 2010; Frayn et al., 1993). Also, about 75% of the insulin‐induced glucose uptake is taken up into the skeletal muscle, whereas this is substantially reduced in patients with type 2 diabetes and obesity (Baron et al., 1988).
If only a glucose bolus is given to healthy individuals, the rise of insulin release to the blood is sharp, and its existence is termed as the first GSIS phase. This is followed by the second GSIS phase with about a half amplitude and prolonged decay up to 1 h (Villard et al., 2018). At the progressed type 2 diabetes, the first phase is often missing, whereas the second phase is enhanced and more prolonged.
What are the major consumers of glucose and why is insulin declining? The reason for declining glycemia is insulin-induced glucose uptake via the GLUT4 transporters, recruited to plasma membranes of skeletal muscle, WAT, heart, and other tissues. The liver, which receives blood through the portal vein from the intestine, is affected by the largest change of glucose levels. Because hepatocytes also express GLUT2, blood glucose is equilibrated with the intracellular glucose concentration. As a result, hepatocyte glycogen phosphorylase is inactivated, therefore blocking the glycogen breakdown, whereas glycogen synthetase is activated. This event contributes to the glucose consumption from the blood, and it lasts for period of 1–2 h after the initial meal load. At the same time, gluconeogenesis is not inhibited. Nevertheless, the resulting glucose cannot leak to the blood, as glucose-6-phosphate is mostly consumed by the activated glycogen synthesis.
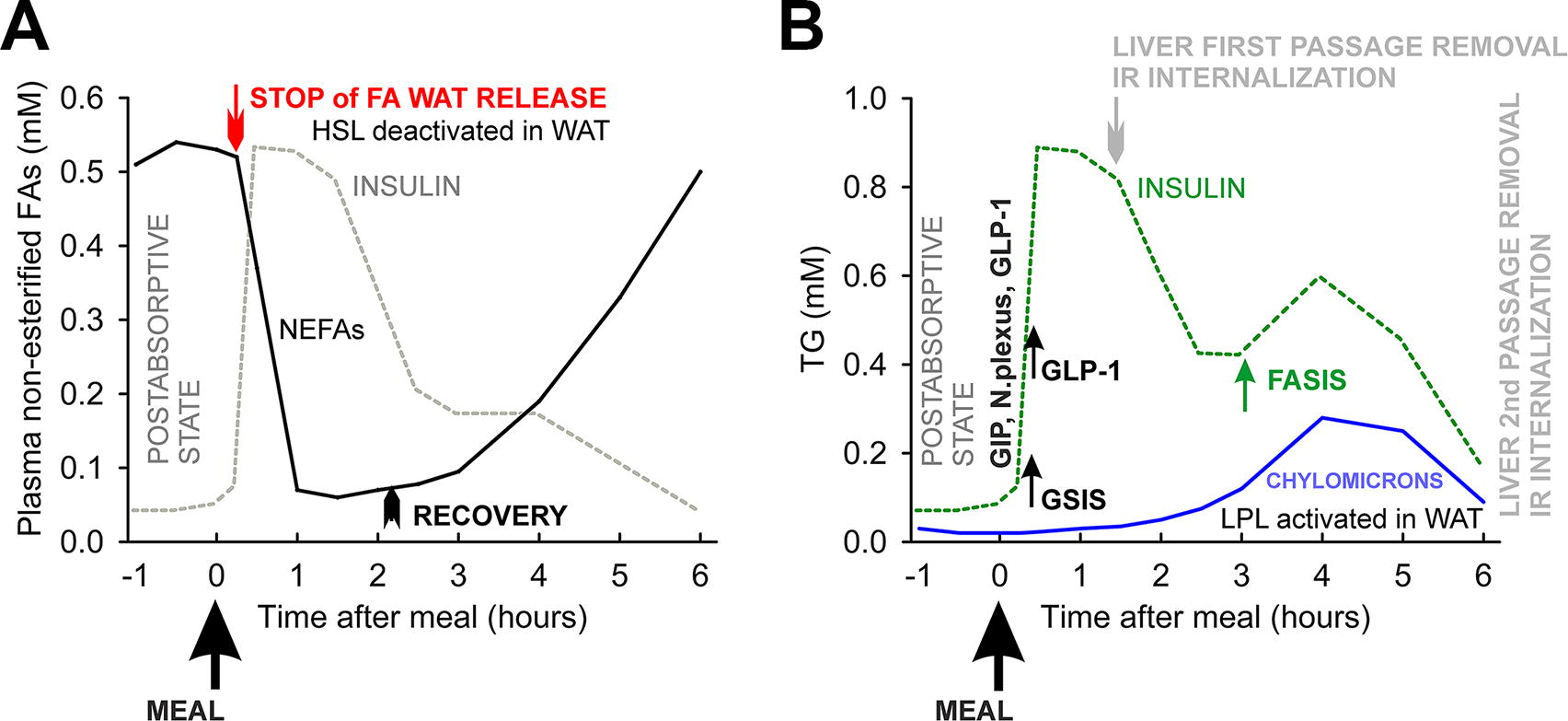
A parallel insulin stimulation of GLUT4-mediated glucose uptake in the skeletal muscle, WAT, and heart also changes the metabolism of FAs in these tissues. Increasing insulin secretion inhibits FA release from WAT (Fig. 2C, D; Fig. 4) and ketogenesis in the liver (3-hydroxybutyrate and acetoacetate release to the blood). Insulin is removed from the circulation by internalization after binding with insulin receptors on the cell surface. After internalization, insulin is proteolytically degraded. About 70% of the insulin reaching the liver is removed in its “first passage” (Frayn, 2010). This is modified when there is more fat in the meal and these situations will be discussed after the description of general FA physiology.
Lipids in circulation
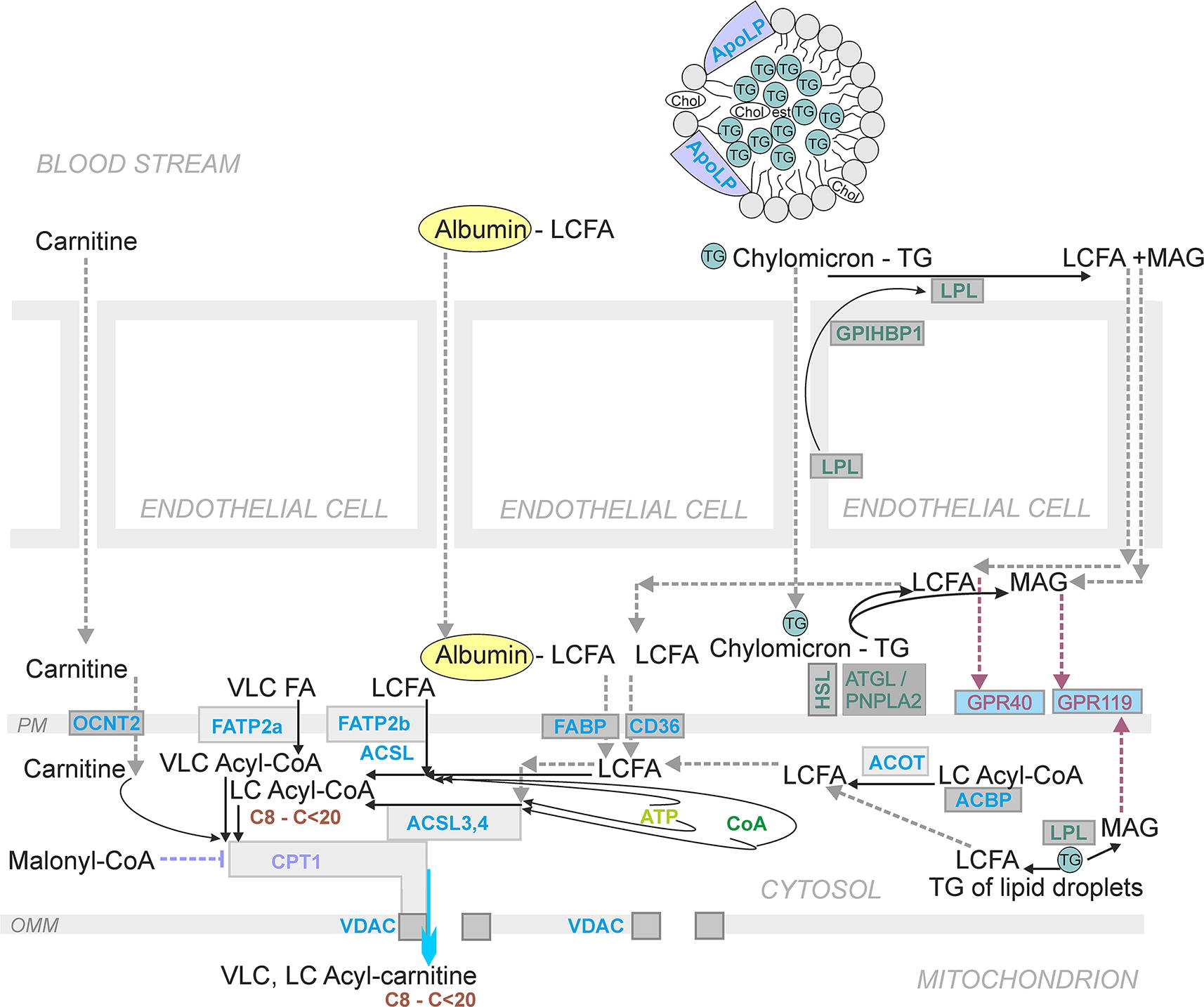
FAs are either NEFAs bound to albumin or serve as constituents of triglycerides (TGs) in lipoprotein particles, among which the largest are chylomicrons. Other lipoproteins of distinct sizes exhibit their specific physiological function. Neglecting individual variations, TG amounts ∼1 mM after overnight fast and almost none is present in the form of chylomicrons (Frayn, 2010) (Fig. 4B). The chylomicrons come late after a fat-containing meal. Surprisingly, NEFAs exhibit a fast turnover in the circulation as inferred from the experiments where a fast turnover of labeled FAs after its intravenous injection was estimated. The label disappeared within a few minutes. Spectrum of NEFA species contains ∼30 different FAs, among which palmitic, oleic, and stearic acid account for ∼78% (Sobczak et al., 2019). In the plasma, NEFAs come mainly from WAT, from which they are mobilized by adipose triglyceride lipase (ATGL/PNPLA2, i.e. patatin-like phospholipase domain-containing protein 2, termed alternatively iPLA2-ζ; Grabner et al., 2021) with a participation of hormone-sensitive lipase (HSL). HSL has a prominent role in hydrolyzing diacylglycerols (DAGs) to monoacylglycerols (MAGs). MAGs can be finally degraded to FAs and glycerol by monoglycerol lipases or ABHD6. NEFAs are bound to albumin, present in ∼0.6 mM in circulation (Nishi et al., 2020). Albumin has up to seven FA binding sites. Therefore, the maximum binding could be ∼4 mM FA bound, existing in an equilibrium with free FAs of ∼200 nM.
In enterocytes of a small intestine, LCFAs in the form of acyl-CoAs and MAGs are condensed first to DAGs and then (with the second acyl-CoA) to TGs. TGs are used to form 0.1–1 μm particles termed chylomicrons when encompassed by apolipoprotein B together with cholesterol. Chylomicrons enter circulation via the lymphatic system up to thoracic duct and thus bypass the liver (Fig. 2D). Because medium-chain (MC) acyl-CoA synthases are not expressed in enterocytes, MCFAs enter the capillary plasma directly.
NEFA levels (i.e., FAs bound to albumin) in circulation are inverse to glycemia, being the highest at fasting state (Fig. 4A). After a carbohydrate meal, their levels fall, with minimum reached after 1 h and rising again after 3 h, being back at the initial maximum levels after 6 h (Frayn, 2010). WAT lipolysis in a fed state is called basal, whereas during fasting, it becomes an induced lipolysis. The switch between them is ensured by regulations of ATGL and HSL at multiple levels, including transcriptional reprogramming, post-translational protein modifications and regulation of enzyme activators and inhibitors for ATGL (Grabner et al., 2021). Plasma adrenaline and norepinephrine of sympathetic nerve terminals in WAT also activate HSL and lipolysis. Thus, the canonical activation of lipolysis in WAT includes the pathway of β-adrenergic receptor, Gαs protein, and cyclic AMP–protein kinase A (cAMP–PKA) pathway. Its deactivation by the insulin receptor pathway is the major way in how insulin suppresses lipolysis. This lipolysis suppression by insulin is withdrawn in WAT when insulin levels are low. Hence, in the postabsorptive state, NEFAs are supplied to circulation by WAT. The uptake of FAs occurs simultaneously mostly into the skeletal muscle, heart, and liver. Only ∼10% of FAs are re-esterified in WAT overnight and when fasting is extended, this part goes to zero.
A half-maximum suppression of WAT lipolysis occurs at 120 pM insulin, that is, before its peak at ∼500 pM is reached 30 min after the abovementioned standard 130 g meal. However, even at peaking insulin, WAT lipolysis is not completely suppressed. NEFAs fall to 0.1 mM. Note that ketone bodies, represented namely by 3-hydroxybutyrate and acetoacetate, decline from 0.1–0.2 mM to 0.02 mM. After 3–5 h at declining insulin, WAT lipolysis is reactivated and plasma NEFAs increase again (Fig. 4A).
Chylomicrons
A meal, with both carbohydrate and fat, results in the production of chylomicron particles, which are slowly transferred into the bloodstream. As a result, the peak in plasma TG concentration occurs at 3–5 h, which is at already declining insulin from GSIS. Note also that the TG fraction of chylomicrons accounts only for ∼18% (Griffiths et al., 1994). The lifetime of chylomicrons depends on TG removal from them in the WAT, skeletal muscle, and heart, where insulin-activated vascular extracellular LPL cleaves TGs of lipoproteins into FA and DAG. Note that insulin also increases the export of lipase expressed in myocytes to the luminal surface of endothelial cells. The peak of lipase stimulation is 3–4 h. Therefore, the maximum LPL activation perfectly coincides with the peak of chylomicrons. LPL in the skeletal muscle is slightly suppressed by insulin to use glucose when insulin is still around but it is stimulated by physical activity. In WAT, most of the FAs liberated from TGs are esterified to form new TGs for storage. In rodents, chylomicrons are expected to come after 30–60 min (Ee et al., 2000; Nauli et al., 2006).
Rodent physiology of insulin secretion
Rodent physiology of insulin secretion
Let’s point out important differences between murine and human anatomy (Alanentalo et al., 2007) and physiology of insulin secretion. Besides more ordered and electrically synchronized β-cells occupying the islet core, mice and other rodents possess typical IGV of a fried egg-appearance in transmission electron microscopy sections. Numerous other differences were described in the excellent reviews (Rorsman and Ashcroft, 2018) (Table 1).
Due to rich innervation (Rodriguez-Diaz and Caicedo, 2014), the ultimate start for insulin secretion in rodents (Fig. 5) and other animals are neurotransmitters released in response to the sight or smell of food (Wiedemann et al., 2022) (Fig. 5E). Note, humans do not have rich innervations of nearly each β-cell within the islet (Rodriguez-Diaz et al., 2011a), and consequently, these effects are less pronounced. Next and earlier than metabolite-stimulating insulin secretion, insulin is released due to the action of incretin peptide hormones (Shilleh et al., 2024), namely GIP (Liskiewicz and Müller, 2024), secreted in the proximal small intestine by K-cells of duodendum (Svendsen et al., 2016) and GLP-1, secreted by the enteroendocrine L-cells of the distal small intestine, that is, jejunum and ileum, plus in the colon (Müller et al., 2019; Nauck and Müller, 2023). Enteral ingestion of carbohydrate, fat, and protein induces the first GIP secretion. As early as digested nutrients leave the pylorus, neuroendocrine reflexes stimulated by GIP trigger GLP-1 secretion (Steinert et al., 2017). Later, when nutrients reach the distal gut and thus directly affect the L-cells, the additional secretion of GLP-1 into circulation takes place (Müller et al., 2019; Steinert et al., 2017). Note that fructose induces GIP and GLP-1 secretion (Seino et al., 2015) and hence indirectly stimulates insulin secretion after subsequent GIP and GLP-1 stimulation of PI β-cells. Chronic fructose also renders increased responses of β-cells to glucose by extrusion of ATP via Panx1 channels with concomitant activation of P2Y1 purinergic receptors, the downstream pathway of which activates insulin exocytosis (Bartley et al., 2019).
GIP stimulates glucagon secretion from pancreatic α-cells. It also accelerates the uptake of glucose and fat, namely TG into WAT. Besides suppressing glucagon secretion and inhibition of gastric emptying, GLP-1 stimulates insulin secretion from pancreatic β-cells. By the conventional terminology, GLP-1 “amplifies” or “potentiates” the nonstimulating effects of fasting low glucose. This accounts for 4–5 mM in humans and 7–10 mM in mice (Frayn, 2010; Li et al., 2009; Plecitá et al., 2020; Remedi et al., 2011). Thus, the net metabolic stimulation comes only as the fourth initiator at the moment, when the plasma glucose content becomes elevated. The incretin effect explains why the intravenous (i.v.) administration of glucose causes lower insulin secretion response than the oral administration. The incretin effect also explains the rather small (∼30%) response in isolated human islets upon glucose increase from 5 to 7.5 mM (Walker et al., 2011), whereas the same in vivo increase in plasma glycemia from 5 to 7.5 mM provides up to a five-fold increase in insulin (Vollmer et al., 2008).
Unlike GLUT1 in humans with a K m of 6, allowing GSIS initiation at 3 mM glucose; GLUT2 in rodents with K m of 11 (but with a high transport capacity) (Thorens, 2015) enables GSIS starting at glucose over 6 mM. In rodents, insulin secretion reaches a half-maximum at 10–12 mM glucose and saturates at glucose concentrations above 20 mM. Supraphysiological experiments with intraperitoneal glucose administration in mice show the clearly distinguished two GSIS phases (Fig. 5A–C). The first phase with peaking plasma insulin and glycemia at ∼10 min with c-peptide peaking at ∼20 min. Within the second phase, insulin declines below 25% of the peak occurs after 1 h with a minor elevation at 2 h, and a final decline up to 4 h to minimum basal values exist. Similar time course exists for c-peptide as a residual from the insulin maturation in IGVs, which could be more resistant to internalization and degradation (Fig. 5C). Glycemia in these experiments exhibits a minor (1.5th phase?) peak at 20–30 min, declining steadily up to 4 h to the initial 5 mM. Even oral glucose administration in mice, which has only <2 mL of blood, will lead to an insulin peak at 10 min and glycemia double peaks at 10 and 20 min. The latter is apparent with a much lower amplitude of 10 mM with saline administration only (Fig. 5A). This reflects the above mentioned cephalic (neurotransmitter) and incretin early fast phases initiating physiological insulin secretion.
Nevertheless, when perifusion of isolated PIs is performed and the rates of insulin release are calculated for each 2 min (Fig. 5D), the rate is maximum immediately at about 5 min, its decline below 50% spontaneously occurs after 6 min, while the rate of insulin secretion is nearly restored after 30 min and returns to the early peak at 40 min. Thus again, the two phases are clearly distinguished as in vivo. Note, however, that physiological postprandial glycemia does not reach values such as 15 mM. When experiments with a stepwise glucose increase were performed, the visible first phase disappeared (Doliba et al., 2012; Otani et al., 2004). Nevertheless, studies of the first phase are important for understanding the triggering mechanism of insulin secretion, as they are typically missing in islets of diabetic subjects (Groop et al., 1991; Hosker et al., 1989). The first and second phases after an initiator triggering insulin secretion concern with the IGV exocytosis. The ready IGVs, residing at the proximity of plasma membranes, undergo exocytosis initially, thus substantiating the first GSIS phase, whereas those required to be recruited from distant deeper positions then substantiate the second phase. The latter process includes also softening of the cytoskeleton barrier for IGVs and is out of scope of this review (but see Refs. Gaus et al., 2022; Izumi, 2023; Vakilian et al., 2019).
Concept of FASIS
Why introduce the concept of FASIS
Concept of independent mechanisms stimulating insulin secretion
The current paradigm assumes that there is a single master mechanism of GSIS with a key role of closing KATP channels and opening of CaV Ca2+ channels. Stimulations with other secretagogues such as FAs and stimulations by incretins are referred to as an amplification (Ashcroft and Rorsman, 2012; Henquin, 2009; Ho et al., 2023; Maechler, 2013; Rorsman and Ashcroft, 2018; Rutter et al., 2015; Seino, 2012; Seino et al., 2017). This traditional concept however cannot withstand the above mentioned description of initiating physiological events (Fig. 5E), when an enteral ingestion of meal induces the first GIP-stimulated insulin secretion (GIP-SIS) in pancreatic β-cells, which are still not in the contact with elevated glucose. Why consider this as an amplification of a master GSIS mechanism?
Next, neuroendocrine reflexes stimulated by GIP trigger GLP-1 secretion (Steinert et al., 2017). After reaching the distal gut, ingested nutrients directly stimulate the L-cells to secrete GLP-1 into the circulation (Müller et al., 2019; Steinert et al., 2017). If this is fast, at the beginning of such GLP-1 secretion, glucose is still lower in the islets. Therefore, we can define a net GLP-1-stimulated insulin secretion (GLP-SIS) mechanism ongoing at low glucose concentration. This mechanism is observed in experimental settings (Ježek and Jabůrek, unpublished; see also Georgiadou et al., 2022). Of course, the canonical GSIS starts as soon as glucose in the plasma becomes elevated and reaches the PIs. Mechanisms of GIP-SIS, GLP-SIS, and the net GSIS synergize and a sum of their exocytosis leads to a higher insulin release, unless the maximum capacity of IGV ready for the earliest triggering (the first phase) is overcome. Various G-protein coupled receptors are involved also in autocrine signaling in pancreatic β-cells, including the pathway of insulin receptor (InsR) (Varney and Benovic, 2024). Autocrine insulin was suggested to elevate arginine-stimulated insulin secretion (Halperin et al., 2022).
Similarly, mechanisms of insulin secretion stimulated by FAs should be considered as GSIS-independent, despite ongoing typically after the physiological GSIS. Note that in the postabsorptive state, where NEFAs are at their peak, there is no significant insulin secretion. Thus any, yet theoretically possible, NEFA interaction with GPR40 metabotropic receptors does not exist. Of course, this fact led to the concept of amplification, where effects of FAs on insulin secretion were superimposed onto GSIS and incretin-SIS. However, there is experimental evidence for FAs acting at low glucose concentrations (Fig. 6) (Jabůrek et al., 2024; Ježek et al., 2015) and for the stimulation of insulin secretion with high-affinity nonmetabolizable GPR40 agonists (Jabůrek et al., 2024). The existence of these phenomena allows the definition of FASIS (fuel plus nonfuel) and its independent receptoric (nonfuel) branch, respectively.
Concept of FASIS
The concept of FASIS comes from the general view that all the above described postprandial metabolite/hormonal timing, including the metabolite pattern in the postabsorptive state, affects pancreatic β-cells as well. In the postabsorptive state, β-cell resting metabolism is based on the utilization of low glucose concentrations plus partly on FA β-oxidation (speculatively including FAs of lipid droplets [LDs]). FAs for β-oxidation could come also from albumin (the NEFA fraction), but these NEFAs evidently do not stimulate FASIS. This is because despite the plasma, NEFA content is highest at the postabsorptive state, and only minimum basal insulin secretion takes place. The real physiological FASIS occurs when either intracellular LPL (Nyrén et al., 2012) cleaves intracellular TGs, such as stored in LDs; or when extracellular LPL cleaves TGs of lipoproteins, including those of chylomicrons, after its transendothelial recruitment to the lumen of islets capillaries (Cruz et al., 2001; Marshall et al., 1999; Nyrén et al., 2012; Winzell et al., 2006) (Fig. 7). Also, adipose TG lipase (ATGL/PNPLA2) may cleave TGs in β-cells (Peyot et al., 2009). LPL of endothelial cells is recruited upon its transport to the luminal surface of islet capillaries. The transport is enabled by glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1; Davies et al., 2010). Hence the liberated TG cleavage products, free FAs, and MAG may diffuse toward β-cells (Fig. 7).
When free FAs are excessively liberated or if the GPR40 metabotropic receptor can compete with albumin, the GPR40 pathway should also be partially activated. However, a resting regime of GPR40 should be involved in the postabsorptive state, when only the basal insulin secretion takes place. Later, in humans presumably after 3 h from the standard meal, when chylomicrons are peaking, (Fig. 4), FAs must restore their effects to GPR40 while being cleaved by LPL in islets capillaries. However, the FAs cleaved from TGs of chylomicrons are also taken up by β-cells and metabolized by the restored FA β-oxidation. Because such liberated FAs are also capable of stimulating insulin secretion, such an additional insulin should be secreted, representing the physiological FASIS. Note that this additional insulin due to FASIS is secreted at glycemia of 5.5 mM, that is, very close to the glycemia of the postabsorptive state.
Independently of how we call this phenomenon, we should discuss the role of insulin at glycemia approaching its low (fasting) values and investigate the molecular mechanisms of insulin secretion, hereafter termed FASIS.
Experiments in mice leading to the FASIS concept
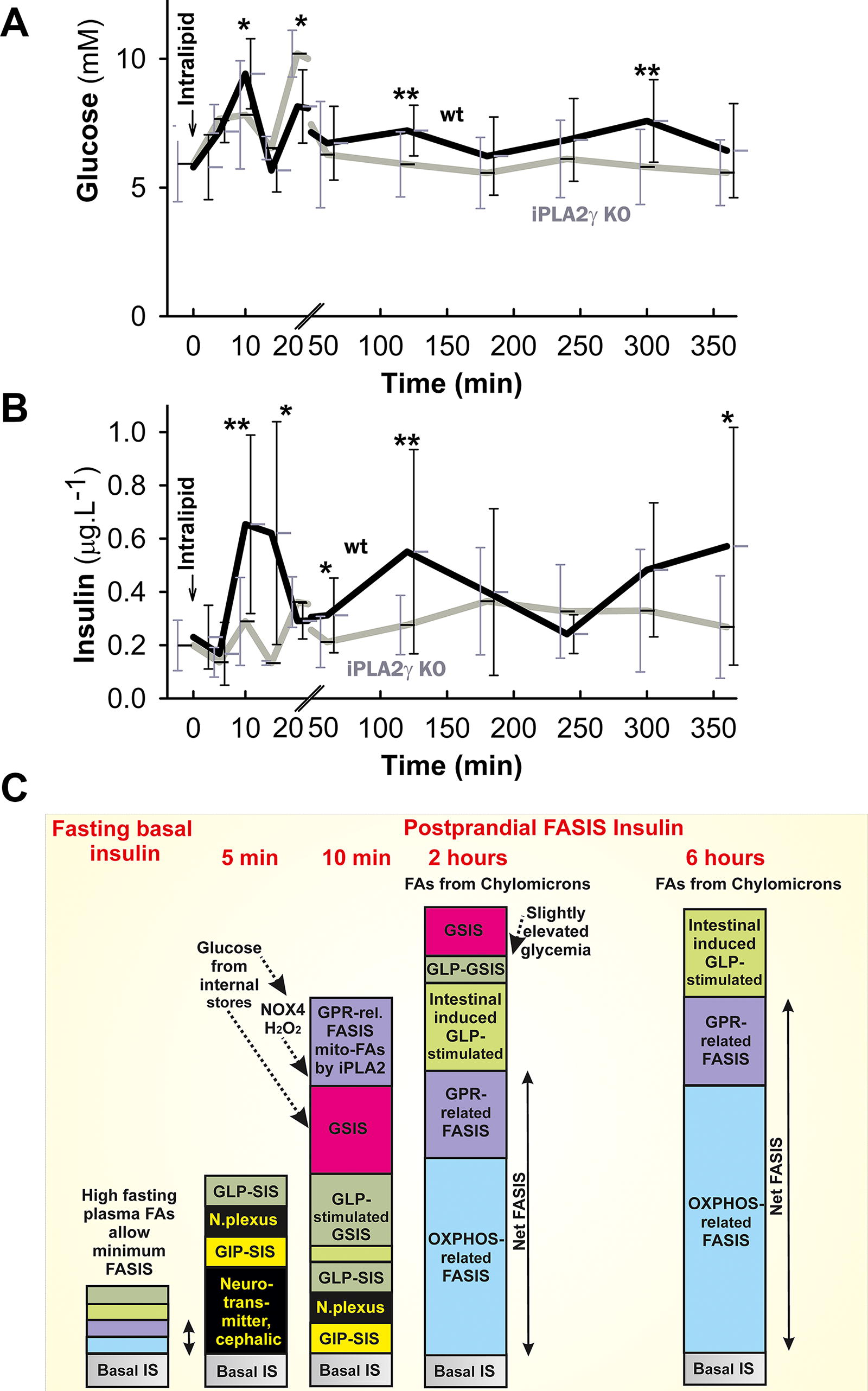
Studies of FASIS in humans were scarce. One can derive some predictions based on analogical investigations with mice or rats. We have employed an intralipid, which is composed of soybean oil and phospholipids, that is, liposomal triglycerides, representing artificial lipid droplet LDs. After oral administration of intralipid to mice, three peaks of insulin secretion were observed for up to 6 h (Fig. 6A, B). Peaks of c-peptide release appeared to be slightly delayed but were roughly synchronous with the insulin peaks (Jabůrek et al., 2024). This reflects that the new insulin was secreted and that its increasing levels do not originate from the decreased internalization by insulin receptors. Glycemia values fell to nearly initial fasting values after 3–4 h, coming back at 6 h (Fig. 6A).
Figures 5E and 6C then predict the hypothetical components of the insulin secretion involved for nonfat and fat-containing carbohydrate meals, respectively. For GLP-1 responses, we distinguish the first coming GIP and GLP-1 stimulated insulin secretion at low glucose (GIP-SIS and GLP-SIS, respectively; Fig. 6A) due to the early intestine presence of nutrients and “GLP-stimulated GSIS,” which is by mechanism independent from GSIS but is traditionally referred to as “amplifying” glucose responses, that is, amplifying GSIS. The increase in insulin exocytosis is substantial with incretin action. For fat-containing meals, we also consider the restored secretion of incretins due to the enhanced fat and TG presence in the meal, which can be delayed, based on accelerated GLP-1 secretion due to later fat digestion into FAs and MAG and concomitant support by bile acids. We called them additional incretin secretion and the additional secreted insulin as intestinal induced GLP-stimulated insulin secretion (Fig. 6C). Controversies exist of how long the lifetime of GLP-1 in circulations is. This is beyond the scope of this review.
In the fasting state, one could expect the two incretin/GLP-1 components to be minimum. But also, a FASIS dependent on FA metabolism and metabotropic receptor responses is expected to be minimum because albumin-bound FAs do not stimulate insulin secretion. Also, a tight NEFA binding to albumin does not allow these bound FAs to be released and interact with metabotropic receptors, as otherwise GPR40 and insulin secretion would also be activated during fasting. Nevertheless, all these components comprise the nonzero fasting insulin levels (basal levels), which are increased at prediabetes, type 2 diabetes, and metabolic syndrome in general.
Immediately after offering the meal to rodents, cephalic (neurotransmitter) initiation of insulin secretion begins and an incretin component is the major one, acting in the resulting insulin secretion (second column of Fig. 5E and Fig. 6C schemes). In our supraphysiological experiment with oral liposomal TG (intralipid) administration to mice (Fig. 6A, B), we assumed the appearance of chylomicrons containing TGs in the blood. Their lymphatic drainage bypass of the liver and subsequent presence in the circulation probably initiates the LPL-mediated cleavage of TG into DAG and MAG and free FAs, with their subsequent activation of metabotropic receptors GPR119 for MAG (Hodge et al., 2016; Moran et al., 2014a) and GPR40 (and GPR120) for FAs, comprising the receptoric branch of FASIS. The nascent free FAs, being locally excessive in the intersticia, interact better with GPR receptors or the GPR40 (and GPR120) receptor is preferentially activated by endogenous FAs.
We found that this GPR40 stimulation of insulin exocytosis is predominantly enabled by mitochondrial FAs cleaved by phospholipase iPLA2γ (Jabůrek et al., 2024). This has been demonstrated in isolated islets as well as in vivo, studying differences between wild-type and iPLA2γ knockout mice (Jabůrek et al., 2024). Moreover, iPLA2γ is redox-activated by excessive H2O2 (superoxide), created by enhanced FA β-oxidation in mitochondria (Jabůrek et al., 2024; Ježek et al., 2015). This fact also explains why external, albumin-bound NEFAs do not activate the GPR40 pathway but iPLA2γ-cleaved mitochondrial NEFAs do.
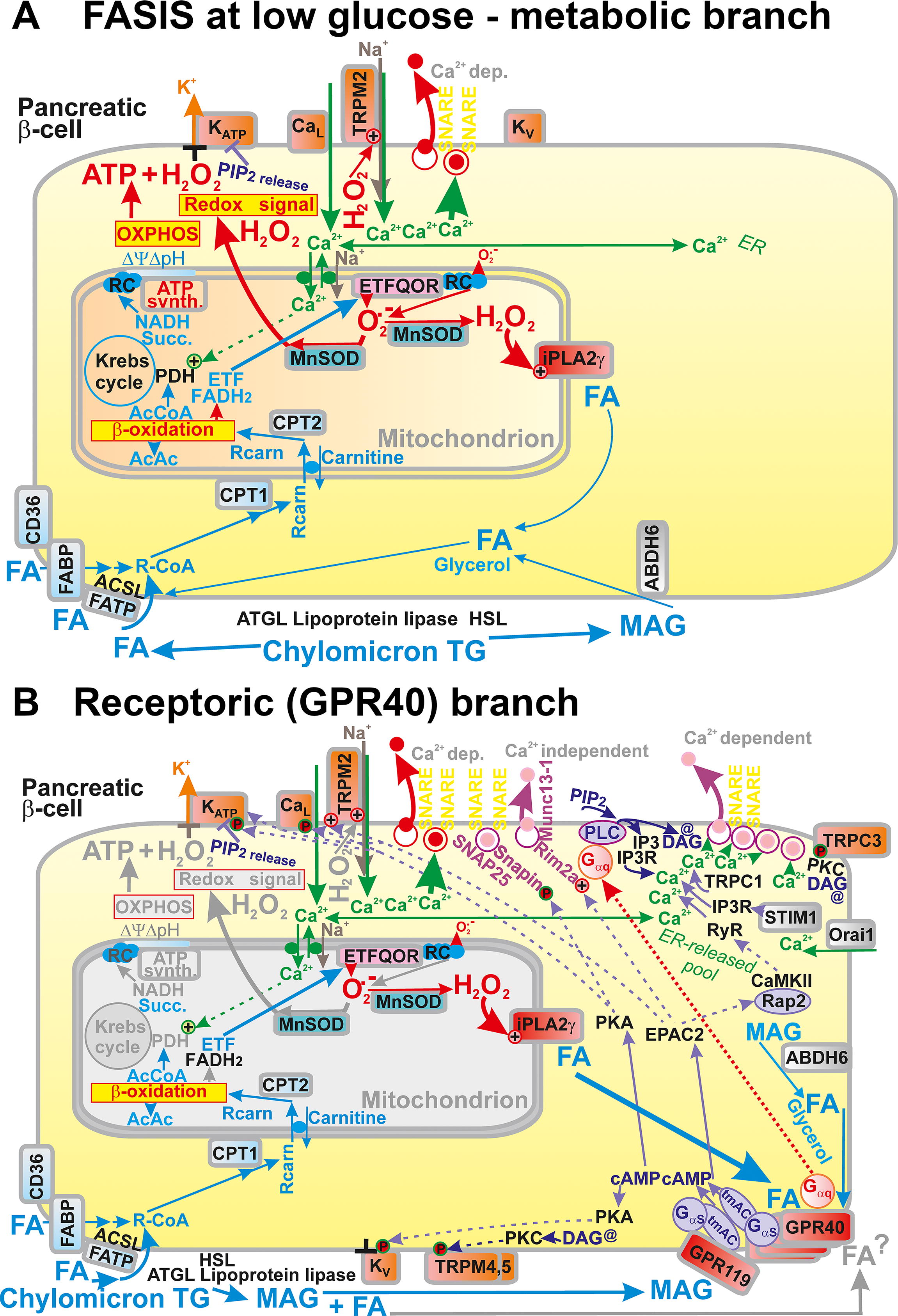
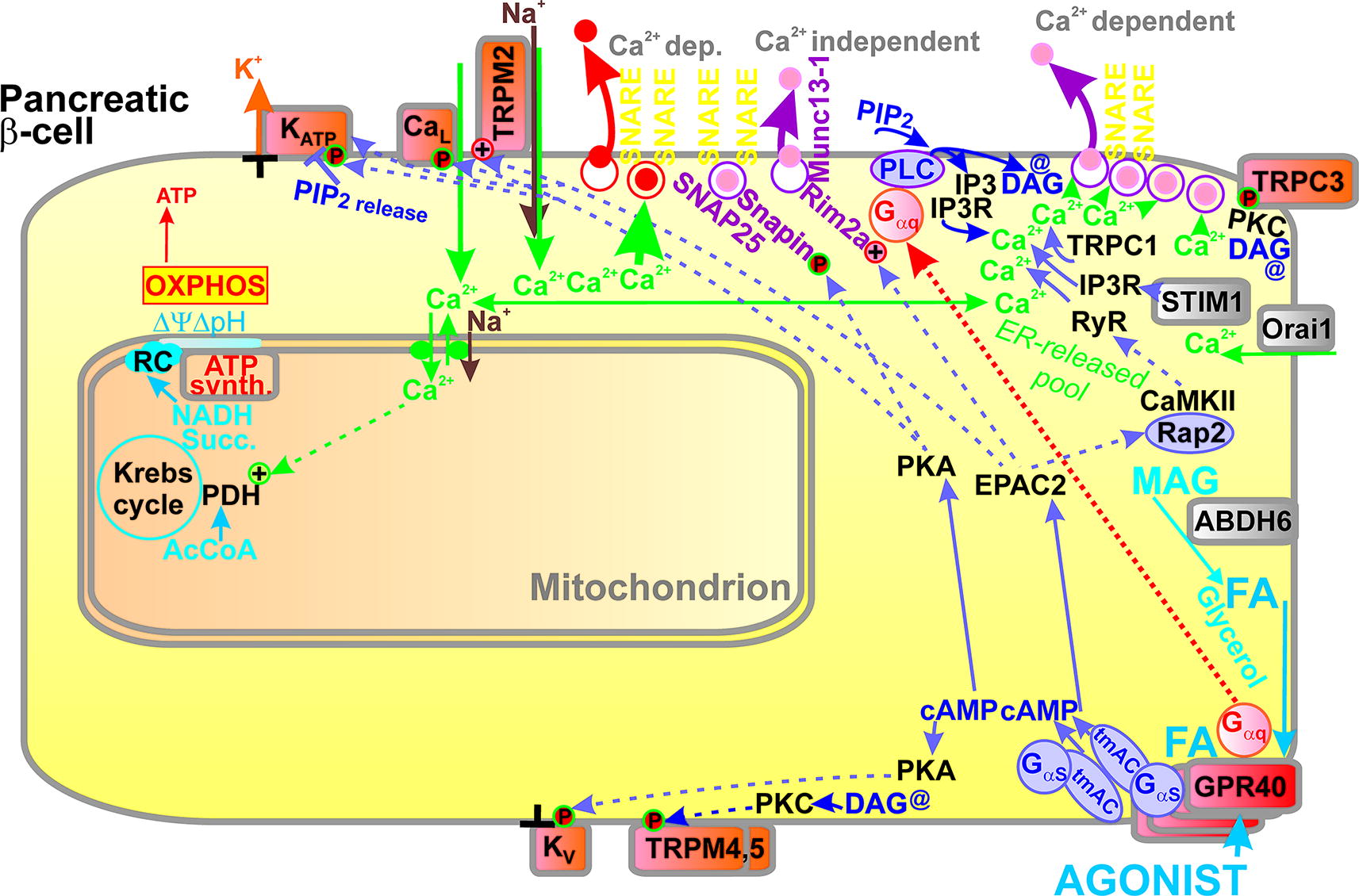
One could view this as a mutual dependence on the receptoric (nonfuel) and metabolic (fuel) branch of FASIS (Fig. 8, 9). Naturally, because FAs of any origin are being metabolized, and metabolic triggering of insulin secretion takes place, being dependent on mitochondrial redox signal substantiated by the H2O2 (superoxide) created by enhanced FA β-oxidation and elevated ATP/ADP (Fig. 1C, D) (Jabůrek et al., 2024). This is similar to the GSIS mechanism in which NADPH oxidase isoform 4 (NOX4) provides the redox signal (Plecitá et al., 2020) (Fig. 1A; Fig. 10).
In vivo, glycemia is elevated as a response to the orally administered intralipid (TG) to mice. Hence, the component of the true GSIS is present as well (Fig. 6C). Nevertheless, all the initiating events are rather fast, leading to the first FASIS phase peaking at 8–10 min. In vivo meal-induced glycemia of 10 mM ceasing up to 1 h prevents any observation of FASIS up to 1 h in the absence of insulin-stimulating glucose doses. However, experimentally, for example, with islet perifusion, such a situation could be set up (Jabůrek et al., 2024). Due to such initial elevated glycemia in vivo, a portion of the released insulin comes from FASIS induced by mitochondrial FAs, cleaved by iPLA2γ responding onto the NOX4 redox signal (“GPR-rel. FASIS mito-FAs by iPLA2 “in Fig. 6C).
Unlike with oral glucose (Fig. 5), after the oral liposomal TG (intralipid) administration to mice, the second phase insulin peak at 2 h has almost the same magnitude as the peak of the initial first phase (Fig. 6B), despite the true GSIS component being diminished due to the declining glycemia (Fig. 6A). Based on our other experiments with perifused islets (Jabůrek et al., 2024), we also hypothesize that the specific additional GLP-1 secretion and the corresponding insulin secretion component is still present; or these could be even more intensive at this stage (Fig. 6C). Moreover, the assumed incoming of chylomicrons at this moment allows the additional LPL-mediated TG cleavage into DAG and MAG, liberating free FAs; and, consequently, this induces FASIS, that is, glucose-independent insulin secretion, enabled by the additional FA metabolism and also involves the action of the metabotropic receptor pathway.
We do not know why following the 2-h peak, there is a decline of insulin for up to 4 h. At least the internalization of insulin bound to its receptors together with their internalization (Hall et al., 2020) could be expected to explain this as well as a decline of the general incretin response. Nevertheless, our findings of insulin peaking at 6 h after the oral liposomal TG (intralipid) administration could be explained only as the net physiological FASIS. This is because chylomicrons should still be present, but glycemia has declined to its original fasting values. So, no GSIS component could be present either. In further text of Chapter IV, we will explain the involved molecular mechanism in more detail.
Physiological meaning of FASIS
FASIS regulation of glycemia
Physiological FASIS seems to be unnecessary when proceeding with lower glycemia. Why should glycemia be lowered even further? Why should such FASIS cause hypoglycemia? Nevertheless, prolonged insulin action could be beneficial, as it allows WAT to utilize a full capacity of chylomicrons. However, FASIS comes much earlier at a high-fat containing meal and at overfeeding (vide infra, Chapter VC and Fig. 17), when it quickly follows the initiating events of insulin secretion. The repeated nearly synchrony, that is, the overlap of GSIS and FASIS, should contribute to the development of prediabetic pathology and must be further investigated. Formally, it is the earliest glucolipotoxicity event causing partial hyperinsulinemia.
Involvement of insulin to facilitate FA uptake into WAT
Another physiological relevance of FASIS might be deduced from the findings of possible insulin requirement for transport of FAs mediated by the insulin-recruited FATP1 into the adipocytes. FASIS would promote such transport and provide the same logical regulatory loop as glucose for the glucose uptake into cells of major peripheral tissues via the recruited GLUT4 to the plasma membrane. This hypothesis is based on the finding that the FA transport protein, isoform FATP1, is highly expressed in the skeletal muscle, heart, and WAT (Anderson and Stahl, 2013). Insulin facilitates its recruitment into the WAT plasma membrane (Jain et al., 2009; Stahl et al., 2002). Interestingly this mechanism might provide an autocrine loop in pancreatic β-cells (Henquin, 2021). In the skeletal muscle, however, FATP1 was reported to be a mitochondrial protein, which complicates relevant interpretation (see below). Nevertheless, mitochondrially localized FATP1 could export mitochondrial FAs cleaved by iPLA2γ.
Past contradictions to the FASIS concept
A previous consensus introduced “initiators” of insulin secretion as those compounds ultimately stimulating insulin secretion without any supportive effects of other metabolites or factors (Ashcroft and Rorsman, 1989; Ashcroft and Rorsman, 2012). Besides glucose, amino acid leucine was considered to be a self-standing initiator and “substances that stimulate metabolism of endogenous nutrients” (Ashcroft and Rorsman, 1989; Henquin et al., 2003; Rorsman and Ashcroft, 2018). Pharmacological initiators are the antidiabetic sulfonylureas (Feingold, 2000), such as glibenclamide, closing the ATP-sensitive K+ channels (Ashcroft, 2023). Other nutrients, which were recognized to stimulate insulin secretion, but apparently were ineffective without the above initiators, were called amplifiers. Besides most amino acids, but leucine (Henquin and Meissner, 1981), FAs were also considered amplifiers or potentiators (Ashcroft and Rorsman, 1989).
The purpose of this review is to modify this view and describe the effects of FAs and MAG by their self-standing effects, ongoing on the background of the “resting” metabolism of glucose. To consider that such metabolism is “amplified” or “potentiated” is unnecessary. A self-standing GPR40 agonist acting at “resting” low glucose concentrations exist and such agonists stimulate insulin secretion (Jabůrek et al., 2024) (Fig. 9); thus, there is no need to describe their downstream pathways as amplifying. Mathematically, the amplification of zero is still zero. Note that this is the same case with incretins, the action of which as initiators is not denied.
Also, the idea of acetylation (Panten et al., 2016) or palmitoylation of proteins (Dong et al., 2023; Yang et al., 2023) was used as an argument for the nonexistence of FASIS. However, the palmitoylated ATP-sensitive K+ channel cannot be closed (Yang et al., 2020). Also, palmitoylation of the IGV protein SCAMP1 prevents the hypersecretion of IGVs (Dong et al., 2023).
Molecular Mechanism of Insulin Secretion
Molecular mechanism of GSIS and redox signaling involved
Molecular mechanisms of GSIS
Prior to the description of molecular mechanisms for FASIS, we recapitulate those for GSIS (Fig. 1A; Fig. 10A). The mechanism of GSIS has been traditionally explained to rely on the ATP/ADP elevation in the peri-plasma membrane space (Ashcroft and Rorsman, 2012; Henquin, 2009; Ho et al., 2023; Maechler, 2013; Rorsman and Ashcroft, 2018; Rutter et al., 2015; Seino, 2012; Seino et al., 2017). This was considered to be sufficient for closing the entire population of the ATP-sensitive K+ channels (KATP) and setting a threshold plasma membrane potential to
However, recently, we have revisited these essential conditions for GSIS and found that besides the elevated oxidative phosphorylation (OXPHOS), providing periplasmic ATP/ADP elevation, the equally essential is the redox signal, mediated by NOX4 (Ježek et al., 2021; 2022; Plecitá et al., 2020) (Fig. 1A; Fig. 10A). Together with ATP, the cytosolic redox (H2O2) signal enables closing of KATP channels (Plecitá et al., 2020). The H2O2 release has been readily semiquantified in murine PI exterior, when it disappeared upon NOX4 ablation (Fig. 5J in Jabůrek et al., 2024). Monitoring was based on the Amplex UltraRed plus horse radish peroxidase. After the instantly increased glucose, this NOX4-mediated H2O2 burst is enabled by the NADPH supply to the constitutively expressed NOX4. The supply is provided by the two enzymes of the pentose phosphate pathway (PPP), that is, glucose-6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH) (Spégel et al., 2013). Up to 10% of glucose and G6PDH utilization is indeed diverted to PPP (Huang and Joseph, 2012; Lu et al., 2002; Schuit et al., 1997; Spégel and Mulder, Spégel and Mulder, 2020).
A certain portion of NADPH can be also provided by the simultaneously activated pyruvate redox shuttles (Ježek et al., 2022; Jitrapakdee et al., 2010; Joseph et al., 2006; Plecitá-Hlavatá et al., 2020). Indeed, ∼50% of glucose-derived pyruvate is carboxylated by pyruvate carboxylase (MacDonald et al., 2005) to oxaloacetate, enabling the pyruvate–malate shuttle. The redox shuttles combine enzyme activities with transport in and out of the mitochondrial matrix. As a result, these shuttles do not allow the synthesis of maximum NADH. For example, one less NADH molecule is formed in the mt matrix, and instead, one NADPH molecule is formed in the cytosol (Plecitá-Hlavatá et al., 2020). Two of these redox shuttles were linked with GSIS, the pyruvate–malate shuttle and the pyruvate–isocitrate shuttle (consuming matrix NADPH) and confirmed their described function (Joseph et al., 2006) by 13C-tracing (Plecitá-Hlavatá et al., 2020; Zhang et al., 2021). Other schemes for shuttles were also reported (Merrins et al., 2022). Note that NADPH provided by these redox shuttles is not essential for GSIS as PPP is its primary source, and individual shuttles can rescue the insufficiency of the other one. That is why, for example, deletions of citrate carrier (Bauchle et al., 2021) and malic enzyme 1 (Ronnebaum et al., 2008) did not affect GSIS.
Besides powering NOX4, NADPH activates glutaredoxin via reduced glutathione by glutathione disulfide reductase, which further reduces cysteine disulfides of sentrin/SUMO-specific protease 1, thus triggering the deSUMOylation of several proteins implicated in granule trafficking, including the Ca2+-sensing protein synaptotagmin VII (Dai et al., 2011).
The relatively easy spread of redox changes in pancreatic β-cells is enabled by a rather weak antioxidant defense system and low capacity of redox buffers (Lenzen et al., 1996; Robertson, 2024). Such a delicate redox homeostasis is then disturbed by a rather weak insult. Nevertheless, the content is relatively high for thioredoxins and glutaredoxins (Ivarsson et al., 2005; Reinbothe et al., 2009) or peroxiredoxins and other proteins capable to spread redox signals (Ježek, 2023; Ježek et al., 2020; Woo et al., 2010). Expression and activity of antioxidant enzymes is low in rodent β-cells as compared with other organs (Lenzen, 2008).
Note also that signs of redox signal presence have been reported some time ago, such as the effect of antioxidants upon exhausted glutathione in pancreatic β-cells, demonstrating an unspecified link between GSIS and external H2O2 (Pi et al., 2007). Also, the inhibition of an unidentified NOX isoform with an antisense p47PHOX oligonucleotide was reported to attenuate GSIS (Morgan et al., 2009). Diphenyleneiodonium (DPI), as a nonspecific NOX inhibitor, was also reported to affect insulin secretion (Imoto et al., 2008; Syed et al., 2011), similarly to an inhibitor of the two specific NOX isoforms (Saadeh et al., 2012). Concerning the two-phase GSIS appearance in the experimental settings with rodents or human volunteers, the KATP‐dependent and KATP‐independent (mostly receptoric) mechanisms participate in both phases (Ashcroft and Rorsman, 2012; Henquin, 2009; Merrins et al., 2022; Prentki et al., 2013; Rorsman and Ashcroft, 2018; Rutter et al., 2015).
The role of mitochondria-generated phosphoenolpyruvate (PEP) was also emphasized. Thus, in addition to glycolysis, PEP is formed by mitochondrial PEP carboxykinase (PCK2) at high glucose (Zhao et al., 2020). Since PCK2 knockout mice exhibited attenuated GSIS, it has been suggested that a fraction of pyruvate kinase (PK) in the peri-plasma membrane loci provides the localized glycolytic ATP formation ready to close KATP (Abulizi et al., 2020).
Glycolysis in pancreatic β-cells is enabled by a specific hexokinase IV, termed glucokinase, which does not respond to product-inhibition by glucose-6-phosphate and has a half-maximum at 8 mM and 4 mM in mice (Doliba et al., 2012) and humans (Liang et al., 1994), respectively. Glucokinase is one of the rate-limiting enzymes of glycolysis (Matschinsky and Wilson, 2019). Synergy of GLUT2 and glucokinase enables a 10-fold rise in the glucose metabolism rate between 1 and 10 mM glucose. Other rate-limiting enzymes of glycolysis are phosphofructokinase-1, PKM2, and an indirect rate-limiting enzyme, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3.
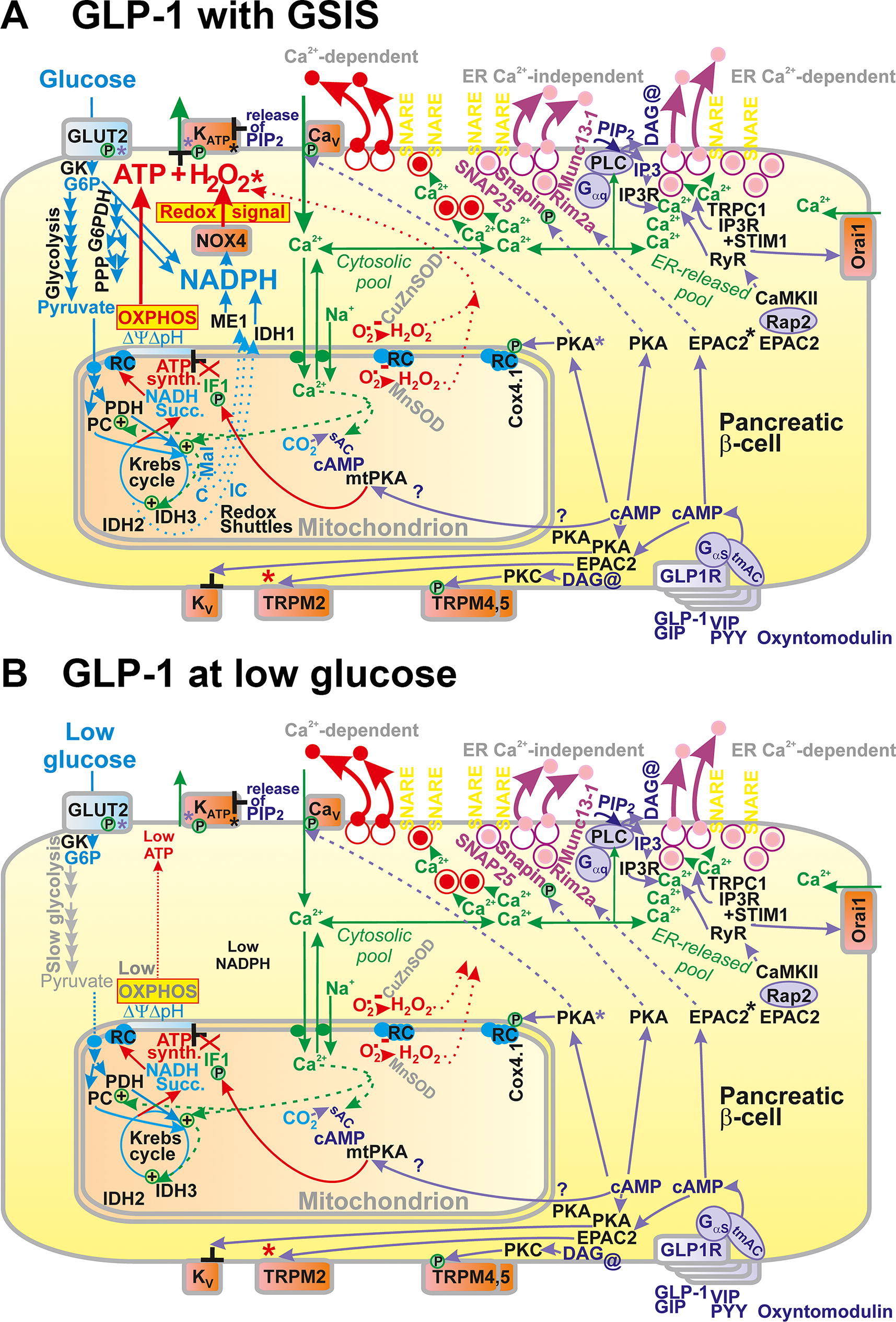
Physiological action of incretins
The two most important incretins are GLP-1 (Müller et al., 2019) and GIP (Itoh et al., 2014; Nauck and Müller, 2023; Zandvakili and Perez-Tilve, 2024). The physiological action of incretin peptide hormones is superimposed onto the net GSIS mechanism (Fig. 10A) and possesses a high impact on the resulting glycemia and insulin postprandial time courses. Nevertheless, incretins can act independently at low glucose concentrations (Fig. 10B), as described above. Thus the early postprandial GIP secretion and GLP1 release stimulated by the early neuroendocrine reflex are the events preceding the insulin secretion stimulation by incoming glucose in circulation. Therefore, the mechanisms of insulin secretion stimulation by GIP receptor (GIPR) and GLP-1 receptor (GLP1R) pathways must act at low glucose and hence independently of GSIS triggering (Fig. 10B).
Following these early events, the apparent amplification of GSIS by the GLP1R pathway occurs, however, these triggering pathways could still be independent, each contributing by its own mechanisms. Physiologically, GLP-1 in human circulation peaks after 30–60 min postprandially at <10 pM levels for fat-free meal (Kuhre et al., 2015) and after 120 min with fat-containing meal (Herrmann et al., 1995). Note, that this is a little bit earlier before chylomicrons are peaking but dependent on the fat amount. Thus, the overlap of GLP1R pathway with FASIS cannot be excluded.
Pathways of GLP-1 and GIP receptors
The main pathway for GIPR and GLP1R receptors depends on Gαs G-protein, which increases the cAMP generation (Furman et al., 2010; Lefkimmiatis and Zaccolo, 2014; Müller et al., 2019) by plasma membrane transmembrane adenylate cyclases at the expense of ATP (Mourad et al., 2010) (Fig. 10A, B). The lifetime of cAMP in the cell generally depends on action of phosphodiesterases (11 families, some are specific for cGMP) degrading cAMP. In β-cells, PDE3B contributes to termination of GLP1R pathway (Härndahl et al., 2002). Soluble adenylate cyclases, including those in the mitochondrial matrix (Chen et al., 2000), also contribute to the cAMP pool, being potentiated by Ca2+ and bicarbonate.
cAMP effects namely the cAMP-dependent PKA (Ould Amer and Hebert-Chatelain, 2018; Taylor et al., 2012; Zhang et al., 2016), which consists of a hetero-tetramer of two distinct catalytic subunits and a dimer of a regulatory subunit. Three distinct catalytic (Cα, Cβ, and Cγ) and four isoforms of the regulatory subunit exist (RIα, RIβ, RIIα, and RIIβ), which bind cAMP (Taylor et al., 2012). The cAMP binding leads to the dissociation of catalytic subunits
PKA typically amplifies the Ca2+-dependent IGV exocytosis (Fig. 10A, B). It acts on channels as well as proteins of IGV exocytotic machinery. PKA-mediated phosphorylation and hence activation of the CaV β2-subunit belongs to the former category, together with concomitant phosphorylation of KATP channels, which increases the ATP concentration range required for their closure (Bünemann et al., 1999). PKA phosphorylation also inhibits Kv channels, which otherwise terminate plasma membrane depolarization, hence this prolongs the already more intensive Ca2+ influx via phosphorylated CaV channels and hence prolongs/intensifies the exocytosis of IGVs (MacDonald et al., 2003).
Concerning the IGV exocytotic machinery, snapin is phosphorylated by PKA, which allows its interaction with the other proteins of the IGV and potentiates namely the first phase of insulin release (Song et al., 2011). Snapin participates in tethering IGVs to the plasma membrane by coiled-coil interaction with a lipid-anchored protein, the synaptosomal nerve-associated protein 25 (SNAP-25) (Somanath et al., 2016).
Another and parallel cAMP-induced pathway is the enhanced signaling via exchange proteins directly activated by cAMP 2 (EPAC2) (De Rooij et al., 1998; Gloerich and Bos, 2010; Kang et al., 2008) (Fig. 10A, B). EPAC2 protein possesses a guanine nucleotide exchange activity and directly activates transient receptor potential cation channels, subfamily M (TRPM2 channels), essential for enabling depolarization shift of up to
The EPAC pathway also affects IGV proteins and hence facilitates IGV exocytosis. Rim2α GTPase, located at the cytosolic (inner) plasma membrane surface and on the IGV membranes, provides an intermediate step in this action, priming the mammalian uncoordinated homology 13–1 protein (Munc13-1), which subsequently opens syntaxin 1, enabling its interaction with IGVs and IGV fusion with the plasma membrane (Kashima et al., 2001; Ozaki et al., 2000; Yasuda et al., 2010). Interacting with another GTPase of IGVs, Rab3A, the Rim2α–Rab3A complex allows such IGV docking (Gheni et al., 2014).
Independence of GIPR and GLP1R pathways on GSIS
As explained above, both PKA and EPAC2 pathways contain branches that directly affect IGV exocytotic machinery (Fig. 10B). Therefore, these actions could be executed also at low glucose, hence in the absence of GSIS. The existence of such a channel-independent insulin secretion must be studied in further detail. Moreover, the existence of KATP-independent insulin secretion when PKA and EPAC2 pathways ensure CaV opening independent of the closure of KATP should be investigated. If GIPR and GLP1R receptors are promiscuous and interact also with Gαq/11 (vide infra), the closure of the entire KATP population would be ensured by withdrawal of PIP2 from the binding sites in KATP due to Gαq/11-induced phospholipase-C (PLC) activity in a synergy with either TRPM2 activated by EPAC2 or TRPM4 and TRPM5 activated by protein kinase-C (PKC) due to the PLC DAG production.
Other main regulations of insulin secretion
Regulators of insulin secretion can be classified as positive and negative ones. The positive regulators either induce self-standing stimulation of insulin secretion (e.g., GPR40, GLP1R) or accelerate certain critical points within the pathways of these stimulants. Negative regulators act against these points and in frequent cases, induce Gαi with further consequences, namely causing repolarization of β-cells (Fig. 11). The significance of negative regulators can be viewed when they counteract the excessive insulin secretion causing hypoglycemia. Hence, the negative regulators defeat these adverse hypoglycemic effects.
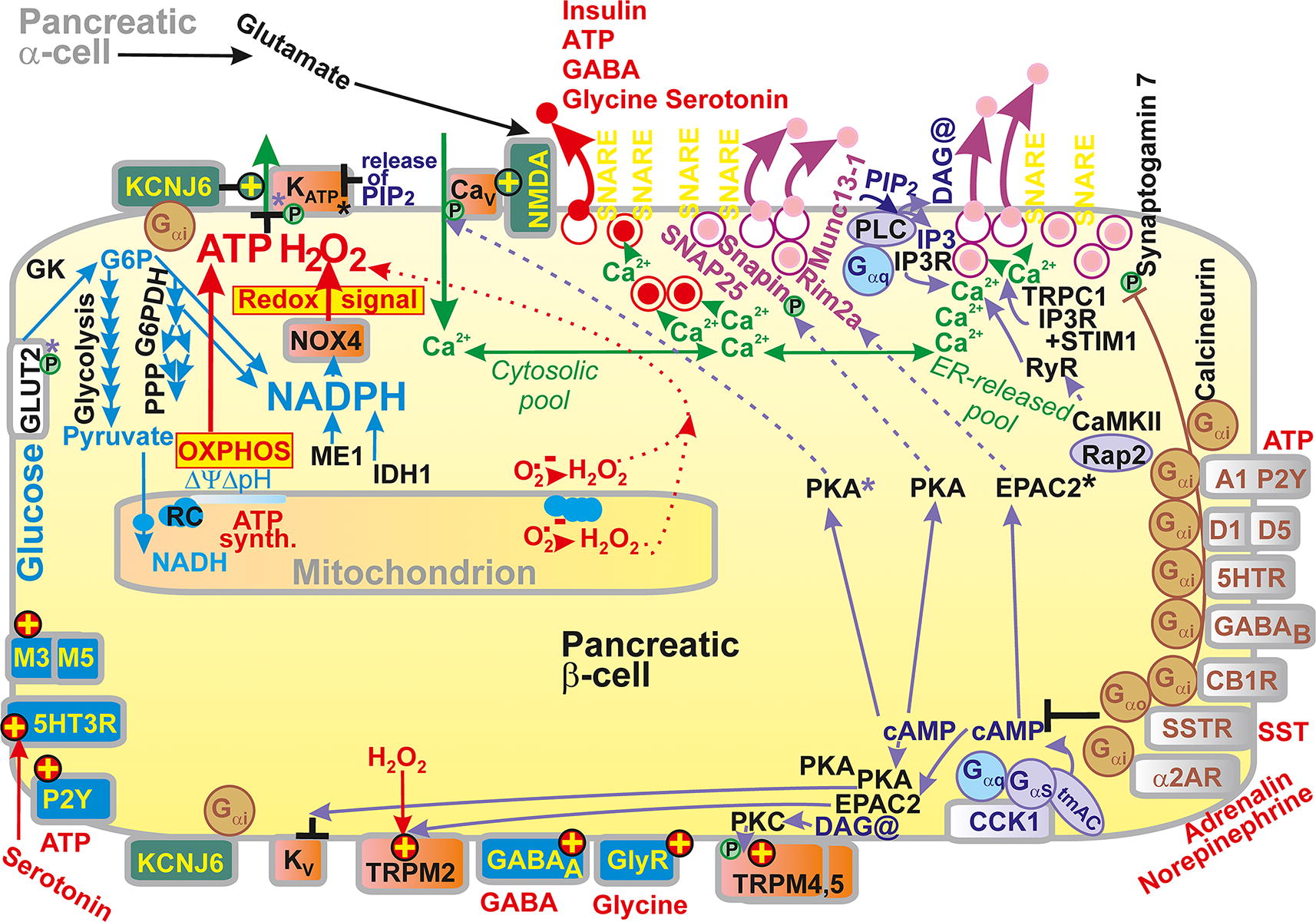
Among negative regulators acting via Gαi, somatostatin could be considered as the most important (Blondel et al., 1994; Rorsman and Huising, Rorsman and Huising, 2018), acting via the somatostatin receptor (SSTR) (Kailey et al., 2012). Adrenalin is similarly important (Drews et al., 1993), termed epinephrine as a neurotransmitter, acting via α2 adrenergic receptor ARα2 in mice. Also, peptide galanin is inhibitory (Nilsson et al., 1989). The inhibitory Gαi pathway suppresses depolarization to a
Somatostatin released by islet δ-cells as an SST-14 peptide that inhibits the release of insulin and glucagon in a paracrine manner. Somatostatin could reach the islets from the gut as an SST-28 peptide (Ensinck et al., 1989). Whereas in mice, SSTR3 receptor is the major one expressed in β-cells, in humans multiple isoforms exist, such as SSTR1, SSTR2, SSTR3, and SSTR5. However, physiologically relevant seems to be mainly SSTR5 (Braun, 2014). Via a certain SSTR pathway, somatostatin also affects IGV exocytosis, not only by decreasing cAMP but also by the activation of the protein phosphatase calcineurin (Renström et al., 1996), which dephosphorylates Ser in synaptogamin 7 protein. The latter is otherwise phosphorylated upon activation of GLP-1R (Wu et al., 2015).
Epinephrine acts both ways in human β-cells, inhibitory effects being mediated by ARα2 receptor, but stimulation of insulin secretion proceeds by activation of ARβ1 and ARβ2 receptors (Hellman et al., 2014; Lacey et al., 1990; Zhang et al., 2024). The ARα2 receptor pathway also leads to activation of GIRK channels. Also, the inhibition of cAMP-activated TRPM2 channels may contribute (Ito et al., 2017).
Cannabinoid 1 receptor (CB1R) is also expressed in pancreatic β-cells and is coupled to the Gαi protein, causing a negative regulation of endogenous signaling. Complying with this, mice with β-cell-specific CB1R deletion exhibited increased GSIS and cAMP levels, increased β-cell viability and proliferation, reflected by the increased islet area in pancreas sections (González-Mariscal et al., 2018).
Leptin, an adipocyte hormone, diminishes insulin secretion by recruiting KATP to the plasma membrane, counteracting their closure (Chen et al., 2013; Wu et al., 2017). Leptin also decreases pre-proinsulin gene expression (Laubner et al., 2005). Another adipocyte hormone, adiponectin acts as a facilitator of β-cell proliferation (Ye et al., 2014).
Neurotransmitters also act as regulators of all islet cell types, the topics being beyond the scope of this review. Nevertheless, their effects have been briefly discussed below. Let's remain that neurotransmitters act via the G-proteins or ligands of gated channels. Thus, extracellular glutamate originates from α-cells in islets (Pan et al., 2022). Glutamate is loaded into glucagon secretory granules by VGLUT2 glutamate transporter. In β-cells, glutamate acts via N-methyl-
Gamma-aminobutyric acid (GABA) is released together with insulin. In β-cells, GABA possesses the equal density of GABAA ligand-gated Cl− channels and GABAB GPR receptors as in CNS. GABAA activation diminishes depolarization but can function similarly as TRPM2 in synergy to reach the
Serotonin, 5-hydroxytryptamine (5-HT), is produced by tryptophan hydrolase in the CNS and intestines and is also released in IGVs together with insulin. Autocrine action via the 5-HT3R serotonin-gated channels increases insulin secretion (Kim et al., 2010; Ohara-Imaizumi et al., 2013). Other serotonin receptors in β-cells, acting via G-proteins, can increase insulin secretion to compensate for situations when metabolic stress is prevalent, such as during pregnancy, overfeeding, or conditions stimulating tryptophan hydroxylase expression. When such stress ends, serotonin receptors also facilitate recovery (Kim et al., 2010; Ohara-Imaizumi et al., 2013). Dopamine such as norepinephrine inhibits insulin and promotes glucagon secretion. In β-cells, dopamine acts via D1 and D5 receptors (Aslanoglou et al., 2021). Also, histamine effects on β-cells inhibit insulin secretion (Pini et al., 2016).
Acetylcholine is released by cholinergic nerve terminals, but in humans, it comes also from α-cells. Muscarinic M3 and M5 acetylcholine receptors are expressed in β-cells and can increase insulin release (Rodriguez-Diaz et al., 2011b). Cholecystokinin (CCK) binding to CCK1 receptors induces the Gαs signaling at high glucose and Gαq/11 signaling at low glucose (Ning et al., 2015). CCK-8 active fragment CCK-8 besides stimulating insulin secretion it also protects against apoptosis. Note that CCK formed in the intestine does not act as an incretin. The activation of oxytocin/vasopressin receptor V1b, expressed in β-cells and α-cells, increases insulin during hyperglycemia and increases glucagon during hypoglycemia (Mohan et al., 2019). Ablation of V1b in mice diminished fasting insulin and glucagon levels, lowered fasting glycemia, and thus enhanced insulin sensitivity (Ding and Magkos, 2019).
Purinergic A1 receptor acting via Gαi/o thus inhibits the secretion of insulin and glucagon in β-cells and α-cells, respectively (Ohtani et al., 2013; Tudurí et al., 2008). Six isoforms of P2Y receptors coupled to G proteins were also identified in islets (Tudurí et al., 2008). Moreover, P2X ligand-gated ion channels then stimulated insulin secretion in β-cells. ATP that is contained in IGVs, being secreted together with insulin thus provides an autocrine loop via P2X activation (Jacques-Silva et al., 2010; North, 2002; Ohtani et al., 2011; Richards-Williams et al., 2008).
Molecular mechanisms of FASIS and redox signaling involved
Overview of molecular mechanisms of FASIS
Next, the mechanisms representing a fundamental basis for FA-induced triggering of insulin secretion ongoing even at low glucose (individually not stimulating secretion) have been described. Free FAs have been regarded for a long time to only augment GSIS (Prentki et al., 2013). This terminology assumed that insulin secretion in the presence of FAs requires certain higher glucose concentrations (Carpinelli et al., 2002; Gehrmann et al., 2010; Graciano et al., 2011). In contrast, our group and others have demonstrated that the net FASIS exists at low glucose concentration, which otherwise does not stimulate insulin release alone (Cen et al., 2016; Fernandez and Valdeolmillos, 1998; Jabůrek et al., 2024; Ježek et al., 2015; Ježek et al., 2021; Ježek et al., 2022; Nyrén et al., 2012; Plecitá et al., 2020).
Also, FAs trigger the action potential at a low, insulin nonstimulating glucose concentration in pancreatic β-cells (Fernandez and Valdeolmillos, 1998) as well as cytosolic Ca2+ oscillations (Jabůrek et al., 2024). Thus, insulin secretion is stimulated by free FAs on the background metabolism of low glucose. Moreover, FA β-oxidation provides both an elevated ATP (ATP/ADP) and the redox (H2O2) signal. (Jabůrek et al., 2024; Ježek et al., 2015). The latter is given by the increased mitochondrial superoxide formation upon FA β-oxidation, transformed after dismutation into H2O2, which is subsequently spread up to the plasma membrane (Jabůrek et al., 2024; Ježek, 2023) (Fig. 8A, B). Increasing mitochondrial reactive oxygen species (ROS) were reported to originate from monooleoyl-glycerol while they modulated insulin secretion (Saadeh et al., 2012). One report has suggested mt-derived ROS as obligatory signals for insulin secretion (Leloup et al., 2009).
FASIS is more complex and consists of “fuel” and “non-fuel” components (Fig. 8A, B), hereafter termed metabolic and receptoric components, respectively. The “non-fuel”, that is, receptoric component, is provided by metabotropic receptors, described in detail below. FAs with C >6 interact with and activate GPR40 and GPR120 metabotropic receptors, while SCFAs such as FAs with C <6 interact with and activate GPR41 and GPR43 receptors.
GPR40 receptors sense FAs at the plasma membrane and initiate a complex downstream signaling. Also signaling by MAG exists in β-cells, proceeding via the GPR119 metabotropic receptors, activating Gαs-PKA(EPAC2) pathways. In vivo, of course, the net FASIS and its GPR receptoric branch are typically accompanied by the GPR119 signaling (Husted et al., 2017; Moran et al., 2014a; Tunaru et al., 2018). The most pronounced downstream pathway initiated by GPR40 activation by FAs involves Gαq/11 heterotrimeric G-proteins (Gαq/11-Gβ-Gγ) activating the Ca2+-dependent PLC-mediated hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) into DAG and inositol-3-phosphate (IP3) (Fig. 8A, B). Because PIP2 stabilizes the open state of KATP channels, this branch of the GPR40 pathway, leading to PIP2 degradation, facilitates the KATP closure by PIP2 release from KATP (Yang et al., 2020).
Moreover, the second reaction product of PLC, that is, DAG, stimulates PKC, which phosphorylates, thus opening the TRPM4 and TRPM5 channels (Shigeto et al., 2015). Their opening allows them to more frequently establish the
The pathways of PKA and EPAC2 (described above) serve for MAG signaling via the GPR119 receptor and as so-called biased pathways for certain GPR40 agonists. Note that PKA phosphorylates KATP and CaV channels to ease and make more frequent action potential triggering by establishing the –50 mV threshold. The EPAC2 pathway leads to phosphorylation of TRPM2 (required for setting the
Yet another phenomenon is concomitant to FASIS. In the case of free palmitic acid, it was not the extracellular palmitic acid delivered with albumin that stimulated the GPR40 receptors but their activation was achieved by mitochondrial FAs cleaved from the mt phospholipids by the redox-activated mt phospholipase A2, isoform γ (iPLA2γ/PNPLA8; Jabůrek et al., 2024; Ježek et al., 2015). Silencing of iPLA2γ or its ablation in mice resulted in a profound decrease of FASIS, despite the intensity of the redox signaling reaching the plasma membrane was not attenuated (Jabůrek et al., 2024; Ježek et al., 2015).
LPL, AGTL, and HSL
LPL, which was found to be required also for intact GSIS, might be the ultimate source of FA liberated from chylomicron TGs (Cruz et al., 2001; Pappan et al., 2005) (Fig. 7). In general, FAs for β-oxidation could come from the intracellularly located LPL in β-cells (Nyrén et al., 2012) acting on TG from LDs. Alternatively, extracellular LPL (Wang and Eckel, 2009) should cleave TGs of lipoproteins, including those of chylomicrons. LPL is transported to the luminal surface of endothelial cells in islet capillaries through the interstitial spaces. The transport is enabled by GPIHBP1. GPIHBP1 binds LPL and transports it from the basolateral surface to the luminal surface (Davies et al., 2010). This may proceed by a 2D diffusion on the endothelial cell surface. Immunohistochemistry showed the predominant LPL content in the cytosol of β-cells within the islets with a minor fraction together with GPIHBP1 within the islet capillaries (Nyrén et al., 2012).
Moreover, the LPL expression in β-cells was strictly dependent on leptin. LPL should be regulated to an optimum, as both β-cell-specific overexpression and β-cell-specific deletion led to a diabetic phenotype; while the former decreased glucose metabolism, the latter increased it (Pappan et al., 2005). Islets from LPL-deficient mice exhibited enhanced insulin secretion and lower fasting glycemia (Marshall et al., 1999). With age, these mice developed glucose intolerance and decreased first GSIS phase (Ding et al., 2010). Other lipases such as AGTL and HSL are also involved in cleaving β-cell TG stores (Peyot et al., 2009). Note that AGTL was found to be predominant in the HSL knockout mice (Mulder et al., 2003).
FA uptake into cells
Due to their high partition coefficient, FAs can intercalate into the bilayer membrane lipids of plasma membrane. Nevertheless, the transport mechanisms were developed by Nature to ease the entry into the subsequent metabolic pathways (Fig. 7). However, their research seems to be only beginning. The first candidate to be considered is a class B scavenger receptor, protein CD36/SR-B2, which binds free FAs and concentrates them on the respective plasma membrane domains to increase their translocation across the plasma membrane (Clavelo-Farrow and Thomas, 2023; Glatz and Luiken, 2018; Pepino et al., 2014). Thus, CD36 could be one of the facilitators of the FA uptake in pancreatic β-cells (Khan and Kowluru, 2018; Noushmehr et al., 2005). However, only two studies demonstrated its presence in the respective cell models (Noushmehr et al., 2005; Wallin et al., 2010). Moreover, CD36 overexpression deteriorated palmitate- and oleate-mediated “amplification” of GSIS and prevented the inhibition of CPT1 by high glucose (Wallin et al., 2010).
Mechanistically, CD36 exposes its hydrophobic binding pocket to the extracellular surface of the plasma membrane, into which FAs relocalize after their debinding from albumin. In the next step, FAs concentrate in the nearby plasma membrane domain, from which they are captured by the fatty acid transport proteins (FATP) (Clavelo-Farrow and Thomas, 2023). An alternative model predicts a direct “sliding” of FAs along the C-terminus exposed into the cytosol to the long-chain acyl coenzyme A synthetase (ACSL), ensuring the first step of FA β-oxidation (Clavelo-Farrow and Thomas, 2023). Note that certain FATP isoforms were reported to provide also the ACSL activity and certain isoforms exclusively only this activity. FATP proteins thus could bind to ASCL enzymes.
FATP could be also involved in the channeling of FAs directly to the proper metabolic enzymes (Fig. 7). The FATP2a,b isoforms are the major ones expressed in rat INS-1E β-cells (Ahowesso et al., 2015) and were found to be upregulated in humans by high glucose (Schrimpe-Rutledge et al., 2012). Interestingly, FATP2b exhibits activity as an FA transporter as well as acts as long-chain ACSL (having the similar domain structure as true ACSL) (Li et al., 2022a). In contrast, FATP2a has exclusive ACSL activity predominantly acting on very-long-chain FAs (VLC-FAs).
Another category of proteins dealing with incoming FAs from plasma albumin or FA moieties of lipoprotein TGs are fatty acid binding proteins (FABP). FABP3 is expressed by pancreatic β-cells. Its silencing diminished FA uptake, LD formation, and the expression of DAG O-acyltransferase 1 (DGAT1), which is required for lipid accumulation (Hyder et al., 2024). FABP3 silencing also prevented artificial lipotoxicity induced by palmitic acid, including inflammation via nuclear factor kappa-light-chain-enhancer of activated B cells (NFκΒ). FABP3 binds ferulic, cleomaldeic, caffeic, sinapic, hydroxycinnamic, 4-p-coumaroylquinic, quinoline-2-carboxylic, chlorogenic, 6-hydroxykynurenic, and rosmarinic acids, all of which could act as FABP3-specific inhibitors (Hyder, 2023).
The first step of FA β-oxidation (Fig. 12; Fig. 13) involves long-chain ACSL proteins. ACSLs have been shown to play a role in facilitating LCFA transport in mammalian cells under physiological conditions (Ansari et al., 2017). Independently whether ACSL is considered as a single unit containing FATP activity or whether heterodimers of the FATP and ACSL exist, in both cases they ensure an instantaneous conversion of FA molecule traversed across the membrane into acyl-coenzyme A. This conversion dramatically increases the aqueous solubility of the molecule. The local concentration of free FAs or albumin-bound FAs externally to the plasma membrane then forms a higher gradient, when ACSL constantly converts the transported FAs.
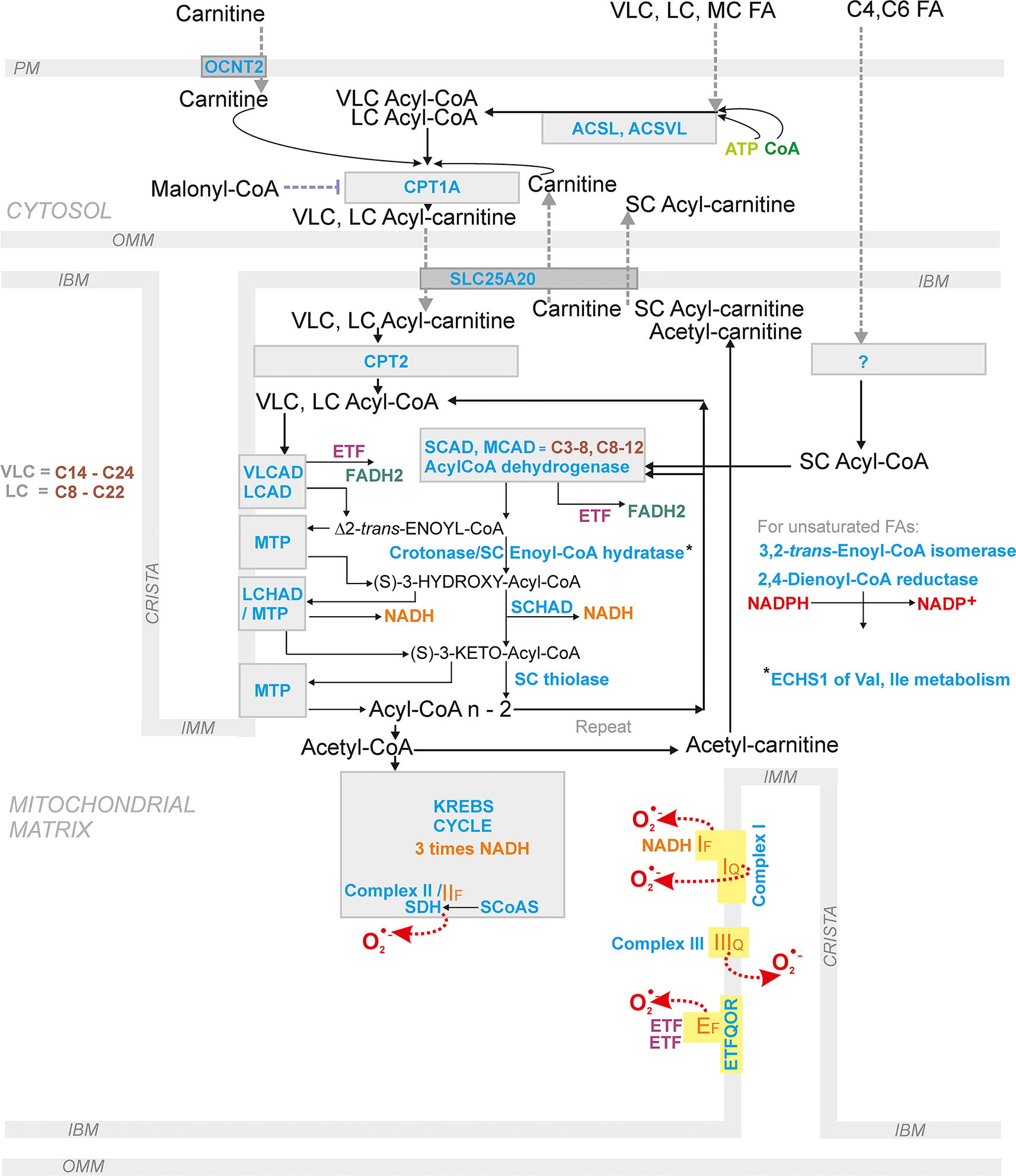
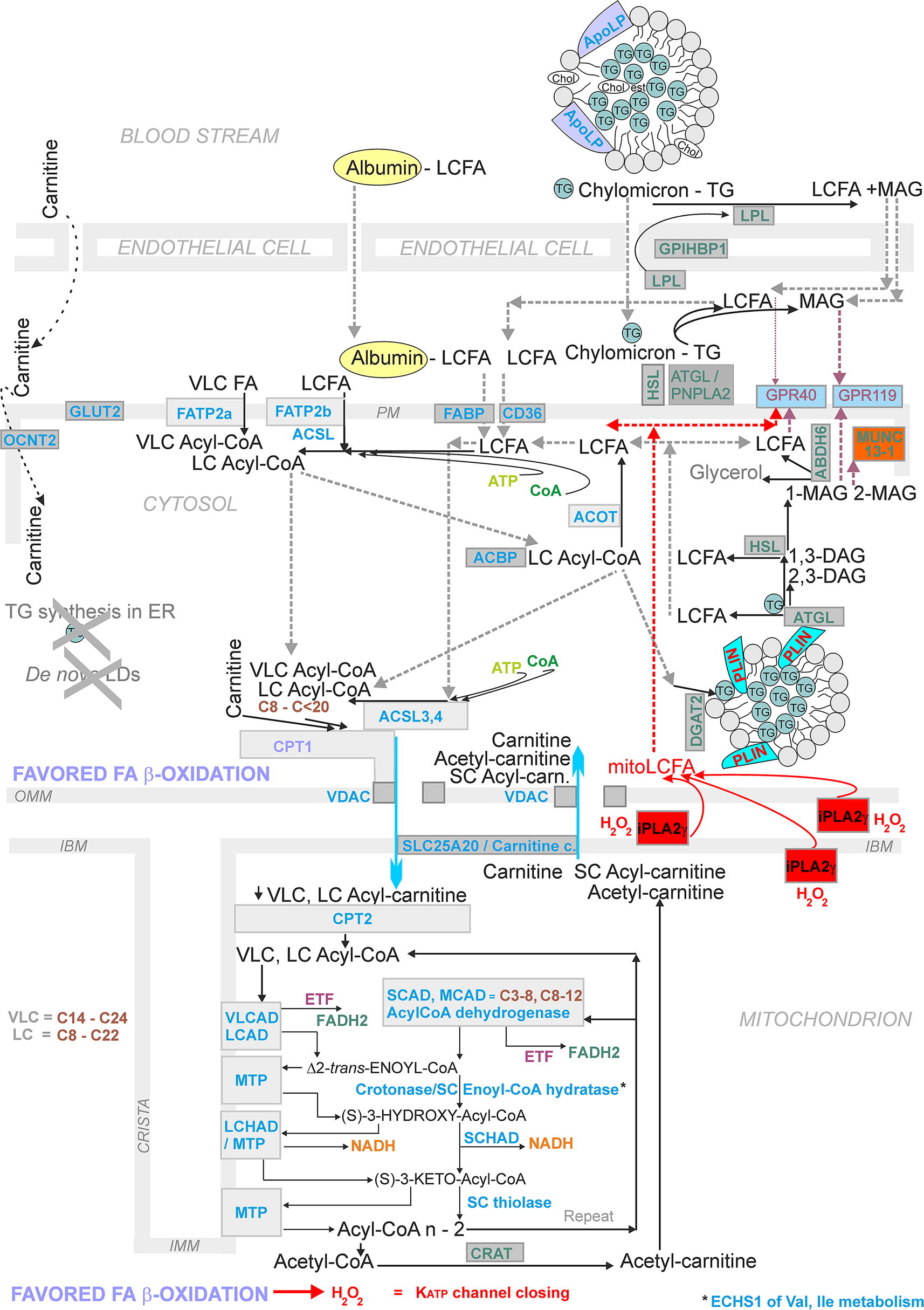
FA β-oxidation in β-cells
Pancreatic β-cells utilize FA β-oxidation at low insulin-nonstimulating glucose. Typically, FA β-oxidation begins with FA conversion to LC-acyl-CoAs by ACSL (Fig. 12; Fig. 13). This is followed by the next step, when carnitine palmitoyl transferase (CPT1, alternatively named carnitine acyltransferase 1, CAT1) transfers VLC-acylCoAs and LC-acyl-CoAs to VLC-acyl-carnitines and LC-acyl-carnitines (Houten et al., 2016; Ribas and Vargas, 2022). Their uptake into the mitochondrial matrix is realized by the carnitine carrier (SLC25A20; carnitine-acylcarnitine translocase, CACT) in an exchange reaction, which needs to be investigated in detail. The carnitine carrier is able to import LC-acyl-carnitines into the mitochondrial matrix, only exchanging them for carnitine. Exchange for acetylcarnitine, and probably also for short-chain SC-carnitines, has also been suggested (e.g., Gotvaldová et al., 2024). The latter possibility should still be demonstrated in pancreatic β-cells. Nevertheless, for pancreatic ductal adenocarcinoma cells, facilitation of FA β-oxidation by the matrix branched-chain SC-carnitines was observed when SC-carnitines are derived from branched-chain amino acids such as valine, leucine, and isoleucine (Gotvaldová et al., 2024).
In the matrix, mitochondrial carnitine palmitoyl transferase (CPT2, alternatively named carnitine acyltransferase 2, CAT2) converts (VLC-) LC-acyl-carnitines back to (VLC-) LC-acyl-CoAs. The own β-oxidation reaction cascade involves the four main steps shortening the FA acyl chain by two carbons and yielding acetyl-CoA. This includes reactions of acyl-CoA dehydrogenases, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and β-thiolase. A central to these reactions is the mitochondrial trifunctional protein (MTP). MTP is a hetero tetramer composed of two α subunits and two β subunits, encoded by the HADHA and HADHB genes, respectively. The HADHA protein exhibits LC-enoyl-CoA hydratase activity and LC-3-hydroxyacyl-CoA dehydrogenase activity. The HADHAB ensures the LC-chain 3-ketoacyl-CoA thiolase activity.
Each of these β-oxidation reactions are ensured by several enzymes, depending namely on the length of a given acyl-CoA. At a certain acyl chain length, even two enzymes could compete on the common substrate. As a rule, enzymes processing VLC FAs and LCFAs are integral part of the inner mitochondrial membrane or are attached to it, processing FA derivatives embedded into the lipid bilayer. The first in the cascade is VLCAD, followed by reactions of acyl-CoA dehydrogenases, short-chain (EC 1.3.8.1), medium-chain (EC 1.3.8.7), long-chain (EC 1.3.8.8), and VLC acyl-CoA dehydrogenase (EC 1.3.8.9) (Fig. 12; Fig. 13).
Metabolic branch of FASIS
However, reactions of FA β-oxidation not only ensure sufficiently elevated ATP, formed by OXPHOS concomitant to the accelerated Krebs cycle (relative to its rate at low glucose), but also provide the redox signal in the form of the excessive superoxide formation with an instant dismutation into the H2O2 (Fig. 1C, D; Fig. 12; Fig. 13). One can only speculate why excessive superoxide is formed and where. The candidate site is the so-called flavin site IF of Complex I, well known to increase superoxide formation at a higher mt-matrix [NADH]m/[NAD+]m ratio (Fig. 1D). However, if superoxide was formed under the precisely same conditions at the 2-oxoglutarate dehydrogenase site OF, this is experimentally indistinguishable, unless certain mutations of the relevant protein subunits are studied. Both these sites form superoxide when the additional NADH is present due to a higher Krebs cycle turnover induced by a surplus of acetyl-CoA as well as the additional NADH input supplied by 3-hydroxyacyl-CoA dehydrogenase.
Moreover, the increased electron input from FADH2 to the respiratory chain is the second cause for the increasing superoxide formation (Fig. 1D). It needs to be investigated how this proceeds mechanistically. Indeed, all mitochondrial flavoprotein acyl-CoA dehydrogenases, short-chain (EC 1.3.8.1), medium-chain (EC 1.3.8.7), long-chain (EC 1.3.8.8), and VLC acyl-CoA dehydrogenase (EC 1.3.8.9) (Fig. 12; Fig. 13), all donate electrons via electron-transfer flavoprotein (ETF) to the inner mitochondrial membrane enzyme, ETF-coenzyme Q oxidoreductase (ETFQOR) (Brand, 2016; Goncalves et al., 2020).
ETFQOR reduces Coenzyme Q, that is, ubiquinone (Q), to QH2. This reaction is coupled within the ETFQOR protein to the oxidation of FADH2 to FAD (although two ETF molecules are required for two transfer steps of single electrons to ETFQOR) (Fig. 1D). ETFQOR itself could produce superoxide, as suggested for skeletal muscle (flavin site EF), but its major reaction product (QH2) is the same as the product of Complex I. Consequently, the additional QH2 may effectively retard Complex I electron transfer by a feedback and thus elevated superoxide formation might occur. Anyway, FA β-oxidation in pancreatic β-cells provides superoxide/H2O2, which is detectable even in the INS-1E cell and islet exterior (Jabůrek et al., 2024).
Note that LC and SC acyl-CoA-esters alone, or malonyl-CoA were considered to be “factors for nutrient stimulus-insulin secretion coupling” (Prentki et al., 2002). Nevertheless, assaying a closure within a population of the ATP-sensitive K+ channels (KATP) and Ca2+ oscillation in the INS-1E cell cytosol upon FA β-oxidation at the blocked GPR40 pathway in the presence of the GPR40 antagonist GW1100, it has been demonstrated that even such metabolic components are able to stimulate insulin secretion (Ježek et al., 2015). This is ongoing by the same canonical pathway as GSIS with the exception that the redox/H2O2 signal is provided by mitochondria, whereas for GSIS, NOX4 is the major H2O2 source.
Molecular mechanisms of FASIS—receptoric branch
In pancreatic β-cells, the PKA pathway is involved in the signaling of incretin (typically GLP-1 and GIP) receptors described above and makes a minor contribution to signaling from metabotropic receptors, such as GPR40, sensing FAs. GPR receptors, particularly four of their isoforms, serving as free FA GPR receptors, play multiple roles. However, with an artificial nonmetabolizable agonists of a high-affinity, the GPR40 pathway stimulates a self-standing fuel-independent stimulation of insulin secretion at low non-stimulating glucose (Jabůrek et al., 2024) (Fig. 9). This is similar to the GLP-1 receptor pathway.
In contrast, when GPR41 and GPR43 interact with Gαi/o-proteins, adenylate cyclase is inhibited. As a result, cAMP signaling is counteracted. Finally, the predominant interaction of GPR40 and GPR120 is with Gαq/11G-proteins (Briscoe et al., 2003; Schröder et al., 2010), activating Ca2+-dependent PLC, which splits PIP2 into DAG and IP3 (Fig. 8A). DAG activates a plethora of isoforms of PKCs, whereas IP3 induces Ca2+ efflux from ER to the cytosol. Because novel PKCs (nPKCs) are activated only by DAG, but not by Ca2+ (Shigeto et al., 2015), the GPR40 pathway activated by the high-affinity nonmetabolizable agonist is able to trigger insulin secretion without the previous Ca2+ entry via CaV channels. Note also that certain PKC isoforms induce a synthesis of 5-diphosphoinositol pentakisphosphate (5-IP7), which in synergy with elevated Ca2+ enable the synaptogamin-dependent exocytosis of IGVs.
Among metabotropic GPR receptors, GPR40/FFAR1 and GPR120/FFAR4 are activated by LCFAs, having 12 or more carbons, and MCFAs (having 6–12 carbons). In turn, GPR41/FFAR3 and GPR43/FFAR2 are activated by short-chain FAs (SCFAs, having fewer than 6 carbons) (Briscoe et al., 2006). GPR40 is abundantly expressed in pancreatic β-cells. Upon LCFA binding, GPR40 typically activates Gαq/11G-proteins, but a biased activation depending on the ligand exists with GαS, Gα1 /13 and Gαι/o proteins. Let us remain again that the Gαq/11 proteins promote the Ca2+-dependent PLC-mediated PIP2 hydrolysis into DAG and IP3 (Feng et al., 2006; Graciano et al., 2013; Hauge et al., 2015; Husted et al., 2017; Kristinsson et al., 2015; Latour et al., 2007; Sabrautzki et al., 2017; Tunaru et al., 2018; Yamada et al., 2016). As described above, since PIP2 stabilizes open KATP, its hydrolysis promotes the closing of KATP (Yang et al., 2020) and thus contributes to triggering mechanism for plasma membrane depolarization. DAG, via stimulating PKC iso-enzymes, enables phosphorylation of TRPM4 and TRPM5 channels (Shigeto et al., 2015), cooperating on setting plasma membrane depolarization, analogously to TRPM2 channels (Kakei et al., 2016; Yosida et al., 2014).
DAG might hypothetically come independently of PLC from the so-called glycerol/FA cycle (Prentki et al., 2013) (Fig. 14), if this cycle was operating at “fasting” glucose, as nPKCs are activated only by DAG but not by Ca2+ (Shigeto et al., 2015). Such activation would be independent of the GPR40-signaling. The theoretical possibility that elevated [Ca2+]c would be independent of (but acting simultaneously with) the GPR40-signaling implies the existence of the Ca2+-induced Ca2+ release from ER, conducted by the IP3-receptor (IP3R). However, for pancreatic β-cells, IP3 formation upon FA stimulation was found to be independent on GPR40.
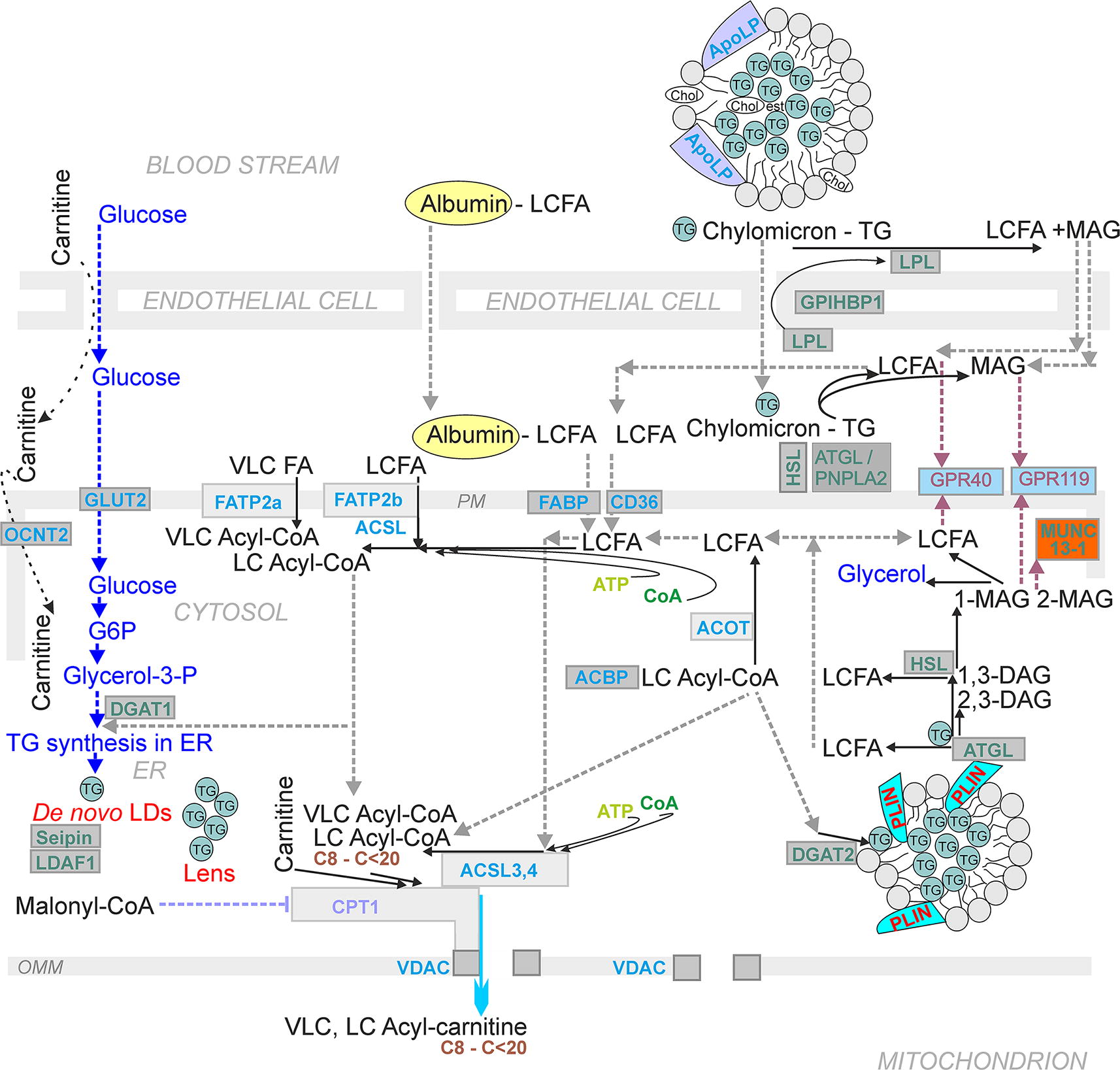
DAG also potentiates the GSIS second phase by promoting protein kinase D phosphorylation, which induces F-actin depolymerization, hence easing IGV access to the plasma membrane and activating the insulin receptor gene (InsR) transcription (Ferdaoussi et al., 2012; Zhang et al., 2010). Also, GPR120 is expressed in murine islets (Kebede et al., 2009) interacts with Gαq/11 and Gαi/o proteins (Carullo et al., 2021; Milligan et al., 2017), so its downstream pathways contribute to elevated cytosolic Ca2+ (Moran et al., 2014b). Deletion of both GPR120 plus GPR40 did not affect glucose homeostasis in vivo in mice (Croze et al., 2021a; Du et al., 2022). Note that GPR120 activity in pancreatic δ-cells also modulates islet functions (Croze et al., 2021b).
Biased agonism, that is, the ability of a ligand to activate a subset of receptor’s signaling cascades, means that the ligand acts at the same GPR (receptor) but provides distinct cellular signaling profiles. Such ligand preferentially stabilizes different active conformational states of the receptor. For GPRs, such signaling is represented by either the β-arrestin-mediated signaling on one hand or Gα-dependent signaling on the other hand, but not by both pathway types simultaneously as would occur with a natural (traditional) ligand. Note that β-arrestins initiate signaling via the proximal MAP kinase, IκB-kinase, and Akt pathways (Dalle et al., 2011).
The human GPR40 structures with selected ligands distinguished two principal positively cooperating binding sites; one for partial agonists such as MK-8666, TAK875, and AMG837; the second one for AgoPAM types such as AM1638 (Lu et al., 2017). In newborn mice, GPR40 promoted GSIS and functional islet maturation and hence is likely needed for adaptive offspring insulin secretion (Lorza-Gil et al., 2023).
SCFAs—receptoric branch
Murine and human primary β-cells and β-cell lines also express GPR41/FFAR3, coupled to Gαi/o, and GPR43/FFAR2, coupled to Gαi/o, Gαq/11, and Gα12/13 proteins (Fernández-Millán and Guillén, 2022). Predominant interaction of both with Gαi/o was suggested by the increased insulin secretion and improved glucose tolerance in mice with deleted both GPR41 and GPR43 (Prentice and Wheeler, 2015; Tang et al., 2015). The Gαi/o pathway decreases cAMP levels and acts against GPR40 and GPR120 pathways. In contrast, propionate and acetate increased insulin secretion in human islets acting on GPR43 (Pingitore et al., 2017, Pingitore et al., 2019). This phenomenon might be regarded as FASIS, when GPR43 acts in the same way as does GPR40. Acetate potentiation effect on insulin secretion, that might be also regarded as FASIS, was found in wild-type β-cells but not after GRP43 deletion when an inhibition occurred (McNelis et al., 2015). Hence, the latter could be dominated by the GPR41 action. Note that 3-hydroxybutyrate is an important GPR41 antagonist (Lorza-Gil et al., 2020).
I emphasize here that SCFAs, notably the most abundant acetate, propionate, and butyrate are produced by gut microbiota in cecum and colon (Fernández-Millán and Guillén, 2022; Rosli et al., 2023). Typical SCFA content in the body is 60% acetate, 25% propionate, 15% butyrate, and the remaining 5% is left for valerate, isobutyrate, and isovalerate (Cummings et al., 1987). SCFA levels are micromolar in circulation, such as 34 µM butyrate in portal vein, 14 µM in hepatic vein, and 4.5 µM in peripheral vein (Cummings et al., 1987), even though butyrate is locally consumed by colonocytes. Most of the acetate, propionate, and other SCFAs also enter the portal vein. SCFA transport into the cell can be realized as flip-flop of neutral SCFA across the membrane or as anion transport by monocarboxylate transporters. Downstream GPR43 receptor pathways, similarly as those of GLP-1 receptor, increase pancreatic β-cell proliferation. Thus, high-fat diet-fed mice have increased GPR43 population in β-cells as well as acetate (McNelis et al., 2015). These effects were missing in mice with deleted GPR43. SCFAs have also a supporting role for β-cell metabolism, promoting survival under cell stress (Hu et al., 2020).
Molecular aspects of FASIS beyond β-cells
Intestinal secretion of GIP, cholecystokinin, peptide YY, and GLP-1
Ingestion of carbohydrates, proteins, and lipids stimulates enteroendocrine cells to secrete hormones along the length of the gastrointestinal tract. GIP is secreted by K-cells in the duodenum, CCK by I-cells in the duodenum and jejunum, and GLP-1 by L-cells in the jejunum, ileum, and colon (Gribble, 2012). Peptide YY (PYY) is expressed in a minority of L cells in the upper small intestine, but in nearly all L cells in the distal ileum and colon. GIP and GLP-1 secretion responses are strong on glucose and fat, whereas CCK is predominantly stimulated by fat and protein.
Emphasizing here, stimuli for GLP-1 secretion by enteroendocrine L-cells, one can recognize that L-cells possess similar mechanism for stimulating GLP-1secretion as pancreatic β-cells for their receptor downstream pathways. All three major nutrient components stimulate the GLP-1 secretion in L-cells. Specifically, monosaccharides, FAs, and MAG stimulate GLP-1 secretion, as well as aromatic amino acids and bile acids. Monosaccharides provide the stimulation via the sodium-glucose cotransporter (SGLT1)-dependent induction of small inward currents (Gorboulev et al., 2012; Mace et al., 2012; Moriya et al., 2009).
Other nutrients provide stimuli via the metabotropic receptor pathways. FAs activate GPR40, MAG activates GPR119, aromatic amino acids activate the pathway of calcium sensing receptor (CaSR), and bile acids act via the TGR5, bile acid receptor (Ekberg et al., 2016; Husted et al., 2017; Müller et al., 2019. FAs stimulate Ca2+ influx via appropriate Ca2+ channels. Galanin and somatostatin are inhibitory. Neuronal stimulation proceeds via neuromedin C and its receptor. Protein and amino acid stimulation of GLP-1 secretion has also been confirmed in rodent (Diakogiannaki et al., 2013; Tolhurst et al., 2011) and human cells (Chen and Reimer, 2009; Hirasawa et al., 2005), using perfusion of ileum or colon (Dumoulin et al., 1998; Plaisancie et al., 1995), as well as in vivo in rodents (Clemmensen et al., 2013; Hira et al., 2009; Kato et al., 2017) and humans (Greenfield et al., 2009; Lejeune et al., 2006).
It must be also noted that both GIP and GLP-1 induce β-cell proliferation via increasing transcription factor Pdx-1 and promote resistance to apoptosis. In this way, they elevated β-cell mass (Li et al., 2005). Thus, for example, impaired regeneration of β-cell mass was reported for mice with deleted GLP-1 receptor (Li et al., 2003).
FA stimulation of incretin secretion
Some authors reported that the lipid- or FA-mediated secretion of GLP-1 by intestinal L-enterocytes is instead instantaneous (Moss et al., 2016). This contrasts with the findings of peaks in circulation for biologically active GLP-1 in human plasma 30–60 min after carbohydrate or protein intake and 120 min after the ingestion of lipids (Herrmann et al., 1995). Hence it needs to be verified for humans whether the peak of GLP-1 coincides with the peak of chylomicrons in circulation, or whether GLP-1 precedes the peak of chylomicrons. The same is needed for rodents. This is important, because knowledge of precise timing can distinguish between GLP-1-amplified GSIS at intermediate glucose from the net FASIS; or else GLP-SIS independent of GSIS from the net FASIS at low glucose. At least FAs mobilized from WAT stores should not coincide with the above phenomena, as lipolysis is inhibited by secreted insulin. Also, the intake of saturated FAs induces insulin secretion by elevating GIP in healthy individuals (Itoh et al., 2014).
Physiological levels of FASIS in rodents and timing
The lack of courage to call “potentiation of GSIS” as contribution of FASIS can be illustrated in the results of Lorza-Gil et al., 2023. The reported lack of GSIS potentiation by palmitate in PIs of GPR40/FFAR1 knockout mice can be simply ascribed to the missing receptoric part (mechanism) of FASIS. Moreover, pertussis toxin blocking Gαi/o proteins and downstream inhibitory pathways restored GSIS in PIs of GPR40/FFAR1 KO mice, demonstrating that palmitate itself was not “toxic.”
Regulation of FATP proteins
FA transport protein FATP1 (SLC27A1) was found within the plasma membrane of 3T3-L1 adipocytes (Huang et al., 2021; Schaffer and Lodish, Schaffer and Lodish, 1994), 293 cells (Hatch et al., 2002), and brown adipose tissue (Wu et al., 2006b). Insulin stimulation in adipocytes can translocate FATP1 from the cytoplasm to the plasma membrane (Huang et al., 2021; Lobo et al., 2007; Stahl et al., 2002). However, for skeletal muscle, mitochondrial localization of FATP1 was reported (Guitart et al., 2014). The problem is whether this localization concerns the outer mitochondrial membrane or the inner mitochondrial membrane. In the former location, a complex with ACSL3 and ACSL4 could be expected, despite these enzymes reside also in ER, Golgi apparatus, and peroxisomes.
What function would be of an FATP1 transporter in the inner mitochondrial membrane without forming a complex with ACS is unclear. Just having FA in equilibrium with FA− anion in the inner (matrix) lipid leaflet of the inner boundary membrane and crista membrane would not end up in β-oxidation. Predominant mitochondrial localization was reported also for myocytes (Guitart et al., 2009; Sebastián et al., 2009). FATP1 was found to act also in the heart (Chiu et al., 2005). In 293 cells, FATP1 was suggested to preferentially channel exogenous FAs into TGs (Hatch et al., 2002). This role is consistent with the finding that in 3T3-L1 adipocytes, FATP1 deficiency led to the diminished TG accumulation and LD size (Schaffer and Lodish, Schaffer and Lodish, 1994).
Physiological lipid metabolism as essential for β-cell fitness
Relationships of lipid versus glucose metabolism in β-cells
During metabolism of higher glucose levels, that is, upon GSIS, enabling the citrate export from the matrix to the cytosol, conditions are set to promote FA synthesis in pancreatic β-cells. Metabolomics indicated an increase of palmitic acid after the transition from low to high glucose (Spégel and Mulder, Spégel and Mulder, 2020). The FA salvage and de novo FA synthesis are promoted because the citrate export elevates cytosolic acetyl-CoA levels. This is enabled by ATP-citrate lyase (ACL) fed by the citrate efflux from the mitochondrial matrix at the truncated Krebs cycle after the citrate synthase reaction (MacDonald et al., 2007; Mugabo et al., 2017). Acetyl-CoA can be transformed to malonyl-CoA by the reaction of acetyl-CoA carboxylase (ACC) (Guay et al., 2013; Lorenz et al., 2013), when ACC is not phosphorylated by AMPK. Since malonyl-CoA inhibits CPT1 and hence FA β-oxidation, FA synthesis is promoted (Corkey et al., 1989).
The citrate export and concomitant reactions belong to the so-called glycerolipid/free FA cycle (GL/FFA cycle) (see below). Such rerouting is not interfering with GSIS, as silencing of ACL and FA synthase in β-cells did not affect GSIS (Joseph et al., 2007). Thus, mice with depleted FA synthase exhibited unaffected insulin secretion (Chakravarthy et al., 2009). However, the depletion of islet lipid stores (LDs, see further) was accompanied with diminished GSIS, and re-establishment of lipid stores rescued the impaired GSIS (Koyama et al., 1997; Stein et al., 1996).
At low glucose (fasting) conditions, certain low levels of FA β-oxidation producing acetyl-CoA in the mitochondrial matrix occur. Under FASIS at low glucose, LDs play a role together with redox-activated mitochondrial phospholipase iPLA2γ (Fig. 13). Switching to higher glucose (initiating GSIS) concomitant with switching-on pyruvate-based redox shuttles still may preserve matrix acetyl-CoA due to the reaction of carnitine O-acetyl transferase (CRAT), catalyzing interconversion of acetyl-CoA and acetylcarnitine in the mitochondrial matrix. Matrix acetyl-carnitine and SC-carnitine stores could be hypothetically transferred by CRAT to acetyl-CoA (SC-acyl-CoAs would be converted to acetyl-CoA by the required cycles of β-oxidation). One may also speculate that BCKA metabolism may supply SC-acyl-CoAs i.e. BC-acyl-CoAs, as recognized in pancreatic ductal adenocarcinoma cells (Gotvaldová et al., 2024).
These speculations are supported by the observation that the β-cell-specific CRAT deletion in mice led to ∼30% attenuation of GSIS, whereas CRAT overexpression enhanced insulin content and secretion (Berdous et al., 2020). GSIS was also attenuated in CRAT-silenced MIN6B1 cells. Authors concluded that CRAT rather regulated the insulin content than the mechanism of GSIS. Also, since CRAT conversion of acetyl-CoA to acetyl-carnitine releases the acetyl-CoA inhibition of pyruvate dehydrogenase and consequently allows a higher glycolytic pyruvate input into the Krebs cycle, the lack of this effect after CRAT ablation would explain GSIS decline. Possibly lower pyruvate input has been confirmed by respiration studies (Berdous et al., 2020).
Glycerol lipid/free FA cycle
The glycerol lipid/free FA cycle (GL/FFA) cycle is an important pathway in pancreatic β-cells at intermediate, high, and excessively high glucose (Fig. 14). This cycle provides a crosstalk between FA and glycerol metabolism and consists of lipogenic and lipolytic arms. As mentioned above, under conditions switching to the citrate efflux from the matrix, ACL generates cytosolic acetyl-CoA, which is subsequently converted to malonyl-CoA (Antinozzi et al., 1998; Corkey et al., 1989; Mulder et al., 2001), providing even more feedback inhibition of CPT1 and FA β-oxidation. The increasing ATP does not allow AMPK-mediated phosphorylation to inhibit CPT1. As a result, upon glucose intake, the existing FA pool is not consumed. Rather FA esterification with glycerol phosphate is promoted, yielding TGs. They are synthesized from the glycolysis-derived glycerol-3-phosphate (G3P) and already accumulated LC-acyl-CoAs via consecutive steps including synthesis of lysophosphatidic acid, phosphatidic acid, and final DAG formation.
A simultaneous lipolytic arm is represented by the reaction of TG lipase, hydrolyzing TG to DAG. Next, either HSL or DAG lipase will convert DAG to MAG, releasing glycerol (Prentki et al., 2020; Zhao et al., 2014). For β-cells, the enzymes involved in both GL/FFA cycle arms are not completely characterized. They include acyltransferases (GPAT, AGPAT, DGAT) and lipases (ATGL, HSL, MAGL/ABHD6).
The lipolytic arm of the GL/FFA cycle produces important mediators involved in the mechanism of IGV exocytosis and in activating TRPM channels cooperating in the establishment of
The predominant β-cell MAG lipase, hydrolyzing MAG to glycerol and FA, α/β-hydrolase domain containing 6 (ABHD6), should also have a prominent role in the GL/FFA cycle. Its deficiency increased GSIS, whereas its deletion potentiated GSIS, pointing out the role of MAG in priming Munc13-1 protein of exocytosis machinery for IGVs (Zhao et al., 2014, Zhao et al., 2015).
Physiological lipid metabolism is essential for β-cell fitness
The question has been raised whether a certain minimum level of FA metabolism is even required for GSIS. Indeed, the inhibition of TG lipolysis attenuated GSIS in isolated PIs (Masiello et al., 2002; Mulder et al., 2004) and the first GSIS phase was lower in mice with lipase deleted specifically in β-cells (Fex et al., 2009) or with deleted ATGL (Attane et al., 2016; Liu et al., 2020; Peyot et al., 2009; Roduit et al., 2001; Tang et al., 2013). In contrast, inhibition of lysosomal acid lipase in pancreatic β-cells indirectly potentiates GSIS by activating ATGL and HSL (Pearson et al., 2014). Besides the explanation emphasizing the decreased MAG, results of β-cell-specific ATGL deletion were explained based on a lower availability of PPARδ ligands causing mitochondrial dysfunction (Tang et al., 2013) and reduced palmitoylation and stability of SNARE protein syntaxin 1a (STX1a), which is crucial for insulin secretion (Liu et al., 2020). Moreover, FAs liberated from WAT were indeed recognized to stimulate FASIS after β-adrenergic stimulation (Scheja et al., 1999). Partly deficient adipocyte ATGL or HSL activity also diminished insulin secretion (Heine et al., 2018; Schoiswohl et al., 2015; Schweiger et al., 2017; Wu et al., 2012; Xia et al., 2017).
In turn, the net FASIS induced by palmitate was not affected by such ATGL deletion, despite the ongoing GL/FFA cycle, where the TG synthesis was alternated with TG hydrolysis (Prentki et al., 2013). Note, that such cycle is futile, which means that it consumes ATP. This futile cycling would be, however, useful for further DAG-stimulated signaling pathways. Note also that moderately elevated cytosolic Ca2+ should be required for PLC activity. However, 1,2-DAG originating from the GL/FFA cycle does not require PLC. Also, note that so-called nPKCs are activated by 1,2-DAG alone and again do not rely on [Ca2+] (Lyu et al., 2020). Hence, the activation of such nPKCs could promote IGV-exocytosis even at low glucose.
It should be noted that intermittently released FAs during the GL/FFA cycle would provide FASIS in the presence of GSIS, at least in the intensity allowed by the established malonyl-CoA levels. Experimentally, added FAs to β-cells and PIs in the presence of insulin-stimulating glucose promotes insulin secretion which is higher than that one without FAs (El-Azzouny et al., 2014). Simultaneously, G3P was rapidly esterified into lysophosphatidic acid and several different glycerolipids, partly replenishing cytosolic NAD+. The involved glycerol-3-phosphatase in this case regulates glycolysis, the cytosolic redox state, and probably also ATP production, at least the glycolytic one (Mugabo et al., 2016). The main consequence, however, lies in the concomitant increase in LC-saturated MAG acting on GPR119 (Mugabo et al., 2017; Zhao et al., 2014), as described above.
The GL/FFA cycle was suggested to act as detoxifying the excessive glucose loading (Prentki and Madiraju, 2012). This is because the excessive glucose converted to glycerol is extruded from β-cells by aquaglyceroporins (Matsumura et al., 2007). The glycerol extrusion mechanism is however independent on the GL/FFA cycle, since β-cells contain G3P phosphatase (G3PP), directly converting G3P to glycerol (Mugabo et al., 2016).
Recently, 0.5 mM oleate but not palmitate exposure for 48 h was reported to enhance β-cell proliferation via elevated ROS and peroxiredoxin signaling (Vivoli et al., 2023). Both FAs, oleate and palmitate, enhanced expression of genes important for mitochondria and energy metabolism.
LDs of pancreatic β-cells
LDs belong to intracellular organelles having a monolayer phospholipid membrane decorated with perilipins (PLINs, isoforms 1–5) surrounding the core composed of TG, cholesterol and retinol esters, that is, of neutral lipids. LDs just enable multipurpose usage of FAs, which can be stored in LDs and released to the cell as free FAs. Consequently, metabolism of lipids in cells is ensured by regulated trafficking of lipids into and out of LDs (Fig. 13; Fig. 14). In β-cells, this can contribute to beneficial effects of lipids as well as to diminishing their possible toxicity (Tong et al., 2022). De novo LDs originate from ER (Fig. 14). As LDs can encounter ER by feedback, but also mitochondria, autophagosomes, lysosomes, and nuclei, they could coordinate lipid metabolism in this more efficient way, namely in adipocytes, myocytes, hepatocytes as well as in human β-cells.
In pancreatic β-cells, LDs act prominently in the abovedescribed GL/FFA cycle. Interestingly, PKA as well as GLP-2 receptor pathway led to the increased PLIN5 (Trevino et al., 2015). In turn, PLIN5 overexpression improved GSIS in the presence of PA (i.e., with a parallel FASIS) as well as in mice fed a high-fat diet (Zhu et al., 2019, 2020). However, the most prominent perilipin in β-cells is PLIN2. Its deficiency in INS-1 cells impaired GSIS in both presence and absence of PA (Mishra et al., 2021). The PLIN2 deficiency rerouted more FAs into mitochondria, speculatively causing mitochondrial dysfunction and consequently impaired GSIS. Similar observations were reported for PLIN2 deficiency in EndoC-βH2-Cre cells (Tong and Stein, 2021). In conclusion, LDs appear to be essential for adult human islet β-cell function due to the LDs ability to balance homeostasis of FAs.
Synthesis and degradation of sphingosine-1-phosphate (S1P) was found to regulate LD number and size and thus modulate lipotoxic effects of FAs in β-cells (Tang et al., 2024). The overexpression of S1P phosphatase prevented FA-mediated caspase 3 activation, whereas degradation of S1P by overexpression of S1P lyase decreased LD biogenesis.
Pathological Effects of Higher FA Doses
Type 2 diabetes
Possible etiology of type 2 diabetes
Patients with type 2 diabetes of five clusters were stratified according to the stage of progression and risk of diabetic complications (Ahlqvist et al., 2018). Type 2 diabetes is characterized by insulin resistance (IR) concomitant to a gradual impairment of β-cell mass and function (Zaccardi et al., 2016). In other words, the IR onset is followed by deterioration of insulin secretion (Chen et al., 2017; Swisa et al., 2017). The IR development may be viewed as originating from a shift of macrophages in WAT to their pro-inflammatory M1 phenotype, which results in WAT tissue remodeling (Xourafa et al., 2024). M1 macrophages promote low-grade inflammatory reactions but without the systemic cytokine spillover. On a molecular level, IR is characterized by the impaired function of downstream pathway of InsR, elegantly summarized in reviews such as (Lee et al., 2022; Li et al., 2022b; Mastrototaro and Roden, 2021).
First, humans typically develop a state of impaired glucose tolerance followed by diabetes (Fig. 15). At the initial stage, pancreatic β-cells can compensate for IR by increasing insulin secretion and cell mass. A progressive type 2 diabetes follows as β-cells are unable to secret sufficient insulin to control glucose homeostasis, either due to their functional impairment (Del Guerra et al., 2005; Ferrannini et al., 2005) and/or due to their reduced mass (Hunter and Stein, 2017; Prentki and Nolan, 2006; Spijker et al., 2015; Wigger et al., 2021). Nevertheless, the question remains, whether the primary cause is deterioration of β-cells and IR is the reflection of it; or whether the primary cause is the IR development and β-cell impairment is caused by their inability to compensate for the resulting increased insulin demand. This could even vary between patients. Moreover, low-grade inflammatory reactions can be promoted also by macrophages residing and/or being attracted to islets, with consequences which I describe below.
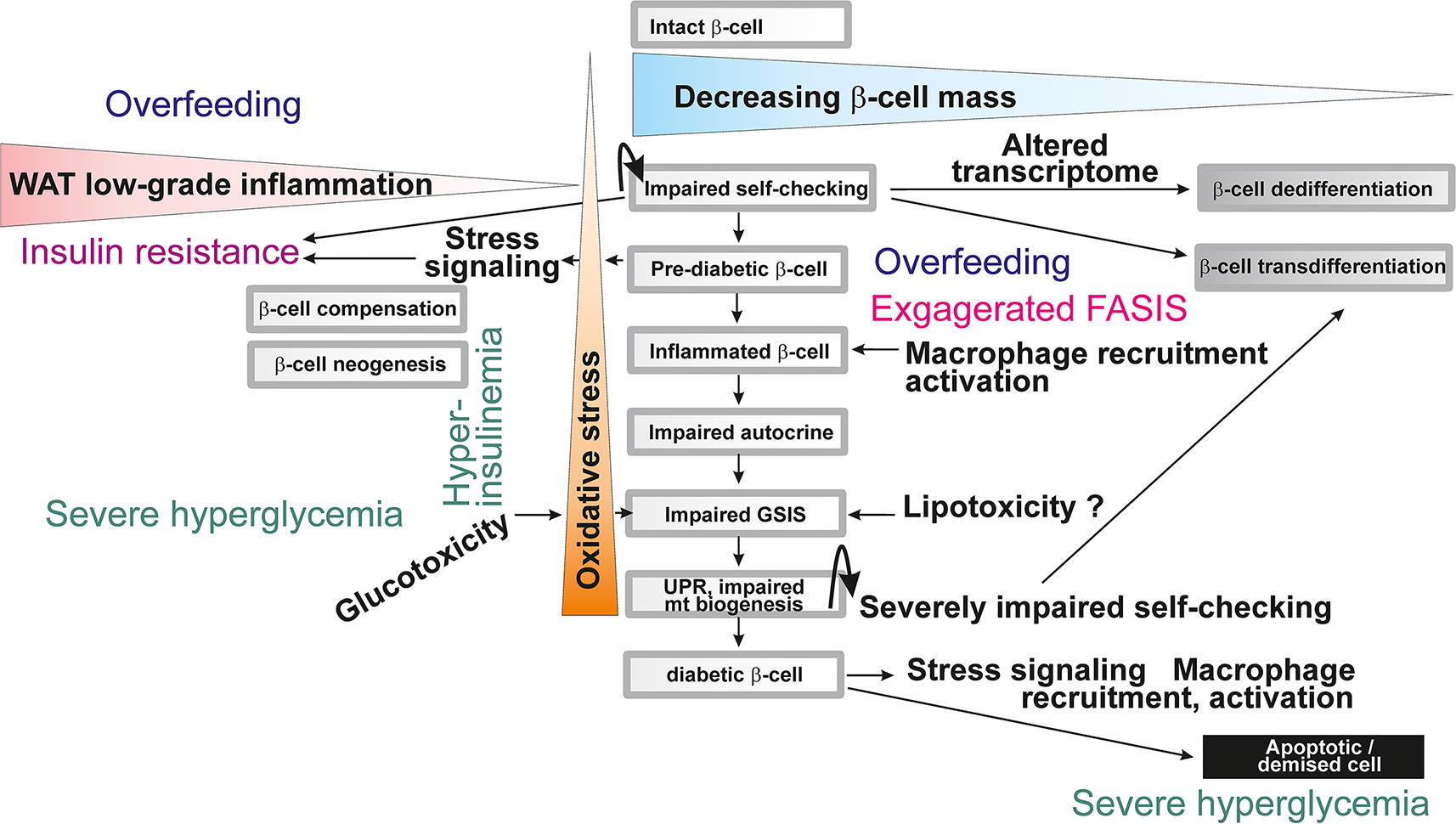
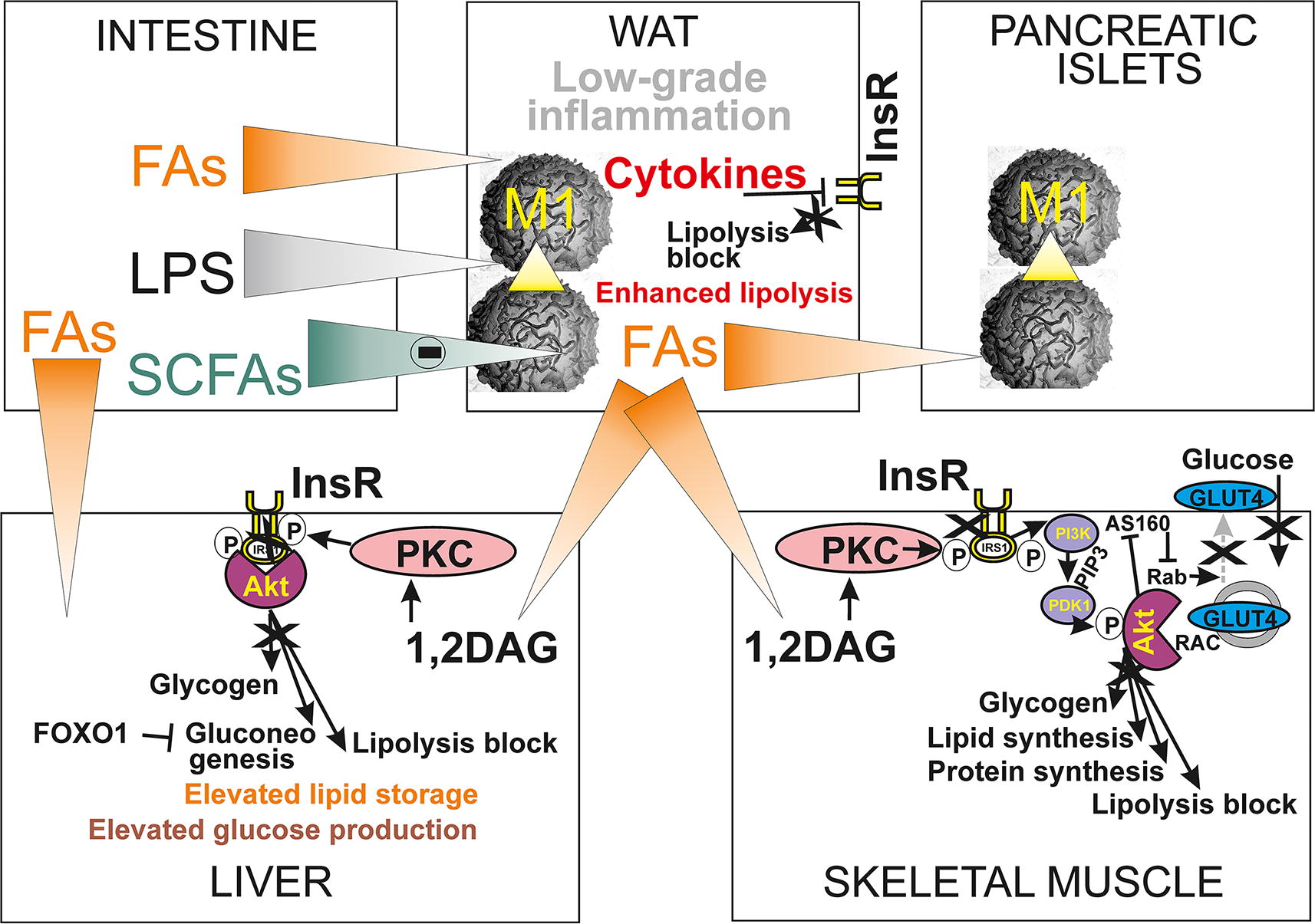
Explanation with IR as the original cause raises an argument that if hyperinsulinemia preceded IR, it would cause hypoglycemia in the absence of IR. Hence, it is considered that β-cells are impaired later after the IR onset. Such a situation is mimicked pharmacologically by sulfonylureas such as glibenclamide provoking insulin secretion. Their administration shortens the period during which patients are able to secrete a sufficient amount of insulin (Feingold, 2000). Nevertheless, a sudden deterioration of β-cell function would not be able to secrete so much insulin to set hyperglycemia and cause hypoglycemia. Also, inhibitory mechanisms for insulin secretion described in Section Other main regulations of GSIS would prevent hypoglycemia. In contrast, the major argument for a combination of IR and β-cell pathology is the possibility to improve type 2 diabetes by transplantation of PIs (Shapiro et al., 2017).
The first GSIS phase is typically lost in type 2 diabetes, correlating with higher glycemia (Pratley and Weyer, 2001). In a progressive type 2 diabetes, β-cell mass is reported to drop to 40%–60% of normal (Butler et al., 2003; Rahier et al., 2008), while the remaining β-cells act on their lower capacity (Ježek et al., 2019; Moin and Butler, 2019; Weir and Bonner-Weir, 2004). A crosstalk exists between IR and β-cells. To maintain normoglycemia after an IR onset, β-cells compensate for the increased insulin demand and attempt to proliferate more to increase their mass. Interestingly, body mass index positively correlates with β-cell mass (Rahier et al., 2008; Saisho et al., 2013). However, an early event is still dysfunction of β-cells, ongoing before the decrease in β-cells (Cohrs et al., 2020). Later, the decreased β-cell mass does not originate from a decreased β-cell proliferation, but rather from apoptosis (Butler et al., 2003; Folli et al., 2018) and other forms of cell death and due to the increased inflammation (Donath and Shoelson, 2011). The IR acts back on islets in a way that induces heterogeneous autophagic flux within the population of β-cells (Aoyama et al., 2023).
How do the elevated FAs and circulating lipids fit into the prediabetes etiology? There exist typically obese individuals who are not diabetic but exhibit high rates of insulin secretion (Camastra et al., 2005; Kloting et al., 2010). These rates most probably compensate for any developing IR due to moderate WAT inflammation. If these individuals for yet unknown primary cause progress to diabetes, their first GSIS phase is decreasing with rising fasting glycemia (Brunzell et al., 1976). However, their FA plasma content does not increase substantially (Carpentier et al., 2000; Lyssenko et al., 2005). Elevated FA release to circulation together with secretion of pro-inflammatory adipokines result in ectopic fat deposits (Ertunc and Hotamisligil, 2016; Hong et al., 2016; McQuaid et al., 2011). The developing low-grade inflammation in WAT (but also in liver and skeletal muscle) induces the impairment of the InsR pathway and hence developing IR.
Obese humans may have abnormal secretion of WAT adipocyte-derived factors, which induce the low-grade chronic inflammation and IR (Scherer, 2019; Stern et al., 2016). In contrast, LPL overexpression in WAT improved GSIS and impaired glucose tolerance in mice (Memetimin et al., 2022). Yet still, high FA levels have been regarded as a major risk factor (Stefan et al., 2003). At first, they could participate in the moderate WAT inflammation so they would be a risk factor at least for IR. When IR further develops, this demands higher insulin secretory responses (Martin et al., 1992).
Glucotoxicity and glucolipotoxicity
Glucotoxicity has been simulated by culturing PIs in media with high glucose (Andersson and Hellerström, 1972), which also reprogrammed their transcriptome, and hence then resembles features of stem cells (Jonas et al., 1999). This phenomenon is called dedifferentiation. These changes are reversible since the restoration of normal glucose levels led to cell revitalization. A surgical procedure where type 2 diabetes is reversed by gastric bypass surgery can be regarded as in vivo revitalization from glucotoxicity (Polyzogopoulou et al., 2003).
Long-term elevated glucose thus suppresses the expression of transcription factors considered to be β-cell-specific, such as Pdx1/IPF1, MafA, or Nkx-6.1, inducing the reversion to progenitor-like cells (Cinti et al., 2016). Glucotoxicity also induces the expression of genes involved in mitochondrial transport (Jimenez-Sanchez et al., 2020), p53 protein (Flores-López et al., 2024), AMPK pathway (Brun et al., 2020), glucose and lipid metabolism, exocytosis, and factors rendering to cell death (Brun et al., 2015; Ottosson-Laakso et al., 2017). High glucose was also reported to increase IL-1β which activated the NF-κB pathway (Maedler et al., 2002). Thioredoxin interacting protein (TXNIP), responding to glucose, is now considered as a central mediator of glucotoxicity in pancreatic β-cells. Note that exaggerated pro-inflammatory cytokine action results in FOXO1 downregulation inducing β-cell dedifferentiation (Nordmann et al., 2017). The expression of orphan nuclear receptor NR4A1 is upregulated by increasing glucose via cAMP/PKA/CREB pathway, which is beneficial for GSIS, as NR4A1 regulates expression of genes such as Glut2, Ndufa4, Ins1, In2, Sdhb, and Idh3g (Herring et al., 2024).
The term glucolipotoxicity has been coined to describe a possible synergy between high levels of glucose and FAs in enhanced damage of β-cells (Lytrivi et al., 2020; Prentki et al., 2002). Inspiration came from the findings of increased malonyl-CoA and LC-acyl-CoAs in different tissues of obese patients with type 2 diabetes (Brun et al., 1996; Prentki and Corkey, 1996). Thus β-oxidation is inhibited at rise of malonyl-CoA, which blocks CPT1 in β-cells. This should lead to cytosolic LC-acyl-CoA accumulation. However, the link to β-cell dysfunction is not known. Similarly, as in human β-cells, such metabolic disbalance could lead to the elevated LD formation observed also in INS-1E -cells (Oberhauser et al., 2022).
Lipid metabolism in prediabetes
Does lipotoxicity exist in humans?
It has been originally suggested that the excessive lipid accumulation induces β-cell death (Shimabukuro et al., 1998). Unlike rat INS-1 823/13 cells, cultured human EndoC-βH1 cells rerouted the added 1 mM palmitate into LDs (Thomas et al., 2022). Palmitate was dissolved in these experiments in 90% ethanol followed by FA-free bovine serum albumin conjugation Nevertheless, even for these cells, monounsaturated erucic acid (C22:1), added analogously, was highly toxic (Plötz et al., 2019). In rat β-cells, exogenous palmitate accumulated into membranes of Golgi apparatus as well as other organelles, leading to a loss of their viability but not to the ER stress. However, the additional oleate supply prevented these changes (Thomas et al., 2022). It has been suggested that lipotoxicity is a phenomenon existing primarily in rodent β-cells (Weir, 2020). In human cells, stearoyl CoA desaturase protects the cells (Oshima et al., 2020).
The central question judging in vivo lipotoxicity is whether the FA levels found in typical nutrient situations are ever high enough to cause harmful effects. Any simulations are obscured by the fact that a pattern of liberated FAs and their local concentrations in the interstitial space surrounding the lipid bilayer plasma membrane of β-cells are not known. Analyzing a large cohort, 0.57 mM circulating FA levels (at 80 µM TG) were found in nonobese subjects after an overnight fast, compared with 0.71 mM FAs (120 µM TG) in obese subjects with a body mass index ≥30 kg/m2 (Arner and Rydén, 2015). Nevertheless, IR and type 2 diabetes led only to a further minor increase among obese subjects. Moreover, lipid infusion was unable to block arginine-stimulated insulin secretion in nondiabetic obese subjects (Carpentier et al., 2001). Rather a genetic disposition played a role, when lipid infusions caused the impaired GSIS in nondiabetic subjects (Kashyap et al., 2003).
LDs in potentiating and/or counteracting lipotoxicity
LDs could play an important role in balance between lipotoxicity and defense against it (Tong and Stein, 2021). TG synthesis in mouse PIs after both high-fat diet and fasting increased together with the expression of perilipins PLIN2 and PLIN5 decorating TG-rich LDs (Faleck et al., 2010; Tong et al., 2020; Trevino et al., 2015). Imaging LDs with size <1.2 µm was achieved in rat and human β-cells (Trevino et al., 2015; Vernier et al., 2012) and PLIN2 was increased in β-cells of subjects with type 2 diabetes (Ji et al., 2019; Tong et al., 2020). Interestingly, ATGL ablation led to an increasing number of LDs and to their larger dimensions (Liu et al., 2020). Intake of FAs and glucose led also to the increased LD number (Tong and Stein, 2021; Vernier et al., 2012). However, the increased LD accumulation was observed also in dysfunctional human β-cells transplanted to mice (Dai et al., 2016).
Counteracting glucolipotoxicity was described for human EndoC-βH1 cells, which highly express stearoyl-CoA desaturase (SCD1). SCD1 catalyzes double bond formation, so to increase the content of oleic acid relative to PA, which subsequently leads to enhanced TG synthesis and biogenesis of LDs (Busch et al., 2005). SCD1-defficient EndoC-βH1 cells were prone to inflammation and ER stress after the PA dosage (Oshima et al., 2020), unlike parental cells exhibiting resistance to stresses induced experimentally by PA (Krizhanovskii et al., 2017; Thomas et al., 2021). As EndoC-βH1 cells contain rich LDs, a consensual conclusion has been raised that human β-cells defend glucolipotoxicity by increasing TG synthesis and expanding the LD pool size. Also, PLIN5 overexpression was reported to prevent PA-induced apoptosis and ER stress in INS-1 cells (Zhu et al., 2019), probably via activation of the nuclear erythroid 2-related factor.
Contradictions concerning lipotoxicity
A problem with the term lipotoxicity can be seen in the fact that it does not represent a unique defined molecular mechanism (Oberhauser and Maechler, 2021), but a general and ex vivo artificial effect in the vast majority of cases. Contrary to glucotoxicity, long-term exposure to excessive FAs does not cause any alternations in transcriptome or does not lead to dedifferentiation in INS-1E-cells and human islets (Oberhauser et al., 2022). Long-term lipid infusions not only in humans but also in rats led to diminished GSIS (Mason et al., 1999). A problem interpreting these results can be seen in the fact that the simulated FA doses far exceeded those found in patients with diabetes (Weir, 2020). In addition, the β-cell dedifferentiation and impaired insulin secretion were found to be independent of lipid level (Kjørholt et al., 2005).
In vitro lipotoxicity of palmitate was ascribed to ER stress and exaggerated ceramide formation. Thus 0.5 mM palmitate (albumin-conjugate) was found to induce apoptosis due to depletion of ER calcium, which activated IRE1, PERK, and ATF6 proteins of the unfolded protein response pathway (Cnop et al., 2010; Cunha et al., 2008). Palmitate also induced palmitoyl transferase and ceramide synthases, leading to elevated ceramide levels by a salvage pathway and causing apoptosis or impairment of β-cell function in MIN6 cells and human islets (Manukyan et al., 2015). Palmitate was also reported to upregulate ER-oxidoreductin-1α, which produced increasing H2O2 and led to lipotoxic ER stress and β-cell failure (Sharifi et al., 2024).
In conclusion, a distinct spectrum of β-cells functions and fitness impairment by more active FAs with C >18 and unsaturated was found. However, it is not clear how these individual mechanisms of, for example, ER stress, initiation of apoptosis and ferroptosis act, and if ever, over decades during the type 2 diabetes development in humans.
Apoptosis and ferroptosis of pancreatic β-cells
Apoptosis can be regarded as the endpoint of β-cell death in type 1 and type 2 diabetes. The extrinsic pathway is induced by FAS/CD95 and tumor necrosis factor (TNF) receptor (TNFR). FAS complex with FADD and FLICE via caspase 8 activates effector caspases 3,6, and 7, which release proteolytic and DNA degrading enzymes (Tummers and Green, 2017). TNFR recruits TRADD initiating caspase 8 and 10 and consequently caspases 3,6, and 7. The intrinsic pathway is mitochondrial, involving mitochondrial outer membrane permeabilization (MOMP; Green, Green, 2022a,b) or permeability transition pore phenomenon (Bernardi et al., 2023) and release of cytochrome c (initiated by pro-apoptotic BAX and BAK and inhibited by BCL-2 and BCL-XL; Green, Green, 2022b) and other proapoptotic components including SMAC/DIABLO, OMI, endonuclease G, and AIF, leading to caspase 2 and 9 activation. Diverse apoptotic pathways are subsequently triggered. Namely, cytochrome c release activates cytosolic APAF1 oligomerization in the presence of dATP, which recruits caspase 9 and forms an apoptosome. Mature caspase 9 subsequently activates effector caspases 3,6, and 7. SMAC and OMI prevent XIAP to inhibit caspase 9. After MOMP endonuclease G cleaves DNA between nucleosomes, while AIF, cleaved by proteases from its tether in the mitochondrial intermembrane space, initiates caspase-independent chromatin condensation and DNA fragmentation.
As mentioned above (Section Glucotoxicity and glucolipotoxicity), besides upregulating numerous genes, increased glucose recruits cell division blocker p53 (guardian of the genome) to mitochondria of model β-cells, RINm5F cells, which leads to elevated superoxide formation at higher membrane potential and to cell death (Ortega-Camarillo et al., 2006), as also observed in rats (Barzalobre et al., 2023). According to these observations, p53 serves in pancreatic β-cells also as mediator of apoptosis. Protein p53 is also regulated by post-translational modifications under hyperglycemia (Flores-López et al., 2024). Its connections to lipotoxicity could be recognized in experiments reporting that excessive palmitic acid inhibited β-cell proliferation by increasing the expression of p53 (Chen et al., 2014) and activated apoptosis through the p38 MAPK/p53 and NFκB pathway (Yuan et al., 2010).
Ferroptotic cell death, among other factors, typically depends on the disbalanced function of glutathione peroxidase 4 (GPX4), which reduces phospholipid and cholesterol hydroperoxides (LPO) within cell membranes in normal conditions. Despite the existence of GPX4-independent ferroptosis (Ma et al., 2022), the typical initiating step of ferroptosis lies in the inhibition of cystine uptake into cells (Dixon et al., 2012). Increased oxidative stress with iron-dependent formation of polyunsaturated FAs (PUFA) side chain hydroperoxides leads to ferroptotic cell death. The iron-containing enzyme lipoxygenase can also induce ferroptosis by enzymatic formation of lipid hydroperoxides, dependent on ACSL4-mediated lipid biosynthesis.
Ferroptosis is considered also to exist in pancreatic β-cells (Elumalai et al., 2022; Krümmel et al., 2021), due to their relative low content of glutathione antioxidant defense (Robertson, 2024). Oxidative stress can be introduced by iron accumulation upon diabetes development leading to β-cell dysfunction and cell death (Simcox and McClain, 2013). When oleoyl residues prevail in lipids, resistance to lipid-induced ferroptosis is established. Also here could be a key to some reported differences between palmitic and oleic acid effects. But only the content of ω−6-polyunsaturated FAs (ω−6- PUFA) residues leads to a threshold, when the concomitant intensity of lipid peroxidation initiates lipid-induced ferroptosis (Krümmel et al., 2022). In contrast, saturated FAs rather promote apoptosis in β-cells. Ferroptosis may be induced by ceramides. Thus, excessive palmitic acid (albumin-conjugated) indirectly induced lipid peroxidation in β-cells by increasing ceramides. Palmitic acid increased the intracellular Fe2+ content and decreased GPX4 expression via elevating ceramide content and c-Jun N-terminal kinase phosphorylation (Guo et al., 2024).
A novel diabetic lipotoxicity concept—overexpressed CD36 prevents exocytosis
Interestingly, a novel concept how disabled FA metabolism results in diabetes was introduced by Nagao et al., (2022). In obese type 2 diabetic donors, perifusion of islets exhibited impaired GSIS as well as insulin secretion response to static incubations with K+, when compared with obese nondiabetics (Nagao et al., 2020). The latter effect indicates that the defect was downstream of KATP triggering. Next, they recorded plasma membrane capacitance on single β-cells which showed the impaired ability of IGV exocytosis by ∼ 50% in obese diabetics relative to obese non-diabetics (Nagao et al., 2020). These results are in perfect correlation with the fact that the first GSIS phase is typically impaired in type 2 diabetes and with previous findings of impaired IGV exocytosis in diabetics (Gandasi et al., 2018). A decade ago, a long-term palmitate treatment was found to release Ca2+ channels from IGVs (Hoppa et al., 2009).
Moreover, an elevated expression of CD36, as a facilitator of FA uptake, was found in islets of obese diabetics (Nagao et al., 2020). This was parallel by downregulation of the IGV-exocytotic proteins SNAP25, STXBP1, and VAMP2. Thus, the impaired IGV exocytosis was caused by the CD36 overexpression, which in INS-1 cells reduced membrane capacitance and this was not consequence of the changes in inward Ca2+ currents (Nagao et al., 2020). The CD36 overexpression also downregulated the insulin receptor substrate proteins, disabled the insulin-signaling phosphatidylinositol 3-kinase/AKT pathway, and recruited FOXO1 to nucleus. It was suggested that prevention of exaggerated CD36 by the corresponding antibodies may lead to curing effects. Also, in Oikawa-Nagao mice representing a model of obesity-induced diabetes, the elevated CD36 was found in parallel with low expression of proteins of the machinery of IGV exocytosis (Nagao et al., 2014). In conclusion, not only the role of CD36 in mechanism of FA uptake and facilitation to enter β-oxidation in β-cells must be further investigated but also effects of excessive FA versus pathologically increased CD36 expression.
Lipids in circulation in prediabetes
Attempts were made to find plasma lipid biomarkers for monitoring prediabetes, specifically deterioration of pancreatic β-cells (Abbasi et al., 2016) as well as to find changes in the β-cell transcriptome (Li et al., 2018b; Solimena et al., 2018; Wang et al., 2011). Dihydroceramides were identified as potential biomarkers for type 2 diabetes (Wigger et al., 2017). Also, reciprocal relations were found for plasma TGs and islets β-cell glucokinase, KATP channel and lipid metabolic enzymes as well as proteins of the insulin receptor pathway machinery (Sánchez-Archidona et al., 2021). TGs thus reflect crosstalk between liver and β-cells, including the liver FA metabolism. Lower β-oxidation in liver, resulting in increasing plasma TGs thus promotes negative changes in β-cell transcriptome. Specifically, phosphatidyl-inositol transfer protein PITPNC1 strongly correlated with TGs. Moreover, low-density lipoproteins have a negative impact on GSIS, in contrast to high-density lipoproteins reported to maintain the β-cell mass or function (Rütti et al., 2009).
Multiple causes of IR
Despite the detailed description of IR development that would require another review, below the role of excessive NEFAs in circulation in IR development is briefly mentioned. In patients with type 2 diabetes, IR is manifested by the impaired glucose uptake into skeletal muscle and other insulin-target tissues, nonexisting insulin-mediated inhibition of endogenous glucose production, insulin-suppressed lipolysis and impaired insulin-stimulation of glycogen synthesis (Mastrototaro and Roden, 2021). A frequent cause could be the excessive food intake. The impaired glucose transport due to the insufficient recruitment of GLUT4 stems from an abnormal InsR signaling on the level of the insulin receptor tyrosine kinase (IRK) and insulin receptor substrate 1 (IRS1) (Fig. 16) (Roden and Shulman, 2019). Also, other post-translational modifications of InsR machinery contribute to IR (Karlsson et al., 2021). An inter-organ crosstalk typically strengthens IR development, being mediated by cytokines, microRNA and by extracellular vesicles (Mastrototaro and Roden, 2021). The research on these phenomena is expanding and is out scope of this review.
Earlier development of low-grade WAT inflammation leads to production of pro-inflammatory cytokines, inhibiting IRK (Hotamisligil et al., 1994). The resulting IR in WAT is then manifested by excessive lipolysis with a consequent increase of plasma NEFAs. In liver and other tissues, toxic lipids are subsequently formed, again inhibiting IRK (Lyu et al., 2020). In the liver, this results in elevated gluconeogenesis and reduced glycogen synthesis. Such IR in the liver is then manifested by almost nonexisting insulin-mediated inhibition of endogenous glucose production.
One can easily conclude that all the above described effects immediately represent glucolipotoxicity affecting pancreatic β-cells. Also, higher glucose in hepatocytes induces higher FA synthesis and de novo lipogenesis, thus increasing intrahepatic lipids (Roumans et al., 2020). Moreover, also gut microbiota could induce IR, when tight junctions are disrupted causing bacterial lipopolysaccharides (LPS) to leak into the systemic circulation (Saad et al., 2016). In contrast, SCFAs produced by the microbiome counteract inflammation and lipid accumulation in WAT (Gancheva et al., 2018). IR is also induced in liver and skeletal muscle by 1,2-DAG via PKC pathways and ceramides (Gancheva et al., 2018). In conclusion, concerning NEFAs, IR development in WAT resulting in increased lipolysis and elevated NEFAs in circulation accelerates IR development in liver and skeletal muscle.
Exaggerated FASIS
Exaggerated FASIS
An exaggerated FASIS can be considered to occur at nonstandard and pathological situations of either excessive lipid intake or as FASIS ongoing under elevated postabsorptive NEFA levels. Molecular mechanisms of FASIS in β-cells will not stop after the excessive fat meal and at excessively elevated NEFAs, but FASIS will be manifested much earlier after GSIS approaching a coincidence of GSIS and FASIS in extreme cases (Fig. 17). This must lead to the development of prediabetic pathology and formally, it can be considered as the earliest glucolipotoxicity event. Indeed, the diablo of the detail is hidden in the progressive transfer from the physiological FASIS mechanism to pathology. The early incoming chylomicrons stimulate early insulin secretion overlapping that one due to glucose and incretins, hence this overlap causes the exhaustion of pancreatic β-cells at initial stages of diabetes development.
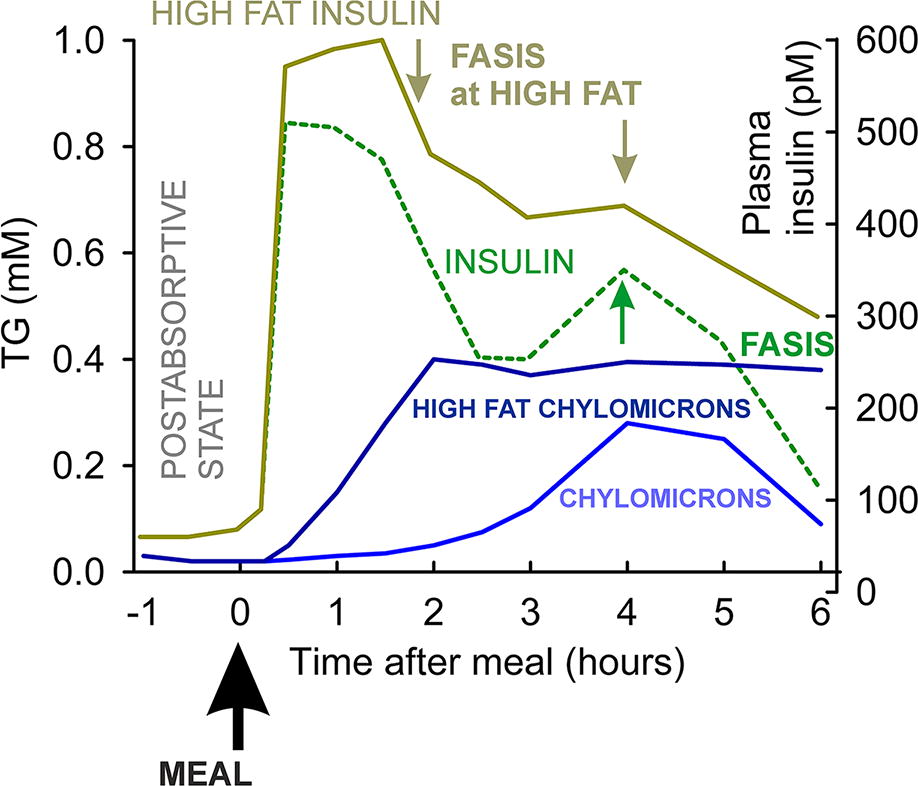
The infusion of intralipid with heparin into normal subjects increased circulating NEFAs three-fold (Paolisso et al., 1995). Tests of GSIS 6 h later showed an apparent increase of such GSIS. Why not to call a surplus portion of insulin secretion as FASIS? However, the GSIS test decreased after 24 h, which might be ascribed to lipotoxicity (Paolisso et al., 1995). Similar results were reported elsewhere (Carpentier et al., 1999; Leung et al., 2004).
One can predict two situations of exaggerated FASIS. The first one is given by the excessive fat intake. Upon a single high-fat dose, an earlier incoming and prolonged contact of chylomicrons with capillaries of pancreatic β-cells is expected, resulting in earlier and prolonged insulin secretion by FASIS mechanism (Fig. 17). A plausible GLP-1 secretion will be prolonged as well. The consequences of such an isolated single overdose should not be serious. However, when repeating frequently, such as under a regime experimentally realized as a type of a high-fat diet, the consequences may lead to a progressive initiation of prediabetic status.
The second situation stems from the already existing diabetic or metabolic syndrome status, when much higher resting NEFA levels exist in circulation in the postabsorptive state (Boden, 2008). Not only impacting β-cells but also other cell types and resident macrophages, FAs at these levels initiate toll-like receptor (TLR)4 and TLR5 pathways, NLRP3-inflammasome formation within the β-cell cytosol (see Chapter V.D. below) and the consequent IL-1β formation in β-cells. Macrophages are thus recruited in higher numbers to the islets and stimulated to acquire the M1 pro-inflammatory phenotype. This has consequences for further deterioration of the β-cell status. The complex phenomena of this outcome were also included under phenomena termed as lipotoxicity or glucolipotoxicity, the latter emphasizing participation of more frequent high glucose presence.
FASIS at high-fat diet as a prerequisite to type 2 diabetes
Overnutrition is the early cause of inducing pancreatic β-cells impairment, specifically due to lipid components. Overnutrition is considered to be the early factor in the type 2 diabetes development (Imai et al., 2020; Unger, 2003). In its progressive state, this leads to the impaired function by these insults collectively termed as (gluco)lipotoxicity and in their further progression. This includes even the loss of β-cell mass by dedifferentiation, trans-differentiation and various forms of the cell death (apoptosis, ferroptosis).
Interestingly, the levels of expressed GPR40/FFAR1 in human PIs correlated with insulin secretion and less developed diabetic status, demonstrating rather a beneficial role of FASIS in rescuing impaired GSIS (Del Guerra et al., 2010; Tomita et al., 2006).
Role of β-cell mitochondria in type 2 diabetes
The mitochondrion has an important role in physiology and pathology of β-cells as the energetic, metabolic, redox, and information hub, controlling also cell death mechanisms as apoptosis and ferroptosis. These roles in peripheral tissues are out of scope of this review, as they could comprise a self-standing review for each tissue. I will now briefly focus on pancreatic β-cell mitochondria (Ježek et al., 2022), besides their role in OXPHOS, redox signaling, and FA plus lipid metabolism.
In progressed type 2 diabetes, hyperglycemia and hyperlipidemia impair β-cells including their mitochondria, causing distorted structural features and consequent abnormal function. For example, early β-cell dysfunction is induced by defects in Complex III (Lang et al., 2023). Also impaired autophagy and specifically mitophagy may contribute to diabetic phenotype (Pearson et al., 2021). Compensation in human β-cells involved increased expression of Complex I and ATP-synthase, despite decreased ATP and ATP/ADP ratio, due to overexpressed uncoupling protein 2 (Anello et al., 2005). In Goto-Kakizaki diabetic rats, we found fragmented mitochondrial network (Dlasková et al., 2010), decreased transcription factor Nkx6.1, and consequent substantial trans-differentiation of former β-cells into δ-cells (Alán et al., 2015; Špaček et al., 2017). Increased apoptosis was found in β-cells of Zucker diabetic fatty rats (Shimabukuro et al., 1998) and after high-fat diet feeding (Sone and Kagawa, 2005). Also, the number of mitochondrial contacts with ER, assayed by in situ proximity ligation as complexes of IP3R2 with VDAC1, was significantly lower in diabetic islets and β-cells of diabetics as compared with controls (Thivolet et al., 2017). As mitofusin MFN2 resides in these contacts, its role together with MFN1 was emphasized (Georgiadou et al., 2022). These authors also documented an independent incretin-stimulated insulin release.
Typically, simulations of glucotoxicity by the increased glucose led to an increased oxidative stress and resulting decreased GSIS (Sakai et al., 2003). Increased oxidative stress does not only affect mitochondrial OXPHOS machinery constituents, including mitochondrial DNA (mtDNA), and increases its oxidation leading to mutations and crippled mtDNA-encoded subunits of respiratory chain and ATP-synthase, but the extreme oxidative stress also effectively hides the redox signaling required for insulin secretion (Plecitá et al., 2020). This essential redox signaling by NOX4 upon GSIS is normally repeatable and may control the identity of pancreatic β-cells (Swisa et al., 2017). When sunk in the overwhelmed oxidative stress, the impaired identity leads to β-cell dedifferentiation or trans-differentiation (Hunter and Stein, 2017; Moin and Butler, 2019). Also, strong antioxidants (Robertson, 2024) should not be beneficial.
The importance of mtDNA can be seen in the observed age-related mtDNA decline resulting in impaired GSIS (Nile et al., 2014; Yousefzadeh et al., 2023). Depletion of mtDNA and increased mtDNA heteroplasmy favoring mutated mtDNA cripple the important constituents of OXPHOS machinery, which inevitably leads to dysfunctional GSIS. Also, damaged mtDNA may have a role in regulating hyperglycemia during type 2 diabetes, as circulating cell-free mitochondria DNA (ccf-mtDNA) contributes to AIM2 inflammasome-mediated chronic inflammation in type 2 diabetes (Bae et al., 2019).
Also, the mitochondrial open-reading-frame of the 12 S rRNA-c (MOTS-c) peptide was reported to have significant effects on pancreatic β-cells (Bień et al., 2024; Dabravolski, Dabravolski, 2023; Kal et al., 2024). MOTS-c belongs to a group of mitochondrial-derived peptides, consisting of eight members. Thus, the MOTS-c sequence is hidden in the 12S rRNA gene (MT-RNR1), while sequences of humanin and small humanin-like peptides 1–6 are portions of the 16S rRNA (MT-RNR2) gene. Because the antiapoptotic, anti-inflammatory, and cardioprotective effects of these peptides have been revealed, they may act as sensitive metabolic sensors, reporting defects due to mtDNA mutations. Their detailed functions in diabetes development have to be established.
Immune system involvement
Attack of immune cells
Low-grade chronic inflammation is caused by the innate immune system upon intake of excessive nutrients and metabolic stress. These events induce IR, which is a hallmark of type 2 diabetes. The process is termed as metabolic inflammation (metaflammation) (Ralston et al., 2017). It may aid also in the deterioration of β-cells and their progressive damage (Patel et al., 2017), leading to a loss of β-cell mass (Kugelberg, 2013). The number of macrophages increases in islets during the progression of type 2 diabetes. Since they produce proinflammatory cytokines, islet inflammation is initiated (Eguchi and Nagai, 2017). Islet macrophages also produce chemokines, which recruit more innate immune cells. Macrophages activated into their M1 type form superoxide and HOCl or HSCN and thus amplify the already existing oxidative stress in β-cell. This is supported by observations that islets of type 2 diabetic human donors exhibit elevated transcription of cytokine and chemokine genes (Taneera et al., 2012). Laser-micro-dissected β-cells from type 2 diabetic donors have elevated expression of cytokines IL-1β, IL-8, and IL-11, and chemokines CCL2, CCL11, and CCL13, but downregulated IL-1α cytokine (Marselli et al., 2010). It is necessary to note that InsR signaling in immune cells contributes to the immune system responses (Makhijani et al., 2023).
Formation of nucleotide-binding oligomerization domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome complex is the typical response of cells of the innate immune system as well as pancreatic β-cells. NLRP3 belongs to cytosolic multiprotein oligomer pattern recognition receptors involved in the recognition of pathogen-associated molecular patterns and danger-associated molecular patterns produced by infected cells. The complex size of NLRP3 inflammasome is up to 1 µm, consisting of core of polymerized ASC proteins (the adaptor apoptosis-associated speck-like protein containing a caspase-recruitment domain) and upper layer of NLRP3 (Glück et al., 2023).
The assembly of the NLRP3 inflammasome in pancreatic β-cells is induced by TXNIP and by oxidative stress of various origin (Zhou et al., 2010), including the H2O2 release by NOX4 (Holendová et al., 2024). The mature complex promotes caspase-1 activation required for the secretion of proinflammatory cytokines IL-1 and IL-18 in response to microbial infection and cellular damage (Wen et al., 2013; Xu et al., 2024). The activated caspase-1 cleaves pro-IL-lβ to mature IL-lβ. This pathway is regarded as a central sensor and mediator in the pathogenesis of metabolic diseases and both type 2 and type 1 diabetes, involving IL-lβ-mediated destruction of pancreatic β-cells (Ralston et al., 2017; Youm et al., 2011).
PIs of NLRP3 KO mice and ASC KO mice exposed to hypoxia or alloxan (inducing their damage) preserved their function and unlike wild-type islets, the ASC KO islets were protected against cell death by these respective insults (Sokolova et al., 2018). ASC KO mice were also protected against the oxidative stress-induced diabetic phenotype caused by repetitive alloxan dosage. The protection was due to diminished macrophage infiltration and activation. Within the diabetic islets, phagocytosis of apoptotic β-cells leads to NLRP3 inflammasome activation due to lysosomal permeabilization by β-cell constituents and ROS generation in macrophages, leading to their cytokine secretion (Ward et al., 2018).
NLRP3 inflammasome in pancreatic β-cells—crosstalk of β-cells and immune cells
Interestingly, a typical pro-inflammatory cytokine IL-lβ in lower concentrations exhibits also physiological roles in PIs upon GSIS, contributing to postprandial insulin secretion (Dror et al., 2017). The source of acute IL-lβ upon GSIS originates from peritoneal macrophages, when their glucose uptake is stimulated by just released insulin. Such macrophage glucose consumption contributes to normalization of glycemia. It may activate an autocrine IL-lβ loop in β-cells. In such a loop, NLRP1 inflammasome could also participate (Ghiasi et al., 2018).
Since pancreatic β-cells exhibit the highest expression of IL-1 receptor of any tissue, higher IL-lβ levels cause islet inflammation (Ježek et al., 2019). Communication via inflammatory cytokines and chemokines between the M1-like macrophages and β-cells provide a vicious cycle progressively amplifying PI inflammation leading to β-cell death (in type 1 diabetes) or at least to excessive macrophage recruitment in type 2 diabetes. As mentioned above, the NLRP3 inflammasome and subsequent IL-lβ formation is induced in β-cells by glucolipotoxicity (typically by FAs at chronically high glycemia via TLR2 and 4 receptors), islet amyloid peptide (IAP), and other insults via an upstream initiation by TXNIP. High glucose promotes the interaction of TXNIP with NLRP3. This involves posttranslational modification of TXNIP by O-linked β-N-acetylglucosamine (Filhoulaud et al., 2019).
Resulting IL-1β exerts an autocrine function by activating its receptor IL-1R, recruiting MYD88 and accelerating its own expression, while the activation of MYD88/NF-κB pathway stimulates the expression of CCL2, CCL3, and CXCL8 chemokines. The chemokines recruit macrophages, which then produce additional IL-1β, TNFα, and INFγ.
Hypoxia induces also in β-cells an inflammatory response and cell death via oxidative stress induced TXNIP-NLRP3 inflammasome formation (Chen et al., 2018). Interaction between NLRP3 and ASC leading to NLRP3 inflammasome formation was directly promoted by oligomers but not mature amyloid fibrils of IAP/amylin (Morikawa et al., 2018). This was prevented by serum C4b-binding protein (C4BP) (Kulak et al., 2017). Interestingly, mice with β-cell-specific CB1R deletion exhibited reduced activation of the NLRP3 inflammasome in β-cells (González-Mariscal et al., 2018). Similarly, long-term glutamate activation of NMDA receptors under high-glucose conditions may contribute to diabetes etiology accelerating β-cell dysfunction dependent on NLRP3 inflammasome (Huang et al., 2017).
FAs versus pancreatic beta cell mass
FAs interact with TLRs activating the NF-kB-dependent production of inflammatory cytokines (Shi et al., 2006). This is concomitant to the NLRP3 inflammasome activation and consequent pro-IL-1β and pro-IL-18 maturation. When exaggerated, it contributes to type 2 diabetes development (Vandanmagsar et al., 2011; Wen et al., 2011). The FA-induced NF-κB plus macrophage migration inhibitory factor (MIF) pathways caused β-cell dysfunction and apoptosis which was attenuated by fenofibrate (Zheng et al., 2017). Excess of saturated FAs (e.g., palmitate) induce chemokine (CXCL1, CCL3) and cytokine expression (IL-6, and IL-8) within islets in vitro (Cnop et al., 2014; Igoillo-Esteve et al., 2010). Palmitate-induced TLR4 signaling in PIs at high glucose elevates expression of the S100 calcium-binding protein A8 (S100A8) (Inoue et al., 2018). In co-cultures with macrophages, S100A8 promoted islet–macrophage interactions, leading to β-cell apoptosis. Palmitate via NF-κB plus MIF pathways was able to induce Fetuin-A secretion. This 2-HS-glycoprotein, normally excreted by the liver, sets “an inflammatory environment,” leading to β-cell dysfunction (Mukhuty et al., 2017).
Future Perspectives
Overview of FASIS mechanism
The basic FASIS mechanism, its “fuel” or “metabolic” branch, is based on the KATP triggering, since its closure requires H2O2, similar to glucose triggering (GSIS), where NOX4 provides H2O2 (Plecitá et al., 2020). The H2O2 release from the islets has been documented for both GSIS and FASIS and ascribed to the elevated NOX4 function (Plecitá et al., 2020) and elevated mitochondrial superoxide formation upon FA β-oxidation (Jabůrek et al., 2024), respectively. The superimposed non-fuel or “receptoric” FASIS branch stems from the downstream GPR40 pathway, which responds mainly to mitochondrial FAs cleaved by iPLA2γ. GPR40 metabotropic receptors do not respond to fasting maximum levels of NEFAs, that is, those bound to albumin. The GPR40 pathway is independent as demonstrated with high-affinity artificial GPR40 agonists, which are not metabolizable, so they act at low ATP (low OXPHOS) (Jabůrek et al., 2024) As iPLA2γ/PNPLA8 is redox activated, incoming FAs from chylomicrons activate it via FA β-oxidation-released H2O2 at the same time as they close KATP. That is why GPR40 is not activated before by NEFAs. Future research should investigate these phenomena in more detail, specifically to derive their time course in vivo.
Future directions of research
Numerous directions of the near future research should uncover details of synergy of all FA transporters and enzymes in pancreatic β-cells, that is, all complex phenomena of FA uptake into β-cells after their cleavage from chylomicrons. It should be investigated how this FA uptake is changed when either maximum NEFAs, that is, FAs bound to albumin, or postprandial minimum NEFA without chylomicrons circulate in β-cell capillaries. Further intracellular phenomena in pancreatic β-cells must be investigated, such as roles of LDs in these FA/lipid incoming events. Situations must be clarified and understood in which LDs act as FA and lipid sink versus those situations when LDs are FA and MAG donors. Moreover, details of interaction of LDs with mitochondria and ER should be uncovered. All these details will help to understand how LDs could prevent otherwise toxic effects of FAs and other lipids.
It should be investigated why maximum levels of NEFAs carried by albumin in the postabsorptive state do not activate GPR40 pathways. The question whether the carnitine carrier exchanges LC-carnitines for SC-carnitines (or MC-carnitines) must be definitively solved, as well as the role of carnitine-O-acetyltransferase in β-cells and specifically in a possible crosstalk between branched-chain amino acid (keto acid) metabolism and FA β-oxidation.
Now, let’s imagine that all these details are already known, not only for pancreatic β-cells but also for adipocytes, hepatocytes, myocytes, enterocytes, and each type of immune cells. Will it then be possible to judge the timeline of postprandial events, phenomena leading to prediabetes and type 2 diabetes, and conclude precautions? Will it be feasible to design optimum drugs to prevent diabetes development?
Hence, investigations on FA and lipid metabolism in the inter-organ crosstalk should follow, comparing healthy states with the impaired states at prediabetes and mild versus progressive type 2 diabetes. As this review briefly discussed, FAs should be considered whenever any molecular mechanisms are investigated. New links between the sole GSIS and FA metabolism should still be uncovered.
Moreover, paracrine and inter-organ crosstalk mediated by the extracellular vesicles such as exomeres, exosomes, ectosomes/microvesicles, migrasomes, and exopheres should be studied, whether it stands for yet uncovered signals between pancreatic β-cells and peripheral tissues; reciprocal signals between peripheral tissues and pancreatic β-cells and in any mutual signals with immune cells, namely concerning resident macrophages in PIs or peripheral tissues. For example, extracellular vesicles carrying cargo such as microRNAs are now well documented. The immune system interaction in islets in healthy states versus prediabetic and progressive diabetic states should uncover crosstalk with a low-grade inflammation in peripheral tissues and its consequences.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This publication was supported by the Grant Agency of the Czech Republic (No. 24-10132S and No. 24-10125S) and by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Program EXCELES, ID Project No. LX22NPO5104)—funded by the European Union—Next Generation EU.