
Abstract
Aims:
NADPH oxidase (NOX)-derived reactive oxygen species (ROS) are critical for platelet activation and thrombus formation. We hypothesized that inhibiting NOX-mediated ROS production with a pan-NOX inhibitor, APX-115, could effectively suppress platelet activation and thrombus formation, potentially serving as a novel antiplatelet therapeutic. This study aimed to explore the effects of APX-115 on human platelet functional responses and ROS-mediated signaling pathways.
Results:
APX-115 inhibited intracellular and extracellular ROS production in collagen-stimulated platelets, suppressing aggregation, P-selectin exposure, and ATP release. By preserving protein tyrosine phosphatase activity, APX-115 reduced tyrosine phosphorylation-dependent pathways inhibition, including spleen tyrosine kinase, LAT, Vav1, Bruton’s tyrosine kinase, and phospholipase Cγ2, leading to decreased PKC activation and calcium mobilization. APX-115 also suppressed collagen-induced integrin αIIbβ3 activation, accompanied by elevated cGMP and vasodilator-stimulated phosphoprotein phosphorylation levels. In addition, APX-115 reduced p38 MAPK and ERK5 activation, leading to diminished phospholipase A2 phosphorylation, thromboxane production, and the exposure of procoagulant phosphatidylserine. These inhibitory effects extended to thrombus development caused by platelet adherence under shear and arterial thrombosis without prolonging bleeding time in murine models.
Innovation:
This study is the first to demonstrate that APX-115 inhibits NOX-mediated ROS production, platelet activation, and thrombus formation. By uncovering its effects on collagen receptor glycoprotein VI-mediated pathways, the work highlights the promise of APX-115 as an antiplatelet and antithrombotic agent.
Conclusion:
Our findings highlight the therapeutic potential of APX-115 in treating thrombotic and cardiovascular disorders by targeting NOX-mediated ROS production to mitigate platelet hyperreactivity and thrombus formation. Antioxid. Redox Signal. 43, 288–307.
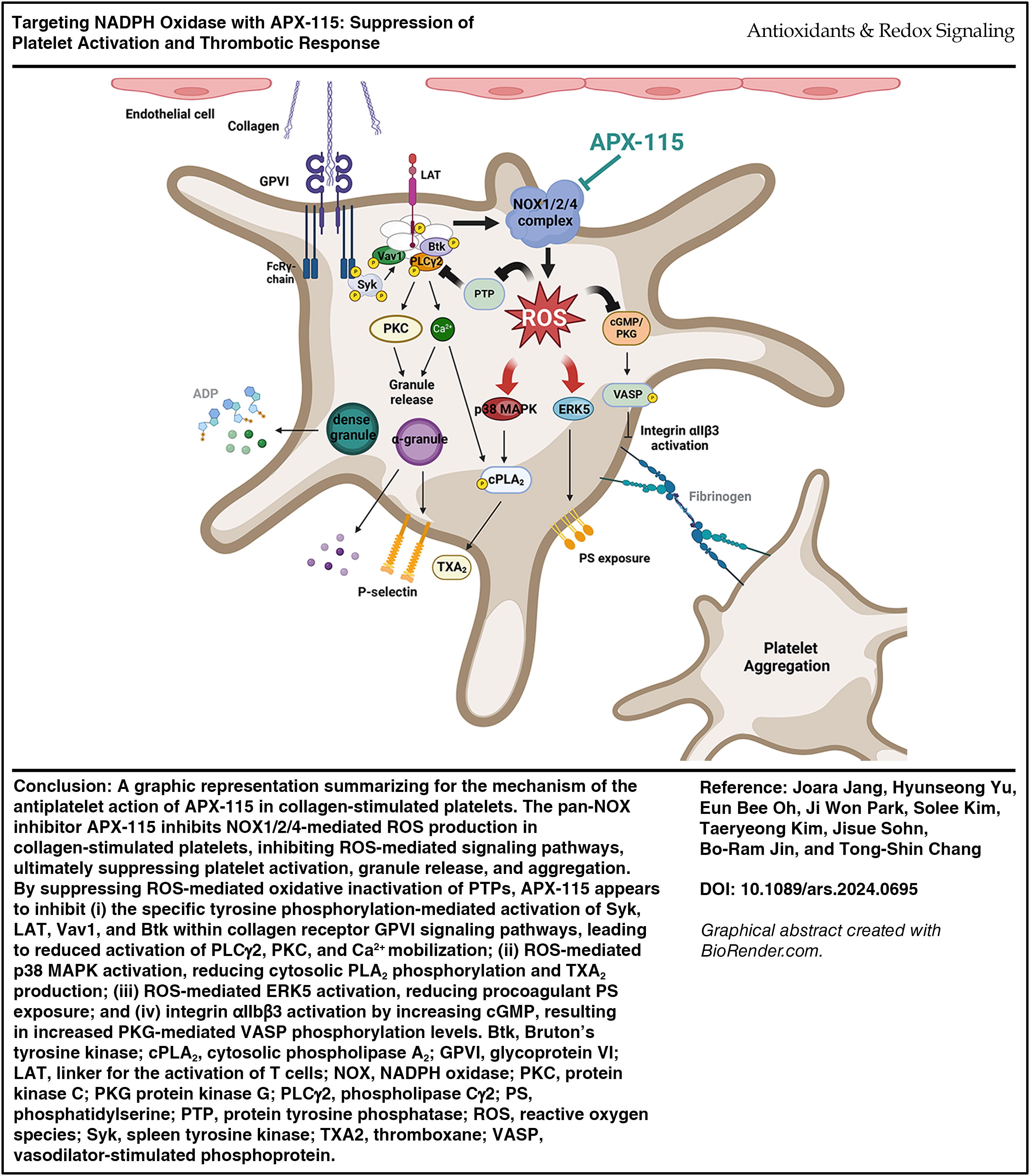
Introduction
Platelets are crucial for the maintenance of normal hemostasis and are also involved in the development of pathological conditions, including atherosclerosis, stroke, and inflammation (Nieswandt et al., 2011; van der Meijden and Heemskerk, 2019). At sites of vascular injury, extracellular matrix components like collagen and von Willebrand factor initiate platelet activation through interactions with glycoprotein (GP) VI and GPIb receptors. This process leads to further platelet recruitment, granule release, and thrombin generation, culminating in a hemostatic plug. Conversely, excessive platelet activation under pathological conditions results in thrombus formation and arterial occlusion, underscoring the need for precise regulation of platelet activity to prevent thrombosis.
NADPH oxidase (NOX) enzymes are significant generators of reactive oxygen species (ROS) in various contexts and contribute to numerous physiological and pathological processes such as atherosclerosis, inflammation, and immunity (Lambeth, 2007). ROS generated by NOX enzymes play a critical role in platelet activation and thrombus formation. NOX-derived ROS modulate platelet signaling pathways, amplifying activation signals and promoting aggregation (Begonja et al., 2005; Carnevale et al., 2014b; Chlopicki et al., 2004; Delaney et al., 2016; Vara et al., 2019, 2021; Walsh et al., 2014). NOX not only plays a role in healthy platelet signaling but also appears to make platelets more sensitive and prone to the excessive response observed in thrombotic diseases (Carnevale et al., 2014a; Delaney et al., 2016; Magwenzi et al., 2015; Vara et al., 2019, 2021).
Innovation
As shown in Figure 1, our findings show that APX-115 effectively blocks the production of ROS triggered by collagen, which in turn prevents the activation of signaling pathways mediated by ROS, which ultimately results in a reduction of collagen-induced platelet aggregation and platelet-dependent thrombosis. Based on these observations, it appears that APX-115 could have potential benefits in preventing hyperreactive platelet-mediated thrombotic disease.
The NOX family includes seven members: NOX1–5 and dual oxidases (Duox1 and Duox2). Human platelets express NOX1, NOX2, and NOX4 (Delaney et al., 2016). The study of mice with triple NOX deletion indicates that NOX1 and NOX2 have prominent roles in platelet function (Vara et al., 2021). NOX1 and NOX2 have an impact on platelet activation and thrombus formation (Carnevale et al., 2014b; Delaney et al., 2016; Vara et al., 2019, 2021). NOX1 primarily contributes to collagen receptor GPVI-stimulated platelet ROS production, aggregation, and granule release, whereas NOX2 is critical for thrombin-induced platelet activation but less so for collagen responses (Vara et al., 2019, 2021; Walsh et al., 2014). In contrast, NOX2 is essential in platelet signaling for both GPVI and G protein-coupled receptor (GPCR) of thrombin or thromboxane A2 (TXA2), while NOX1 has a specific role in GPCR-dependent platelet activation (Delaney et al., 2016). NOX4, in contrast, has a minimal role, with only slight effects on superoxide generation in response to collagen (Vara et al., 2021).
The specific mechanisms by which individual NOX isoforms affect platelet activation are still debated. Nevertheless, it is widely recognized that NOX is essential for the activation of platelets. Therefore, exploring NOX as a therapeutic target holds potential for the development of new antiplatelet and antithrombotic drugs.
APX-115, a pan-NOX inhibitor, has shown promise in reducing ROS production and alleviating oxidative stress in preclinical models (Cha et al., 2017; Joo et al., 2016; Lee et al., 2020). However, its potential to modulate platelet function and thrombus formation has not been explored. This study aims to evaluate the effects of APX-115 on NOX-mediated ROS production, platelet activation, and thrombus formation, with a focus on elucidating its underlying mechanisms and therapeutic potential.
Results
APX-115 inhibits collagen-induced aggregation and ROS production in platelets
The concentration of APX-115 was chosen based on prior studies and experimental validation, demonstrating its efficacy in inhibiting collagen-stimulated platelet aggregation with an IC50 of ∼13.5 μM (Fig. 1A). This aligns with previous findings of its optimal inhibitory effects on ROS generation in other cell types (Han et al., 2022; Joo et al., 2016). These results validate the concentrations used in this study for assessing its effects on platelet aggregation. The concentration of APX-115 required to inhibit platelet activation was higher than the reported IC50 values for NOX enzymes. This discrepancy is likely due to the differences between cell-free NOX activity assays and the complex environment within platelets, where intracellular drug bioavailability and secondary signaling pathways may modulate the effective inhibitory concentration.
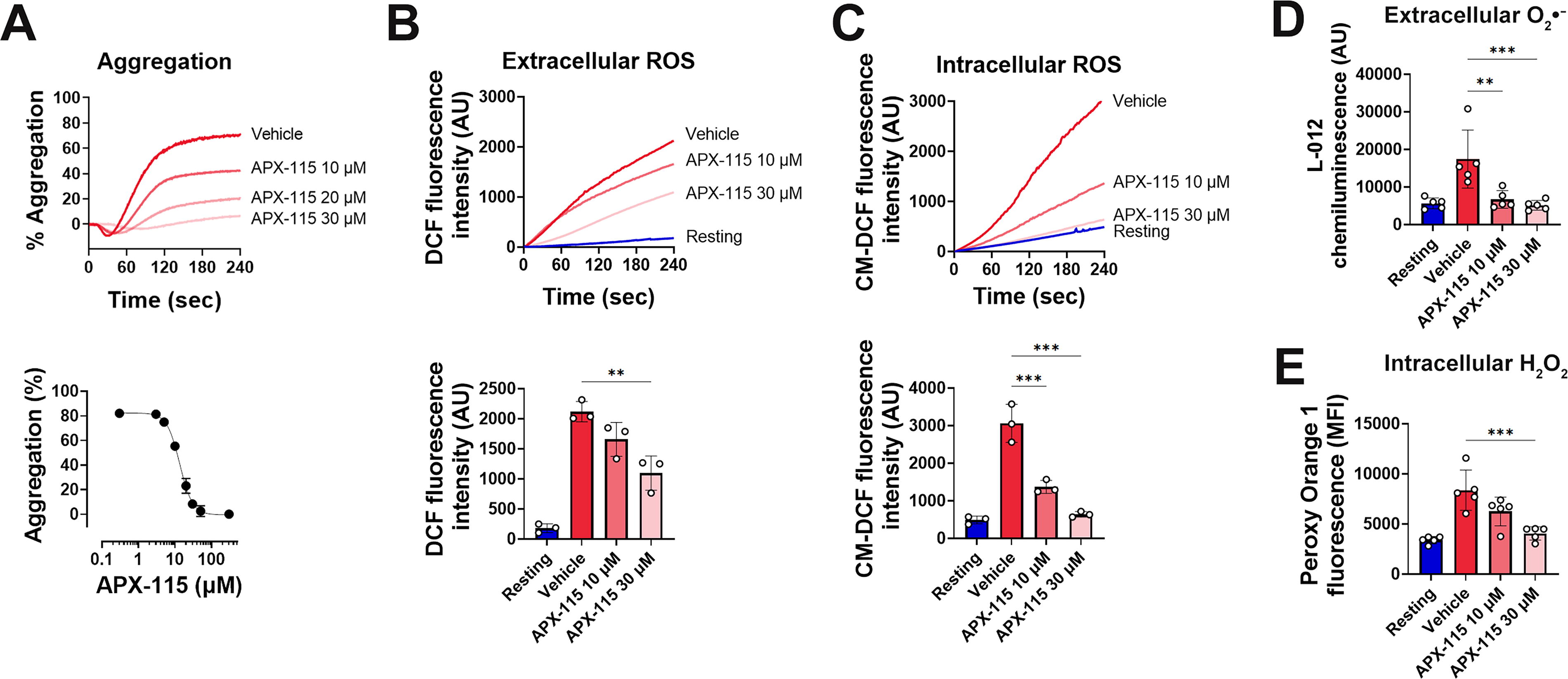
Using the redox-sensitive probe 2′,7′-dichlorodihydrofluorescein (DCFH2) (Reiniers et al., 2017), collagen significantly increased extracellular ROS, a response effectively inhibited by APX-115 (Fig. 1B). Similarly, intracellular ROS levels were reduced by APX-115 in a concentration-dependent manner, as shown by 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) measurements (Fig. 1C). Our findings imply that APX-115 can have an antiplatelet impact by preventing the production of NOX-mediated ROS.
NOX complex-generated superoxide spontaneously dismutates into H2O2 in the extracellular environment (Holmstrom and Finkel, 2014). The negatively charged superoxide anion has limited membrane permeability, but neutral H2O2 easily diffuses into the cytoplasm, increasing intracellular ROS. To overcome the limitations of H2DCFDA and DCFH2 (Rezende et al., 2018; Sies et al., 2022), L-012, a superoxide-specific chemiluminescent probe for superoxide detection and Peroxy Orange 1, for intracellular H2O2 detection (Dickinson and Chang, 2011) was employed. APX-115 significantly inhibited extracellular superoxide and intracellular H2O2 formation induced by collagen (Fig. 1D, E), underscoring its role in suppressing NOX-mediated ROS.
Given that NOX1 and NOX2 deficiency inhibited thrombin-induced aggregation and ROS production in mouse platelets (Delaney et al., 2016; Vara et al., 2019, 2021), we examined the effect of APX-115 on thrombin-induced human platelet aggregation and ROS generation. APX-115 also effectively inhibited thrombin-induced platelet aggregation (Supplementary Fig. S1A) and the increase in ROS, superoxide, and H2O2 (Supplementary Figs. S1B–S1E). These results suggest that APX-115 exhibits antiplatelet effects by targeting NOX-mediated ROS production, critical for GPVI-mediated collagen receptor signaling and GPCR-mediated platelet activation through thrombin.
APX-115 showed broader inhibition compared with isoform-specific inhibitors ML171 (NOX1) and GSK2795039 (NOX2) (Supplementary Fig. S2A, S2B). While ML171 strongly suppressed collagen-induced aggregation, GSK2795039 was less effective. APX-115 demonstrated superior inhibition in both collagen- and thrombin-induced aggregation, highlighting its pan-NOX inhibitory activity and broader therapeutic potential.
A study reported that protein disulfide isomerase-A1 (PDIA1) regulates NOX1-dependent ROS production in platelets (Przyborowski et al., 2022), where PDIA1 inhibition suppresses NOX1 activation, reducing ROS production and downstream signaling. To test if APX-115 affects PDIA1, we evaluated its activity in the presence of APX-115 or isoquercetin, a known PDIA1 inhibitor, using an insulin transhydrogenase assay. While isoquercetin inhibited PDIA1 activity in a dose-dependent manner, APX-115, even at 30 μM, showed no significant effect, indicating that its impact on platelet function is independent of PDIA1 (Supplementary Fig. S3).
A previous report has suggested that extracellular vesicles (EVs) from platelets express NOX1, produce superoxide, and influence intercellular signaling, contributing to extracellular ROS production and platelet activity (Gaspar et al., 2021). To assess whether APX-115 reduces extracellular ROS by inhibiting EV production, we analyzed EV concentration and size. Collagen stimulation significantly increased EV concentrations (Supplementary Fig. S4A), which APX-115 at 30 μM effectively reduced. EV size remained consistent across groups (Supplementary Fig. S4B), indicating that APX-115 decreases EV quantity without altering size. This suggests that the reduction of extracellular ROS by APX-115 is partly due to its inhibition of EV production, highlighting its role in targeting EV-mediated signaling pathways.
To determine if APX-115 inhibits platelet activation by altering the surface expression of key membrane proteins, we measured GPVI, protease-activated receptor 1 (PAR-1), and integrin αIIbβ3 levels using flow cytometry (Supplementary Fig. S5). Fluorescent probe-conjugated antibodies specific to GPVI, PAR-1 (the primary thrombin receptor), and αIIb (a subunit of the integrin complex) were used to analyze their surface expression. APX-115 treatment did not significantly affect the surface expression of these proteins, suggesting that its antiplatelet effects are not mediated through receptor downregulation.
The distinct roles of NOX2 and NOX1 in platelet aggregation remain debated (Delaney et al., 2016; Vara et al., 2021), but platelets from patients with chronic granulomatous disease (CGD) with NOX2 deficiency show impaired ROS production in response to collagen and thrombin, highlighting the importance of NOX2 (Pignatelli et al., 2004). To assess the effects of APX-115 on NOX2, we examined its impact on gp91phox and p47phox, the membrane-bound and cytosolic subunits of the NOX2 complex. Western blot analysis showed no significant changes in their protein levels following APX-115 treatment compared with collagen-stimulated platelets (Supplementary Fig. S6A, S6B), suggesting that APX-115 inhibits ROS formation without affecting protein expression.
APX-115 attenuates collagen-stimulated GPVI signaling in platelets
Collagen-activated GPVI triggers the phosphorylation of linker for the activation of T cells (LAT) tyrosine residues by spleen tyrosine kinase (Syk), which in turn leads to the formation of LAT signaling complexes with important constituents like Syk, Vav1, Bruton’s tyrosine kinase (Btk), and phospholipase Cγ2 (PLCγ2) (Gibbins, 2004; Poole et al., 1997; Watson et al., 2005). Because protein tyrosine phosphorylation cascades are the primary characteristic of GPVI-stimulated signaling events (Gibbins, 2004; Poole et al., 1997; Watson et al., 2005), ROS-mediated oxidation and inactivation of protein tyrosine phosphatases (PTPs) may augment GPVI signaling pathways in collagen-stimulated platelets (Jang et al., 2014; Karisch et al., 2011; Salmeen and Barford, 2005). We then investigated the impact of APX-115 on protein tyrosine phosphorylation in light of the fact that it prevented ROS production in collagen-stimulated platelets. The total tyrosine phosphorylation levels induced by collagen exhibit a substantial increase in comparison with the baseline values (Fig. 2A). Protein tyrosine phosphorylation was suppressed when platelets were pretreated with APX-115 at 30 μM prior to collagen stimulation.
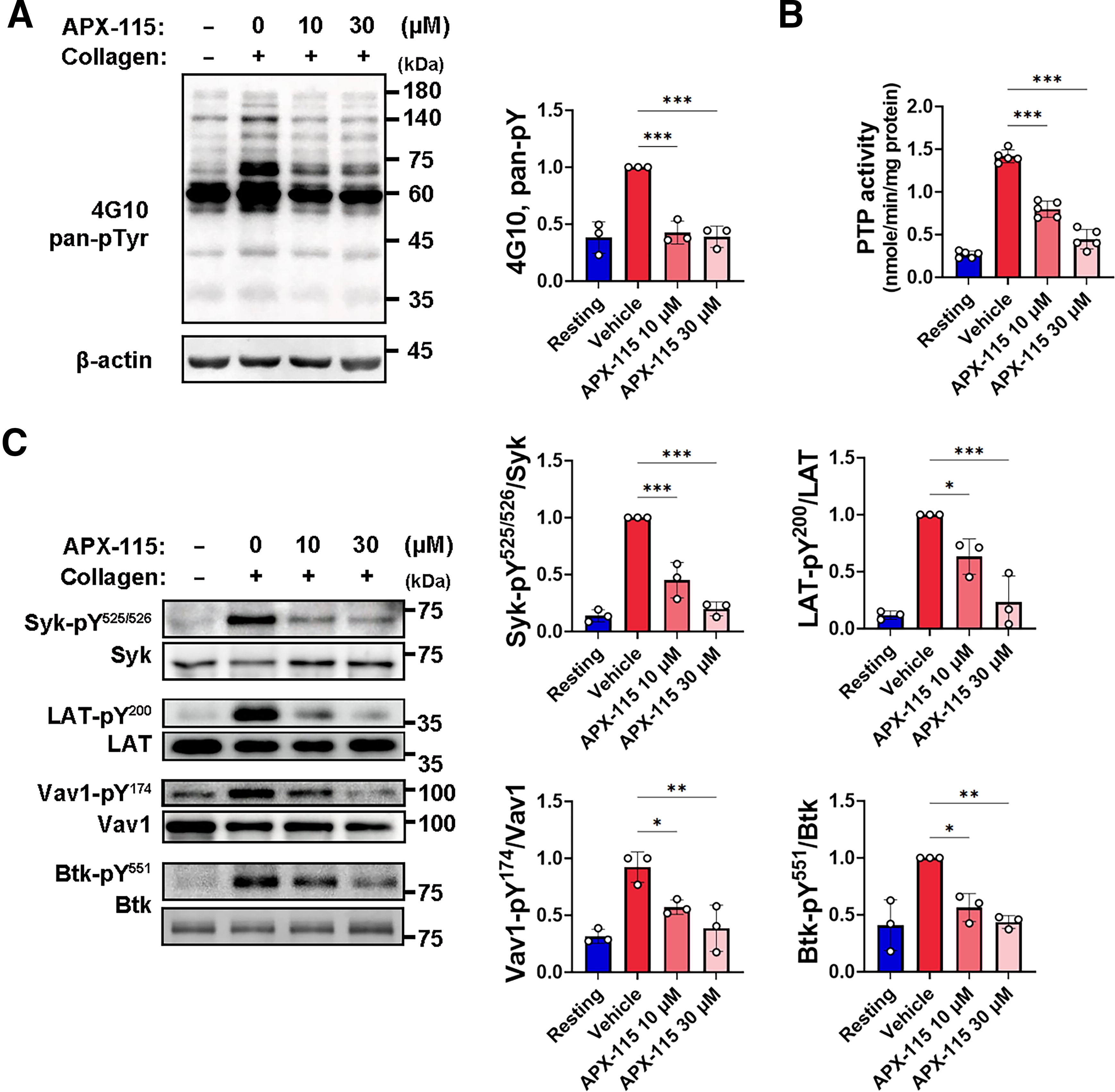
The ability of APX-115 to reduce ROS production and protein tyrosine phosphorylation in collagen-stimulated platelets prompted an investigation into its effect on preventing oxidative inactivation of PTPs. Platelet lysates were treated with cysteine-alkylating agents to stabilize reactive cysteines, followed by dithiothreitol to restore reversibly oxidized cysteines to their active form. PTP activity, measured using a tyrosine phosphopeptide substrate, served as an indicator of oxidation levels. Collagen stimulation increased PTP cysteine oxidation (Fig. 2B), but APX-115 significantly inhibited this effect, indicating its role in protecting PTPs from oxidative damage by blocking NOX-mediated ROS production.
Assembling Syk, Vav1, Btk, and PLCγ2 in the LAT signaling complexes is mainly mediated by the binding module between phosphorylated tyrosine residues and the SH2 domain, ultimately activating PLCγ2 events (Gibbins, 2004; Poole et al., 1997; Watson et al., 2005). We assessed the tyrosine phosphorylation-based activation of Syk, LAT, Vav1, and Btk in order to investigate whether APX-115 controls the activation of the GPVI signaling cascade. Western blot analysis using phospho-specific antibodies demonstrated that collagen stimulation increased the phosphorylation of Tyr525/Tyr526 on Syk, Tyr200 on LAT, Tyr174 on Vav1, and Tyr551 on Btk (Fig. 2C). The activation of every signaling protein depends on its tyrosine phosphorylation, which can also be used to measure the activity of individual molecules (Fujii et al., 1994; Furlong et al., 1997; Pearce et al., 2002; Rawlings et al., 1996). Treatment with APX-115 specifically reduced the activation of Syk, LAT, Vav1, and Btk in collagen-stimulated platelets through reducing tyrosine phosphorylation. These findings imply that APX-115 may protect PTPs from oxidative inactivation by preventing NOX-mediated ROS production, which in turn inhibits the activation of Syk, LAT, Vav1, and Btk.
APX-115 suppresses collagen-induced activation of PLCγ2/PKC/Ca2+ pathway in platelets
Phosphorylating Tyr753 on the downstream target PLCγ2 in response to collagen stimulation is related to tyrosine phosphorylation-based activation of Vav1 and Btk, hence enhancing its activation (Pearce et al., 2002; Quek et al., 1998; Suzuki-Inoue et al., 2004). The upstream critical molecules were blocked by APX-115, which allowed us to examine the phosphorylation of PLCγ2 at Tyr753 in collagen-stimulated platelets. APX-115 also prevented PLCγ2 phosphorylation at Tyr753 that was caused by collagen (Fig. 3A). Together, our findings show that APX-115 prevented the activation of Syk, LAT, Vav1, and Btk, which in turn led to a reduction in PLCγ2 activity via blocking NOX-mediated ROS generation. When PLCγ2 generates diacylglycerol and inositol-1,4,5-trisphosphate (IP3) in collagen-stimulated platelets, PKC is activated and the cytosolic Ca2+ levels are increased due to the Ca2+ mobilization from intracellular stores (van der Meijden and Heemskerk, 2019; Watson et al., 2005). To assess whether APX-115 inhibits PKC activation, we analyzed the phosphorylation of classical PKC target motifs using Western blotting (Nishikawa et al., 1997). Collagen stimulation produced distinct multiband patterns of PKC-phosphorylated proteins (Fig. 3B), which were significantly reduced by APX-115 treatment.
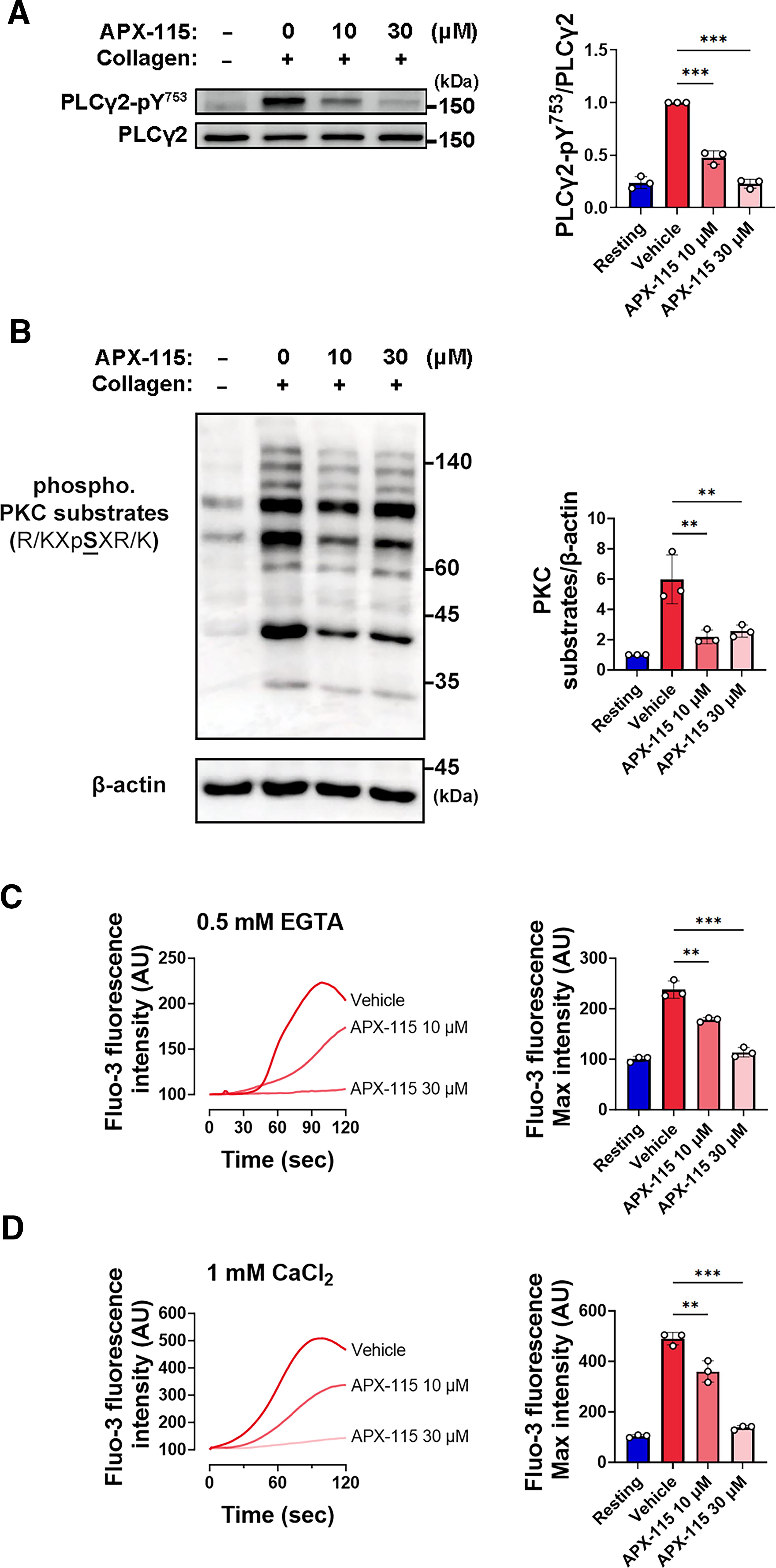
This suppression of PKC activity has broader implications, as PKC mediates the phosphorylation of p47phox at Ser304, a key step in NOX2 activation (Fontayne et al., 2002). APX-115 significantly reduced p47phox phosphorylation (Supplementary Fig. S7), likely by interfering with the PLCγ2/diacylglycerol/PKC signaling pathway (Fig. 3A, B) and impairing NOX2 complex assembly. Additionally, consistent with prior findings (Joo et al., 2016), APX-115 appears to target the NADPH-binding domain of gp91phox, inhibiting electron transfer and ROS production. This dual action on NOX2 inhibition and downstream signaling cascades highlights the antiplatelet effects of APX-115, including reduced membrane translocation of cytosolic NOX components. Future studies using platelet subfractions may provide further insights into the spatial dynamics of NOX assembly and the modulatory role of APX-115.
Next, we used Fluo-3-AM as a probe to measure the levels of cytosolic Ca2+. Because the release of Ca2+ from internal stores is reflected in the rise in cytosolic Ca2+ levels seen in the absence of external Ca2+, cytosolic Ca2+ levels were assessed in the presence of 0.5 mM EGTA (Fig. 3C). APX-115 effectively inhibited the collagen-induced mobilization of Ca2+ from internal stores. Furthermore, APX-115 also successfully prevented the collagen-induced rise in cytosolic Ca2+ even in the presence of external Ca2+ (Fig. 3D). These findings suggest that the antiplatelet action of APX-115 may be influenced by its regulation of Ca2+ mobilization and PLCγ2 activation.
APX-115 inhibits collagen-induced platelet granule release
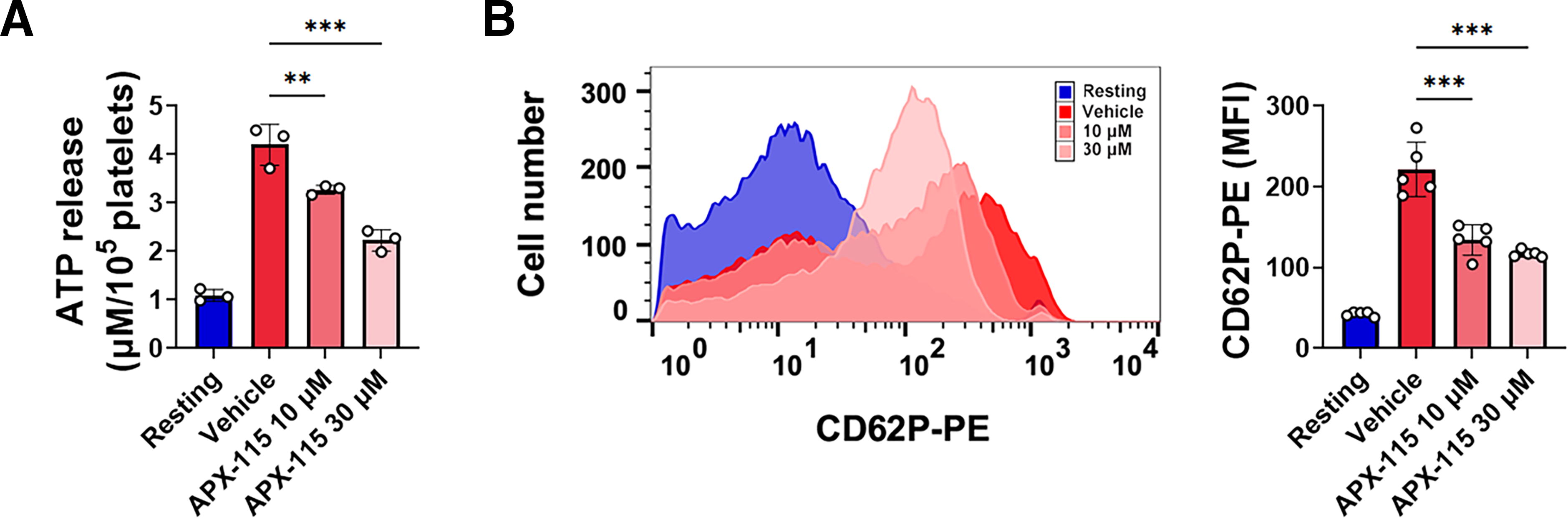
Following activation, the release of active substances from dense and α-granules contributes significantly to the enhancement of platelet activation (van der Meijden and Heemskerk, 2019). Granule release in collagen-stimulated platelets is facilitated by PLCγ2-mediated PKC activation and an increase in cytosolic Ca2+ (Durrant et al., 2017; van der Meijden and Heemskerk, 2019). We ascertained the impact of APX-115 on granule release induced by collagen because it suppressed PLCγ2. By analyzing ATP release, we evaluated the platelet dense-granule release. APX-115 significantly reduced the amount of ATP released from human platelets stimulated with collagen (Fig. 4A). P-selectin serves as an adhesion molecule facilitating platelet-leukocyte contact on the surface of activated platelets that have undergone degranulation of α-granules (Frenette et al., 2000). Flow cytometry using PE-labeled anti-P-selectin antibody (CD62P-PE) demonstrated that collagen increased platelet surface expression of P-selectin, which was potently inhibited by APX-115 (Fig. 4B).
APX-115 attenuates collagen-induced activation of p38 MAPK/cytosolic phospholipase A2/TXA2 production signaling in platelets
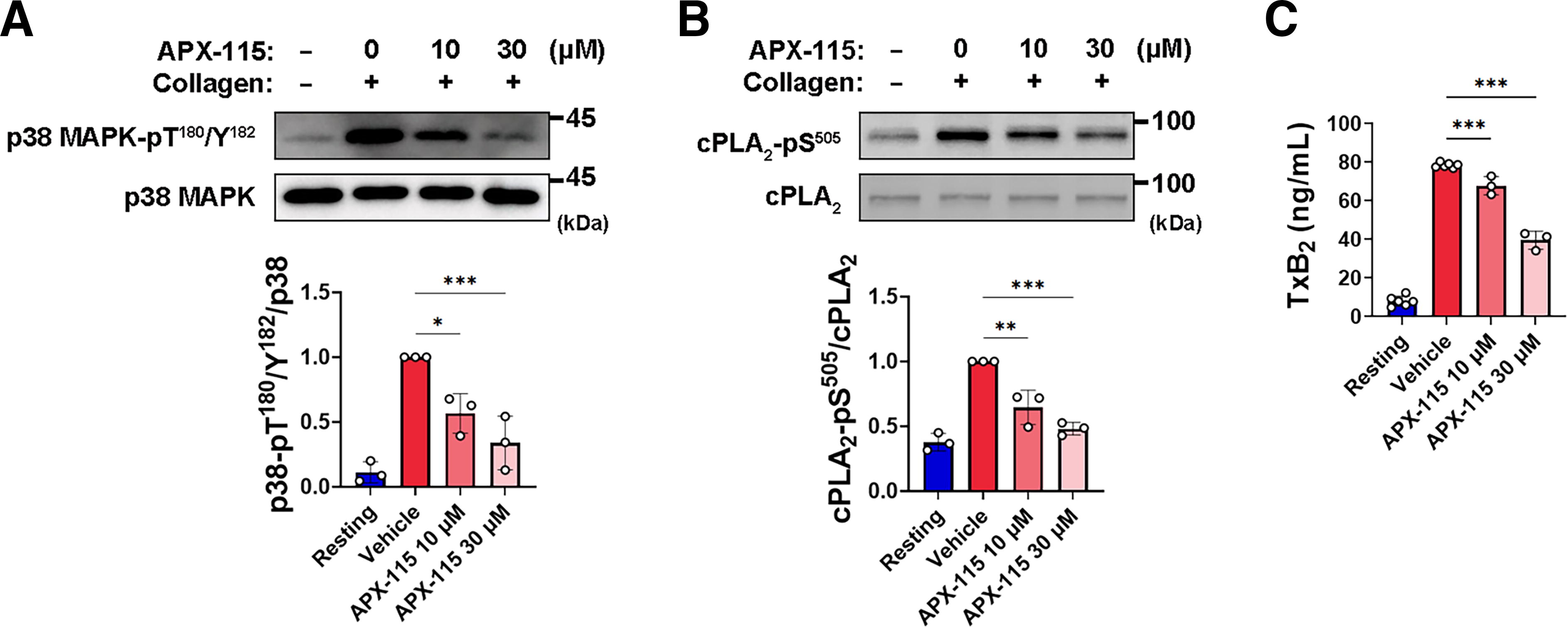
The collagen-induced platelet aggregation process is amplified by TXA2, a secondary-wave agonist (Atkinson et al., 2003). Cytosolic phospholipase A2 (cPLA2) is an essential enzyme in collagen-stimulated platelets, aiding in the formation of TXA2 by liberating arachidonic acid from membrane phospholipids (Wong et al., 2002), and its activity is enhanced by p38 MAPK-dependent phosphorylation at Ser505 (Borsch-Haubold et al., 1997; Lin et al., 1993). It has been shown that the activation of p38 MAPK in GPVI-stimulated platelets is inhibited by selective NOX2 inhibitor or deficiency of p47phox, a regulatory subunit of NOX2 (Akbar et al., 2018; Wang et al., 2020). Furthermore, previous research has shown that in GPVI-stimulated platelets, appropriate p38 MAPK activation and subsequent TXA2 synthesis depend on ROS produced from NOX1 (Walsh et al., 2014). Consequently, we investigated whether APX-115, a pan-NOX inhibitor, has an impact on the capacity of p38 MAPK to activate cPLA2. Collagen-induced platelet activation resulted in the phosphorylation of p38 MAPK at Thr180/Tyr182 and its downstream protein, cPLA2, at Ser510. Preincubation with APX-115 resulted in a considerable reduction of these phosphorylations (Fig. 5A, B). Next, we assessed whether APX-115 affects the production of TXB2, a stable metabolite of TXA2. The generation of TXB2 in collagen-stimulated platelets is significantly inhibited by APX-115 (Fig. 5C). These results suggest that the antiplatelet action of APX-115 is partly due to the downregulation of the ROS-mediated p38 MAPK/cPLA2/TXA2 production signaling pathway.
APX-115 suppresses collagen-induced ERK5 activation and PS exposure in platelets
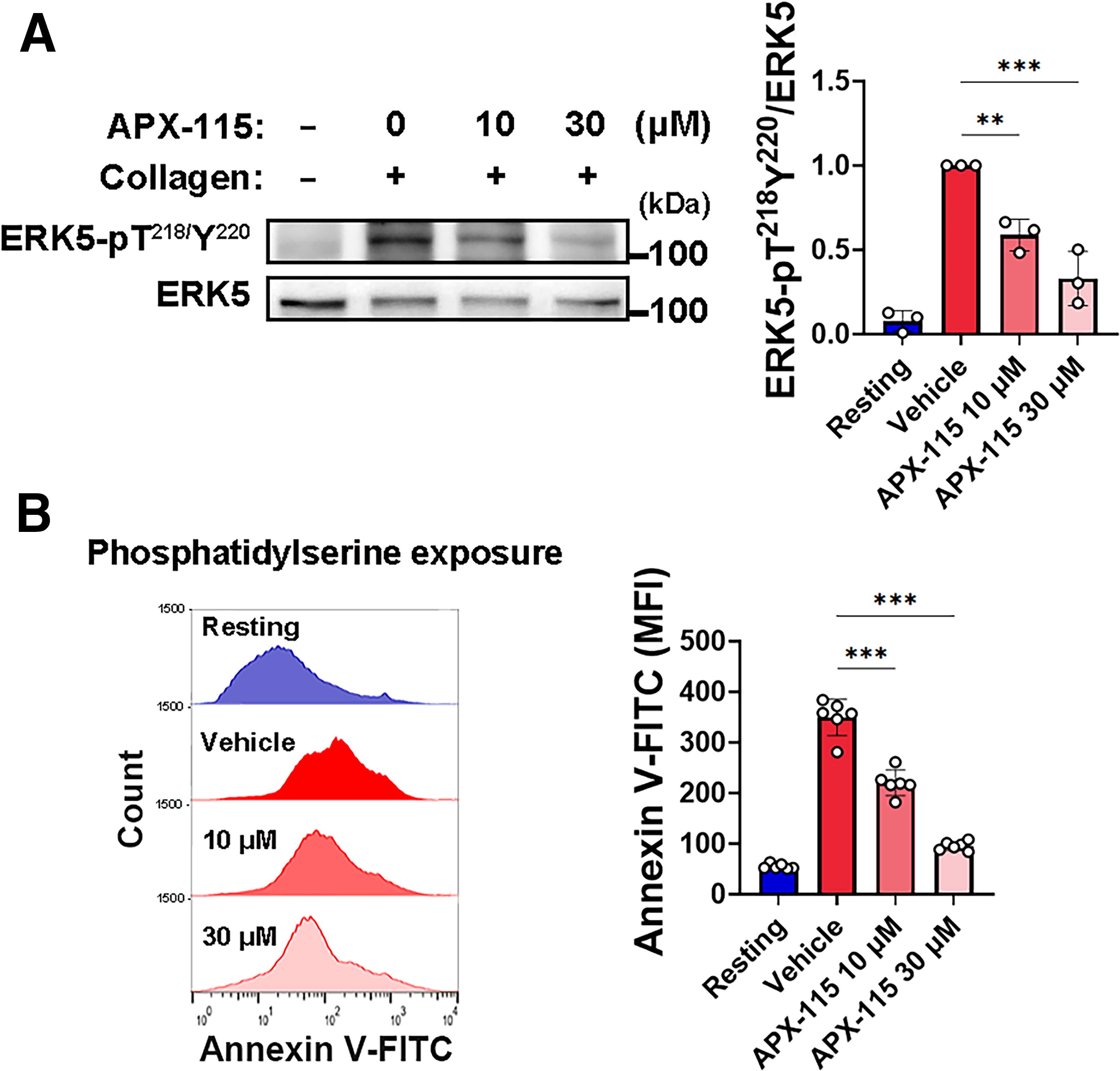
The redox-sensitive MAP kinase ERK5 is stimulated by NOX-mediated ROS in activated platelets (Cameron et al., 2015; Yang et al., 2018). This prompts procoagulant phosphatidylserine (PS) to externalize, which favors fibrin production (Yang et al., 2018; Zheng et al., 2023). Thus, we investigated the possibility that APX-115 influences the capacity of ERK5 to increase PS exposure on the extracellular surface. The phosphorylation of ERK5 at Thr218/Tyr220 was induced by collagen stimulation of platelets and these phosphorylations were considerably inhibited by preincubation with APX-115 (30 μM) (Fig. 6A). Next, using flow cytometry to measure the annexin V-FITC binding to platelets as a marker of PS exposure, we assessed the impact of APX-115 on platelet procoagulant reactions. Collagen treatment resulted in a considerable increase in PS exposure; nevertheless, the proportion of annexin V-positive platelets decreased by pretreatment with APX-115 (Fig. 6B). These results provide credence to the hypothesis that the capacity of APX-115 to reduce ROS generation may aid in the regulation of procoagulant states in cardiovascular disease by blocking ROS-sensitive ERK5 activation.
Platelet-derived EVs contribute to intercellular signaling and procoagulant activity through PS externalization (Owens and Mackman, 2011). We assessed whether APX-115 affects PS exposure on EVs. Collagen stimulation significantly increased EV concentration, size, and PS exposure, as shown by annexin V-FITC fluorescence (Supplementary Fig. S4). APX-115 at 30 μM significantly reduced PS exposure on EVs (Supplementary Fig. S4C). These findings suggest that APX-115 not only reduces the production of platelet-derived EVs but also decreases their procoagulant potential by limiting PS exposure.
Taken together, our results demonstrate that APX-115 effectively reduces ROS-sensitive ERK5 activation, PS exposure on platelets, and procoagulant activity of EVs in collagen-stimulated platelets. By targeting NOX-mediated pathways, APX-115 modulates key events in platelet activation and intercellular signaling, providing mechanistic insight into its antiplatelet and antithrombotic effects. These findings underscore the therapeutic potential of APX-115 in the prevention of procoagulant states and thrombotic complications.
APX-115 inhibits collagen-induced signaling pathways for integrin αIIbβ3 activation in platelets
Platelet adhesion and aggregation are significantly impacted by the inside-out activation of integrin αIIbβ3, leading to the binding of fibrinogen with high affinity (Bennett, 2005). Considering the significant role of NOX-mediated ROS in the activation of integrin αIIbβ3 in GPVI-stimulated platelets (Vara et al., 2021), we assessed the potential effects of APX-115 on αIIbβ3 inside-out signaling. Using flow cytometry and the FITC-labeled antiactive-integrin αIIbβ3 antibody (PAC1-FITC), we observed that APX-115 effectively inhibited the collagen-induced increase in active conformational changes of integrin αIIbβ3 (Fig. 7A). By regulating integrin αIIbβ3 activation, the adaptor molecule vasodilator-stimulated phosphoprotein (VASP) plays a crucial role in preventing platelet activation (Aszodi et al., 1999; Hauser et al., 1999). Phosphorylation of VASP at Ser239 plays a role in inhibiting the activation of platelets by integrin αIIbβ3, and this specific phosphorylation is controlled by the cGMP/protein kinase G (PKG) signaling pathway, which has a negative effect on platelet responsiveness (Aszodi et al., 1999; Smolenski et al., 1998). Triple NOX-deficient mouse platelets showed consistently greater intraplatelet cGMP levels in resting settings and considerably higher levels of VASP Ser239 phosphorylation in collagen-stimulated conditions as compared with the wild type (Vara et al., 2021). Moreover, it was shown that NOX-generated ROS in platelets blocks the cGMP/PKG signaling pathway (Magwenzi et al., 2015). Thus, the impact of APX-115 on VASP phosphorylation and the levels of cGMP within platelets were assessed. As seen in Figure 7B, a noteworthy decrease in the phosphorylation of VASP at Ser239 was observed following collagen stimulation. The addition of APX-115 resulted in a notable decrease in phosphorylation. In addition, under normal conditions, the administration of APX-115 at a concentration of 30 μM resulted in a significant 3.7-fold rise in cGMP levels within the platelets (Fig. 7C). These results suggest that the regulation of the ROS-mediated cGMP/PKG/VASP/integrin αIIbβ3 signaling pathway contributes to the antiplatelet activity of APX-115.
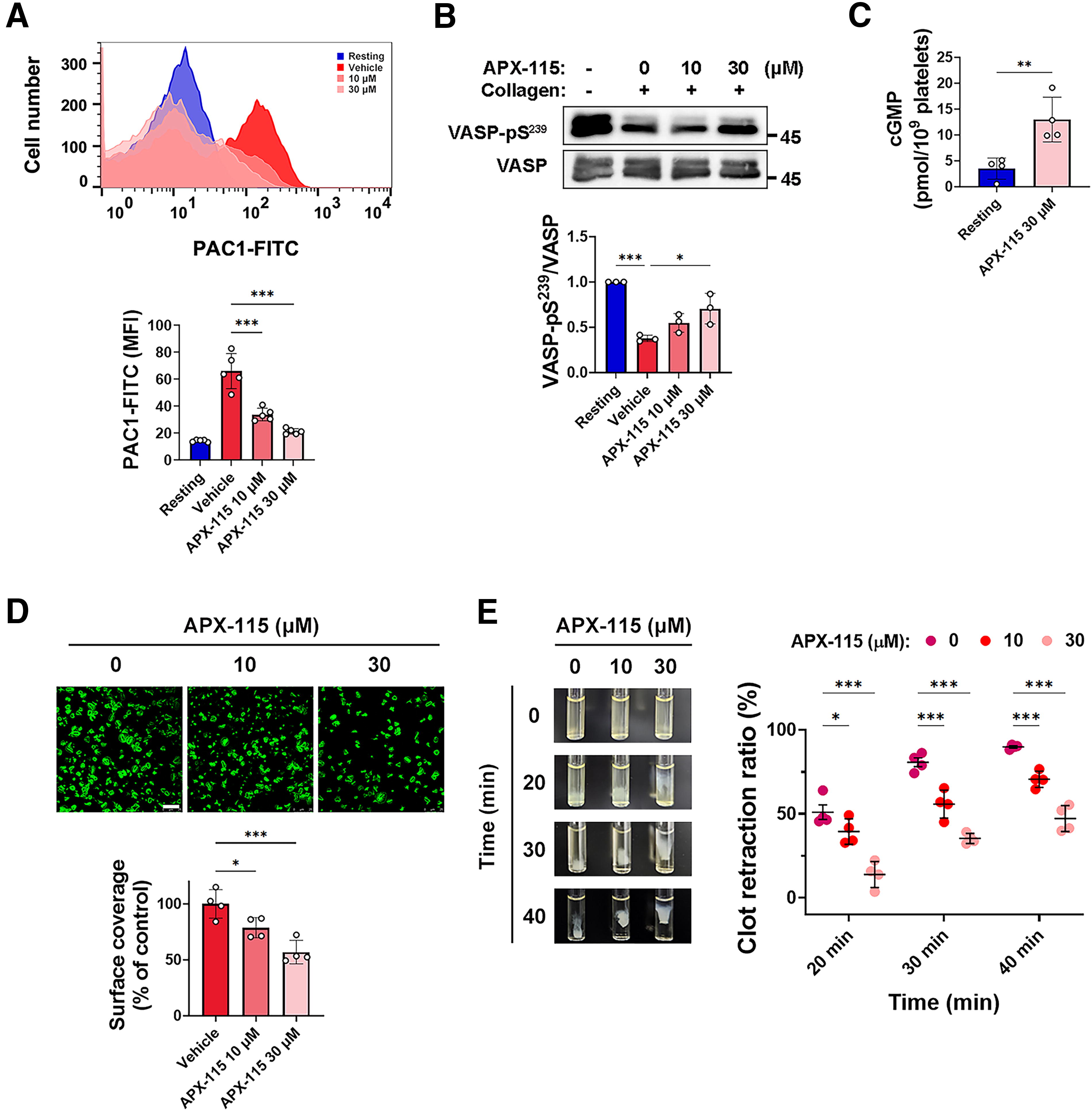
Because APX-115 inhibited collagen-induced integrin αIIbβ3 activation, its effects on integrin αIIbβ3-mediated outside-in signaling were assessed by examining platelet spreading and clot retraction, key processes in thrombus stabilization. While adhesion to immobilized fibrinogen does not require prior activation, spreading and clot retraction rely on outside-in signaling via integrin αIIbβ3. APX-115 significantly reduced platelet spreading (Fig. 7D) and markedly inhibited clot retraction (Fig. 7E), indicating disruption of integrin αIIbβ3-mediated cytoskeletal remodeling. These findings highlight the ability of APX-115 to impair thrombus stability and its potential as an antithrombotic agent targeting outside-in signaling pathways.
APX-115 inhibits platelet adhesion and thrombus formation under flow conditions in vitro and arterial thrombosis in vivo
The primary cause of platelet adhesion and aggregation at the site of endothelium damage, followed by thrombus development, is exposure to subendothelial collagen inflammation (Nieswandt et al., 2011; van der Meijden and Heemskerk, 2019). NOX1 or NOX2-knockout mouse platelets had decreased thrombus volume after platelet adherence to collagen under shear (Vara et al., 2019, 2021; Walsh et al., 2014; Xu et al., 2021). Consequently, we investigated platelet adhesion and thrombus development under shear in a flow chamber covered with collagen and perfused with platelets probed with DiOC6. In platelets treated with APX-115, there was a decrease in both stable platelet adhesion and the diameters of platelet thrombi on immobilized collagen under shear (Fig. 8A). These findings show that APX-115 prevents the growth of thrombus under shear as well as stable platelet attachment.
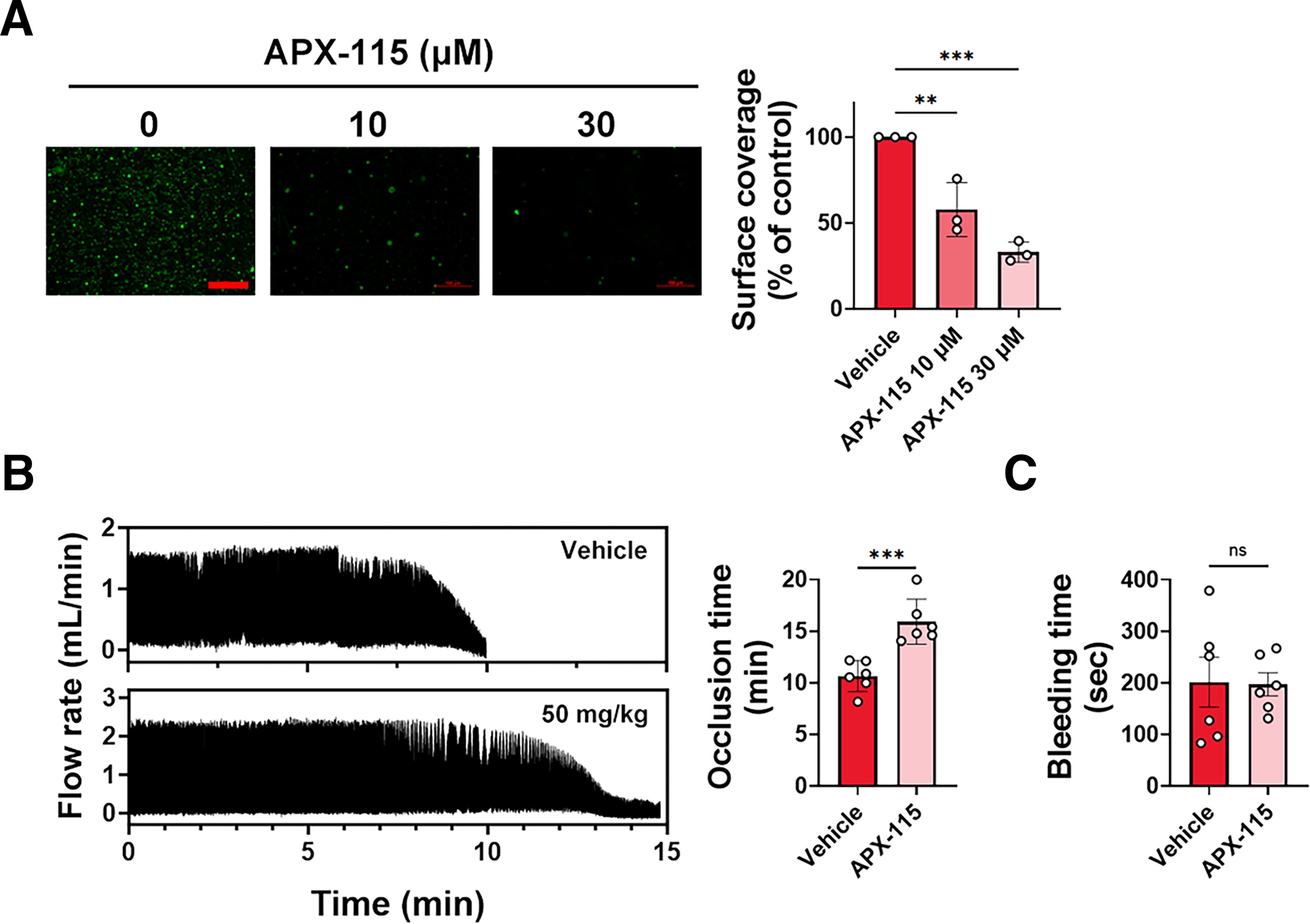
We further assessed the in vivo effects of APX-115 using a FeCl3-induced carotid artery occlusion model. As shown in Figure 8B, APX-115 treatment significantly prolonged the occlusion time compared with the vehicle control group (10.6 ± 1.5 vs. 15.9 ± 2.2 min, n = 6, p = 0.0022). This indicates a potent antithrombotic effect of APX-115, consistent with the inhibition of platelet activation and thrombus formation observed in our earlier in vitro experiments. Notably, tail bleeding time assays showed no significant difference between APX-115-treated and vehicle-treated groups (201.3 ± 48.1 vs. 197.0 ± 22.2 s, n = 6, p = 0.8182), indicating that APX-115 does not impair normal hemostasis (Fig. 8C). In addition, there was no significant change in platelet count following APX-115 treatment (Supplementary Fig. S8), indicating that APX-115 does not induce thrombocytopenia.
Discussion
This study highlights the potent antiplatelet and antithrombotic effects of APX-115, a pan-NOX small molecule inhibitor, by demonstrating its ability to inhibit ROS production, platelet activation, and thrombus formation. By targeting NOX-mediated pathways, APX-115 disrupts key signaling cascades involved in platelet function, providing a strong foundation for its therapeutic potential. These findings align with extensive evidence supporting the role of NOX-derived ROS in platelet activation and thrombosis (Carnevale et al., 2014b; Delaney et al., 2016; Pignatelli et al., 2004, 2011; Vara et al., 2021; Xu et al., 2021).
Our study highlights the critical role of APX-115 in inhibiting platelet activation by targeting NOX-mediated ROS production and preserving PTP functionality. PTPs, which regulate protein tyrosine phosphorylation, are highly sensitive to oxidative inactivation by ROS (Rhee, 2006; Tonks, 2005). APX-115 significantly reduced intracellular ROS levels in collagen-stimulated platelets (Fig. 1) and protected PTPs from oxidative inactivation, as demonstrated by the PTP activity assay (Fig. 2B). This preservation of PTP activity maintained the balance of tyrosine phosphorylation/dephosphorylation, thereby inhibiting the activation of key signaling molecules such as Syk, LAT, Vav1, Btk, and PLCγ2, ultimately suppressing downstream signaling pathways (Figs. 2C and 3A).
NOX-mediated ROS generation amplifies GPVI signaling by inactivating PTPs, which would otherwise dephosphorylate substrates to regulate signaling cascades (Vara et al., 2021). PTPs like LMW-PTP, SHIP-1, PTEN, and SHP-2 are known to attenuate GPVI-mediated platelet activation through substrate dephosphorylation (Chari et al., 2009; Jang et al., 2014; Mancini et al., 2007; Weng et al., 2010). Collagen binding to GPVI initiates a signaling cascade by triggering the phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) on the Fc receptor γ-chain via Src family kinases, which subsequently facilitate the recruitment and activation of Syk, a central mediator of platelet activation (Gibbins, 2004; Poole et al., 1997). Once activated, Syk phosphorylates LAT, a critical adaptor protein in the GPVI signaling pathway. This leads to the assembly of a signaling complex involving Syk, Vav1, Btk, and PLCγ2, with PLCγ2 activation indicated by phosphorylation at Tyr753 (Pasquet et al., 1999; Pearce et al., 2002; Quek et al., 1998; Suzuki-Inoue et al., 2004).
APX-115 disrupted these pathways by preventing ROS-mediated PTP inactivation, thereby counterbalancing kinase activity and reducing phosphorylation of key signaling proteins, including PLCγ2 at Tyr753 (Figs. 2C and 3A). This partially explains the observed reduction in protein tyrosine phosphorylation in collagen-stimulated platelets. In addition, a previous report has shown that ROS can oxidatively modify Src family tyrosine kinase in platelets, enhancing their ability to phosphorylate downstream signaling proteins (Yang et al., 2020). Thus, the observed reduction in tyrosine phosphorylation following APX-115 treatment likely results from a combination of preserved PTP activity and direct inhibition of ROS-sensitive kinase functions. This dual mechanism underscores the unique approach of APX-115 in suppressing platelet activation.
When collagen receptors are stimulated in platelets, PLCγ2 is activated, breaking down phosphatidylinositol 4,5-bisphosphate into diacylglycerol and IP3, leading to PKC activation and intracellular Ca2+ release (Watson et al., 2005). PKC and elevated cytosolic Ca2+ collaboratively promote degranulation and integrin αIIbβ3 activation, essential for platelet aggregation and thrombus formation (Bennett, 2005; Watson et al., 2005). P-selectin on activated platelets interacts with P-selectin GP ligand-1 on leukocytes or endothelial cells, contributing to thrombus formation (Vandendries et al., 2004). Dense-granule-released substances such as ADP and epinephrine further enhance platelet activation through autocrine and paracrine signaling. This study demonstrates that APX-115 effectively inhibits key platelet responses, including cytosolic Ca2+ elevation, granule release, P-selectin exposure, αIIbβ3 activation, and aggregation. These results highlight the ability of APX-115 to suppress the GPVI-ITAM-Syk-PLCγ2 signaling pathway in collagen-stimulated platelets, primarily by inhibiting ROS generation and tyrosine phosphorylation-dependent signaling.
Our findings show that APX-115 effectively inhibits the release of Ca2+ from both internal and external sources of platelets (Fig. 3). GPVI-PLCγ2 signaling triggers Ca2+ release from the dense tubular system, similar to the endoplasmic reticulum, via activation of IP3 receptors (IP3R). Following the stimulus, cytosolic Ca2+ is brought back to resting levels by the sarcoendoplasmic reticulum and plasma membrane Ca2+-ATPases (SERCA and PMCA) (Varga-Szabo et al., 2009). Collagen-stimulated platelets produce high levels of H2O2, which can activate IP3R and inhibit SERCA and PMCA, leading to increased cytosolic Ca2+ (Pignatelli et al., 1998; Redondo et al., 2004). APX-115 inhibits the elevation of intracellular H2O2 levels (Fig. 1E), which may contribute to lowering cytosolic Ca2+ levels by mitigating the oxidative effects on IP3R, SERCA, and PMCA. Further research is needed to determine which of the several NOX-derived ROS specifically modulate intracellular Ca2+ in platelets.
APX-115 effectively inhibits the generation of TXA2, a key pro-aggregatory molecule in platelet activation. TXA2 production depends on cPLA2, which releases arachidonic acid from phospholipids, a rate-limiting step in the pathway. When cytosolic Ca2+ levels rise, cPLA2 is activated and translocates to the membranes via its Ca2+-dependent phospholipid-binding domain (Clark et al., 1991; Kramer and Sharp, 1997). APX-115 suppresses collagen-induced Ca2+ elevation (Fig. 3), linking its suppression of TXA2 generation to reduced Ca2+-dependent cPLA2 activation. In addition, APX-115 blocks the phosphorylation of p38 MAPK (Thr180/Tyr182) and cPLA2 (Ser505), essential steps in TXA2 production (Borsch-Haubold et al., 1997). Apoptosis signal-regulating kinase 1 (ASK1) regulates p38 MAPK activity and is activated by ROS, particularly H2O2, in platelets (Naik et al., 2017; Patel et al., 2019). Collagen-mediated TXA2 production is blocked by scavenging ROS (Chlopicki et al., 2004). APX-115 reduces ROS levels, likely hindering ASK1 activation and attenuating the p38 MAPK-cPLA2-TXA2 axis, contributing to its inhibition of platelet activation.
ROS produced by activated platelets is a key driver of ERK5 activation, a process highly sensitive to redox changes (Cameron et al., 2015; Yang et al., 2018). This activation triggers downstream events, including caspase activation and membrane scramblase activity, leading to PS exposure that facilitates prothrombinase complex assembly and fibrin production (Yang et al., 2018). APX-115 effectively inhibits collagen-stimulated ERK5 activation, resulting in a significant reduction in PS exposure on platelets (Fig. 6). These findings highlight the potential of APX-115 to reduce thrombotic risk by targeting redox-sensitive signaling mechanisms and mitigating procoagulant activity.
Platelet-derived EVs play a critical role in promoting thrombosis by generating ROS and presenting PS as a scaffold for thrombin generation (Gaspar et al., 2021; Owens and Mackman, 2011). Dysregulated EV production amplifies platelet activation and systemic thrombotic risk. This study shows that APX-115 significantly reduces EV concentration and PS exposure in collagen-stimulated platelets. By inhibiting NOX-mediated ROS production, APX-115 suppresses pathological EV biogenesis while preserving normal cellular functions. The reduction in PS exposure further disrupts the assembly of prothrombinase complexes, highlighting the potential of APX-115 to mitigate thrombotic activity linked to EVs. The ability of APX-115 to target EV-mediated signaling suggests its broader applicability in managing thrombotic disorders. Future research should investigate its effects on EV production in other pathological contexts to enhance our understanding of its therapeutic potential.
APX-115 treatment significantly elevated cGMP levels in human platelets (Fig. 7), consistent with findings linking NOX-derived ROS to soluble guanylate cyclase activity and cGMP regulation (Vara et al., 2021). Elevated cGMP activates PKG, which phosphorylates VASP at Ser239, a key regulator of integrin αIIbβ3 activation (Aszodi et al., 1999; Smolenski et al., 1998). By reducing NOX-derived ROS, APX-115 maintained VASP phosphorylation and effectively inhibited collagen-induced integrin αIIbβ3 activation. This supports its role in modulating the cGMP/PKG/VASP signaling pathway to suppress platelet aggregation. In addition, APX-115 disrupted integrin αIIbβ3-mediated outside-in signaling, as demonstrated by reduced platelet spreading and clot retraction, processes crucial for thrombus stabilization. These findings highlight the dual action of APX-115 in targeting both adhesion and stabilization phases of thrombus formation through redox-sensitive signaling pathways.
Platelet adhesion to the extracellular matrix is essential for thrombus formation at sites of arterial injury (Dutting et al., 2012). Both intracellular and extracellular ROS play pivotal roles in facilitating platelet adhesion and activation under shear stress, ultimately driving thrombus development (Delaney et al., 2016; Vara et al., 2021; Xu et al., 2021). APX-115 effectively inhibited collagen-induced ROS production, preventing platelet adhesion and thrombus formation under shear conditions (Fig. 8). These findings are consistent with reduced platelet adhesion and smaller thrombi observed in NOX1- or NOX2-deficient mouse platelets under similar conditions (Vara et al., 2021; Xu et al., 2021). By targeting ROS-dependent processes, APX-115 not only disrupts initial platelet adhesion but also attenuates shear-induced thrombus stabilization, highlighting its potential as a therapeutic agent for arterial thrombosis.
Our in vivo findings confirm the pivotal role of NOX enzymes in platelet-mediated thrombus formation and demonstrate the antithrombotic efficacy of APX-115 (Fig. 8). In the FeCl3-induced carotid artery thrombosis model, APX-115 significantly prolonged occlusion time, consistent with studies showing impaired thrombus formation in NOX1/NOX2/NOX4 triple knockout mice (Vara et al., 2021). Importantly, APX-115 selectively inhibits pathological thrombus formation without prolonging bleeding time, as evidenced by tail bleeding assays. This dual benefit—effective thrombus reduction coupled with preserved hemostatic function—addresses a critical limitation of traditional antiplatelet therapies, which often compromise hemostasis. By targeting NOX-mediated ROS production, APX-115 offers a promising therapeutic strategy with a favorable safety profile.
The ability of APX-115 to target NOX1, NOX2, and NOX4 provides comprehensive inhibition of platelet activation pathways mediated by both collagen and thrombin, surpassing the limitations of isoform-specific inhibitors. Comparative studies with ML171 (NOX1-specific) and GSK2795039 (NOX2-specific) demonstrated that APX-115 exhibits broader and more potent inhibitory effects against both stimuli (collagen and thrombin), highlighting its capacity to inhibit multiple NOX isoforms simultaneously (Supplementary Fig. S2A, S2B). Evidence suggests that NOX1 and NOX2 play complementary roles in GPVI-mediated collagen receptor signaling and GPCR-mediated thrombin receptor activation (Delaney et al., 2016; Vara et al., 2021), respectively. This dual targeting by APX-115 allows it to suppress interactions between NOX isoforms, enhancing its antiplatelet efficacy. While hereditary NOX2 deficiencies in patients with CGD underscore the critical role of NOX2 in ROS generation and platelet activation (Pignatelli et al., 2004), further studies are needed to clarify the interplay between NOX isoforms in human platelets. Such insights could optimize the therapeutic application of pan-NOX inhibitors such as APX-115 in diverse thrombotic conditions.
APX-115 effectively regulates platelet function by inhibiting NOX-derived ROS. However, mitochondrial ROS also plays a crucial role in platelet reactivity and can interact with NOX-derived ROS to amplify signaling pathways (Ajanel et al., 2023; Krotz et al., 2004). This interplay warrants further investigation to fully understand the dynamics of redox signaling in platelet activation. Future research is warranted to investigate the interplay between NOX-derived and mitochondrial ROS, as this may provide additional insights into the comprehensive effects of APX-115 on platelet function and oxidative signaling pathways.
In addition, long-term safety studies are required to assess potential off-target interactions, given the structural similarities between NOX enzymes and other NADPH-dependent proteins. Understanding the balance between ROS inhibition and the maintenance of physiological ROS signaling will be critical for optimizing the clinical use of APX-115.
Ongoing clinical trials evaluating APX-115 in diabetic populations (ClinicalTrials.gov Identifier: NCT04534439) will provide valuable insights into its systemic effects and its ability to mitigate platelet hyperactivity in high-risk groups. Diabetes-associated platelet hyperreactivity, driven in part by NOX-mediated ROS production, significantly elevates thrombotic risks (Vaidya et al., 2021). By targeting these mechanisms, APX-115 offers the potential to simultaneously reduce thrombotic complications and alleviate oxidative stress-related vascular dysfunction. These dual protective benefits position APX-115 as a promising candidate for managing thrombotic and vascular complications in diabetes and other high-risk conditions.
Conclusions
Taken together, our results demonstrate that APX-115 effectively inhibits ROS production and platelet activation, leading to a reduction in thrombus formation while sparing normal hemostatic processes. These findings highlight the therapeutic potential of APX-115 as a novel antithrombotic agent, with the added benefit of reducing bleeding risk.
Materials and Methods
Ethics statements
All animal experiments were performed in accordance with the relevant guidelines and regulations, and the study was approved by the Seoul National University Institutional Animal Care and Use Committee (IACUC; approval number: SNU-220331-6-5). The collection and use of human blood samples from healthy volunteers were conducted in accordance with the relevant guidelines and regulations, and the study was approved by the Seoul National University Institutional Review Board under protocol no. 2206/001-006. Written informed consent was obtained from all participants prior to participation.
Reagents and antibodies
APX-115 (Cat# U109969; Achemblock, Hayward, CA, USA), DCFH2 (Cat# HY-D0940; MedChem Express, Princeton, NJ, USA), CM-H2DCFDA, DiOC6, Fluo-3-AM, (Cat# C6827, D273, F1242) (all from Molecular Probes, Eugene, OR, USA), AEBSF (Cat# A-540; Gold Biotechnology, St. Louis, MO, USA), aprotinin, NaCl, NaHCO3, Nonidet P-40 (Cat# 0332, 0241, 0335, M158) (all from Amresco, Solon, OH, USA), bovine serum albumin, CaCl2, DMSO (Cat# A6003-5G, C5670, D8418), EGTA, FeCl3, glucose, (Cat# E4378, 31235, G7021), KH2PO4, Na2HPO4, Na3VO4, Na4P2O7•10H2O, NaF (Cat# P5655, S9763, S6508, S6422, S7920), iodoacetamide, L-012, N-ethylmaleimide (Cat# I6125, E3876, SML2236), N, N-dimethylacetamide, paraformaldehyde, prostaglandin E1, (Cat# D5511, P6148, P5515), S4641), sodium citrate, fibrinogen, thrombin (Cat# S4641, F3879, T6884), Triton X-100, Tween 80, β-glycerophosphate (Cat# T9284, P4780, G9422) (all from Sigma-Aldrich, St. Louis, MO, USA), collagen (Cat# 385; Chrono-Log, Havertown, PA, USA), citric acid (Cat# 4337; Duksan, Seoul, Korea), EDTA, KCl, leupeptin, MgCl2 (Cat# 75829, 74075, 78436, 75826) (all from USB, Cleveland, OH, USA), FITC-labeled annexin V (Cat# 556419; BD Biosciences, San Jose, CA, USA), HEPES (Cat# 11344041; Thermo Fisher Scientific, Waltham, MA, USA), dithiothreitol (Cat# D1309.0010, Duchefa Biochemie, Haarlem, Netherlands), Peroxy Orange 1 (Cat# 4944; Tocris Biosciences, Ellisville, MO, USA) were purchased from the respective companies.
The following antibodies were used: antiphospho-Btk (Tyr551) antibody (Cat# 44-1355G, Invitrogen, New York, NY, USA), anti-Btk (Cat# sc-81238), anti-LAT (Cat# sc-365626), anti-phospho-PLCγ2 (Tyr753) (Cat# sc-130252), anti-PLCγ2 (Cat# sc-407), anti-Syk (Cat# sc-1240), anti-VASP (Cat# sc-46669), antiphospho-Vav1 (Tyr174) (Cat# sc-101858) antibodies (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), antiphospho-cPLA2 (Ser505) (Cat# 2831), anti-cPLA2 (Cat# 2832), antiphospho-ERK5 (Thr218/Tyr220) (Cat# 3371), anti-ERK5 (Cat# 3372), antiphospho-p38 MAPK (Thr180/Tyr182) (Cat# 9211), anti-p38 MAPK (Cat# 8690), antiphospho-Syk (Tyr525/526) (Cat# 2711), antiphospho-VASP (Ser239) (Cat# 3114) antibodies (all from Cell Signaling Technology, Danvers, MA, USA), antiphospho-LAT (Tyr200) (Cat# ab68139), anti-Vav1 antibodies (Cat# ab40875 both from Abcam, Cambridge, UK), antiphosphotyrosine (4G10) and antibody (Cat# 05-321) (from Sigma-Aldrich, St. Louis, MO, USA), anti-β-actin antibody (Cat# LF-PA0207) (Abfrontier, Seoul, Korea), horseradish peroxidase labeled goat anti-rabbit IgG and anti-mouse IgG (Cat# 5220-0336, 5450-0011; both from Seracare Life Sciences, Milford, MA, USA), PAC1-FITC (Cat# 340507) and CD62P-PE (Cat# 555524) antibodies (both from BD Biosciences, San Jose, CA, USA).
Human platelet preparation
Blood was taken by venipuncture from healthy and drug‐free volunteers and collected in tubes containing anticoagulant acid/citrate/dextrose (22.0 g sodium citrate, 24.5 g dextrose, and 7.3 g citric acid per 1 L) (Cat# 364816) (Becton Dickson, Franklin, NJ, USA). Platelet-rich plasma (PRP) obtained by centrifugation for 15 min at 150 × g was further centrifuged for 10 min at 300 × g to concentrate the platelets. Platelet pellet was suspended in a solution containing Tyrode’s-HEPES buffer (10 mM HEPES [pH 7.4], 129 mM NaCl, 0.8 mM KH2PO4, 8.9 mM NaHCO3, 2.8 mM KCl, 0.8 mM MgCl2, and 5.6 mM glucose), 2 mM EDTA, 10% of citric acid/citrate/dextrose solution, 1 μM prostaglandin E1, and then centrifuged again. The supernatant was discarded, and the platelet pellet was resuspended in Tyrode’s-HEPES buffer to the required concentration. Unless otherwise stated, washed platelets were incubated with 1 mM CaCl2 for 2 min prior to stimulation.
Light transmission aggregometry
Platelet aggregation was measured in a siliconized glass cuvette under continuous stirring at 1000 rpm at 37°C using a 4-channel aggregometer (Chrono-Log, Havertown, PA, USA). Data were collected using Aggrolink software (Model# 700; Chrono-Log, Havertown, PA, USA). Platelet aggregation was measured as percentage change in light transmission, where 100% refers to transmission through the buffer control.
Assessment of extracellular ROS levels
Washed platelets (5 × 108/mL) in Tyrode’s-HEPES buffer containing 1 μM DCFH2 were stimulated in a fluorometer cuvette under constant stirring at 800 rpm at 37°C. The fluorescence of DCF (excitation at 488 nm/emission at 525 nm) was measured using a spectrofluorophotometer as described above.
Assessment of extracellular superoxide anion levels
Extracellular superoxide anion levels were assessed using L-012, a chemiluminescent probe specific for superoxide (Daiber et al., 2004). After washed platelets (5 × 108/mL) in Tyrode’s-HEPES buffer containing 200 μM L-012 were stimulated, luminescence was read immediately for 15 min at 20-s intervals using a Cytation3 microplate spectrophotometer (BioTek, Burlington, VT, USA).
Assessment of intracellular levels of ROS, hydrogen peroxide and cytosolic Ca2+
CM-H2DCFDA and Fluo-3-AM were used to detect the intracellular levels of ROS and Ca2+, respectively. Washed platelets (5 × 108/mL) were incubated in PBS containing 5 μM CM-H2DCFDA or 1 μM Fluo-3-AM for 30 min at 37°C in the dark, pelleted by centrifugation, and resuspended in Tyrode’s-HEPES buffer. Probe-loaded platelets (5 × 108/mL) were stimulated in a fluorometer cuvette under constant stirring at 1000 rpm at 37°C. The fluorescence of CM-DCF (excitation at 495 nm/emission at 525 nm) or Fluo-3 (excitation at 488 nm/emission at 525 nm) was measured using a spectrofluorophotometer (Model# FP-8350; Jasco, Tokyo, Japan). The initial signal was recorded for 20 s prior to collagen stimulation and used as a baseline followed by continued measurement. The fluorescence signal was normalized to the baseline value for each sample.
PTP activity assay
PTP activity was measured as described previously (Wang et al., 2015). Briefly, after stimulation, platelets were lysed in oxygen-free lysis buffer (50 mM Tris-HCl, pH 6.5, 150 mM NaCl, 1% Nonidet P-40, 1 μg/mL leupeptin, 1 μg/mL aprotinin, and 1 mM AEBSF containing 10 mM iodoacetamide and 10 mM N-ethylmaleimide in a glovebox workstation (<1% oxygen; Mbraun, Garching, Germany). The cell debris was removed by centrifugation at 12,500 × g for 10 min at 4°C. Cell lysates were incubated at room temperature in a dark environment for 30 min to achieve complete alkylation of free thiols. The labeling was quenched by adding 50 mM dithiothreitol. The protein concentrations were measured using the Bradford reagent (Bio-Rad, Hercules, CA, USA). PTP activity was measured using tyrosine phosphopeptide (RRLIEDAEpYAARG, where pY represents phosphotyrosine) as the substrate, according to the manufacturer’s protocol (PTP Assay Kit 1, Cat# 17-125, EMD Millipore, Billerica, MA, USA). PTP activity was expressed as nmol of inorganic phosphate released/min/mg protein used for the assay.
Western blotting
After stimulation in a tube under constant stirring at 1000 rpm at 37°C using a Thermomixer, the platelets were lysed in cell extraction buffer (20 mM HEPES [pH 7.0], 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 2 mM EGTA, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, 1 μg/mL aprotinin, and 1 mM AEBSF). Cell debris was removed by centrifugation at 12,500 × g for 10 min at 4°C. Equal volumes of cell lysates with adjusted protein concentrations were used for analysis using specific antibodies, as indicated. Band intensity was analyzed using ImageJ software (NIH, Bethesda, MD, USA).
TXB2 ELISA
After treatment in a tube under constant stirring at 1000 rpm at 37°C using a Thermomixer, the reaction was stopped by adding 5 mM EDTA and 200 μM indomethacin. The platelet reaction mixture (5 × 108 platelets/mL) was immediately centrifuged for 30 s to obtain supernatant. The respective level of TXB2 in the supernatant was detected using ELISA kit (Cat# 501020; Cayman, Ann Arbor, MI, USA) in a Cytation3 microplate spectrophotometer (BioTek, Burlington, VT, USA).
Flow cytometry and analysis
For analyzing the surface P-selectin, activated integrin αIIbβ3, PS exposure, or the levels of intracellular H2O2, platelets were incubated with fluorophore-conjugated antibodies CD62P-PE (0.5 μg/mL), PAC1-FITC (0.5 μg/mL), annexin V-FITC (0.1 mg/mL), or Peroxy Orange 1 (5 μM), respectively. The platelet mixture was stimulated with collagen at 37°C for 5 min in the dark. Each sample was fixed in 1% paraformaldehyde and diluted with PBS. FACS Aurora flow cytometer (Cytek Biosciences, CA, USA) was used for all analyses with a minimum of 5 × 104 cells per sample. Platelet populations were identified and gated based on forward scatter and side scatter characteristics. Data were analyzed using FlowJo software 10.9 (FlowJo LLC, Ashland, OR, USA).
ATP release assay
Platelets were incubated in a white, flat-bottom 96-well plate and then stimulated with collagen. Luciferin‐luciferase reagent of the ATP bioluminescent assay kit (ab113849; Abcam, Cambridge, CB2 0AX, UK) was added to each well, and the 96-well plate was placed in a LB 960 Centro microplate luminometer (Berthold, Bad Wildbad, Germany). An automated protocol was carried out where the sample was shaken and the luminescence was measured and recorded at 30 s intervals for 5 min.
cGMP ELISA
Platelet reaction mixture (5 × 108 platelets/mL) was centrifuged through a 0.8 μm size centrifugal filter (Cat# VK01P042; Sartorius, Elk Grove, IL, USA) at 10,000 × g for 30 s to selectively remove supernatants. The platelets on the filter membrane were lysed in 0.1 M HCl containing 1% Triton X-100. The lysates were centrifuged at 12,500 × g for 10 min at room temperature to eliminate debris. The resulting supernatant was utilized for the assay. The concentration of cGMP in the supernatant was then measured using an ELISA kit (Cat# ADI-900-164; Enzo Life Sciences, Exeter, UK) in a Cytation3 microplate spectrophotometer (BioTek, Burlington, VT, USA).
Platelet spreading on fibrinogen
Microscope cover glasses (Deckglaser, 18 mm, Marienfeld, Germany) were coated overnight with 100 μg/mL in 0.1 M NaHCO3 (pH 8.3) at 4°C, placed into 12-well plates, and blocked with 5% bovine serum albumin in PBS. Washed platelet suspensions (2 × 107/mL) were then aliquoted into the 12-well plates (300 μL/well) and incubated at 37°C for 75 min. The platelet suspension was then aspirated to remove nonadherent platelets and fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS, and stained with Alexa Fluor 488-conjugated phalloidin (Cat# A12379; Invitrogen, Carlsbad, CA, USA). Adherent platelets were observed with a TCS8 confocal microscope (Leica, Wetzlar, Germany). Representative images were acquired from 6 different fields and analyzed with ImageJ software (NIH, Bethesda, MD, USA).
Clot retraction
Blood was taken by venipuncture from healthy and drug‐free volunteers and collected in a blood-collection tube containing 3.8% (w/v) sodium citrate (Soyagreentec, South Korea). PRP was centrifuged at 300×g for 10 min to obtain platelet poor plasma. For clot retraction assays, 0.3 mL plasma was added to 0.1 mL washed platelets in a glass cuvette (Cat# 312; Chrono-Log, Havertown, PA, USA). Clot formation and retraction were initiated by thrombin (1 U/mL) and CaCl2 (1 mM) at room temperature. Digital photographs were taken of the clot retraction progression.
Determination of platelet adhesion and thrombus formation under flow conditions
Washed platelets (1 × 109/mL) were incubated with 1 μM of DiOC6 for 10 min at 37°C. A collagen-coated coverslip (Cat# GG-12-Collagen; Neuvitro, Camas, WA, USA) was mounted in a parallel plate flow chamber (Chamlide CF, Live Cell Instrument, Seoul, Korea). DiOC6-labeled platelets were perfused over a matrix of collagen at 1000 s−1 until 1 mL of sample ran out using a syringe pump (Harvard Apparatus, Holliston, MA, USA). Nonadherent platelets in the chamber were washed with PBS. Platelet thrombus and adherent platelets were fixed with cold 4% paraformaldehyde for 15 min, washed with PBS, and then visualized with a fluorescence microscope (Ts2-FL, Nikon, Tokyo, Japan). Representative images from 5 to 10 different fields were captured. Flow chamber surface coverage was calculated using ImageJ software (NIH, Bethesda, MD, USA).
Assessment of arterial thrombosis
Mice (C57BL/6 J, male) obtained from The Jackson Laboratory (Bar Harbor, ME, USA) were housed in a specific-pathogen-free animal facility at Seoul National University. Mice were provided a standard chow diet (Cat# 2018; Harlan Teklad, Madison, WI, USA) ad libitum. Male mice aged 10–11 weeks were divided into three groups. The mice were orally administered APX-115 dissolved in 10% N, N-dimethylacetamide, 10% Tween 80, and 80% distilled deionized water with oral gavage in a volume of 10 mL/kg body weight, as described previously (Joo et al., 2016). After an intraperitoneal injection containing 100 mg/kg alfaxalone (Jurox, Cat# 520400; New South Wales, Australia) and 10 mg/kg xylazine (Rompun; Bayer Korea, Seoul, Korea) for anesthesia, the right common carotid artery was exposed. Carotid occlusion was performed basically as described previously (Bonnard and Hagemeyer, 2015). Occlusion of the carotid artery was measured by recording blood flow with a perivascular Flowmeter (model TS420, Transonic Systems Inc., Ithaca, NY, USA). A Perivascular Flowprobe (model MA0.5PSB, Transonic Systems Inc.) was placed on the carotid artery, isolated by dissection, and adjusted to ensure baseline flow between 0.3 and 0.9 mL/s. Vascular injury was induced in mice 1 h after APX-115 administration by applying a filter paper (1 × 1 mm) that had been saturated with 20% FeCl3 proximal to the carotid artery. After 3 min, the filter paper was removed, and the flow was measured. Initial and final data points were collected every 0.5 s, and the time to achieve a stable occlusion was calculated. An occlusion was determined to be stable when the flow was reduced to 0 mL/s for at least 30 s.
Assessment of tail bleeding
Mice were administered APX-115 and anesthetized using the same method used to measure arterial thrombosis. The tail was excised at a fixed diameter of 1.5 mm and the remaining tail was immersed in prewarmed isotonic saline at 37°C. Bleeding was followed visually. The time to stable cessation of bleeding was recorded and determined when recurrent bleeding did not occur within 1 min of the time of cessation.
Statistical analysis
Statistical analysis and sample size calculations were performed using GraphPad Prism software (version 10.0.4.1, GraphPad Software Inc., La Jolla, CA, USA). The effect size was estimated based on preliminary data or similar studies, with a power of 0.8 and a significance level of 0.05 (two-tailed). Statistical significance was calculated using the Student’s t-test for two-group comparisons and one-way or two-way analysis of variance tests for multiple comparisons with Bonferroni correction, respectively. p Values less than 0.05 were considered statistically significant.
Footnotes
Acknowledgments
Figure 1 was designed using ![]() , adhering to their academic license agreement.
, adhering to their academic license agreement.
Authors’ Contributions
J.J.: Conceptualization, methodology, software, formal analysis, data curation, visualization, writing—original draft preparation, and writing—reviewing and editing; H.Y.: Methodology, software, and formal analysis. E.B.O.: Methodology, software, and formal analysis. J.W.P.: Methodology, software, and formal analysis. S.K.: Methodology, software, and formal analysis. T.K.: Methodology, software, and formal analysis. J.S.: Methodology, software, and Formal analysis. B.-R.J.: Methodology, software, and formal analysis. T.-S.C.: Conceptualization, methodology, resources, supervision, project administration, funding acquisition, and writing—reviewing and editing.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2022R1A6A1A03046247) and the Brain Korea 21 Plus Program funded by the Korean government (MOE) in the Republic of Korea.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.