
Abstract
Significance:
In all modern urbanized and industrialized societies, noncommunicable diseases, such as cardiovascular disease (CVD), are becoming a more important cause of morbidity and mortality. Classical risk factors for CVDs, such as hypertension, are reinforced by behavioral risk factors, e.g., smoking and diet, and environmental risk factors, e.g., transportation noise and air pollution.
Recent Advances:
Both transportation noise and air pollution have individually been shown to increase the risk for CVD in large cohorts. Insights from animal studies have revealed pathophysiologic mechanisms by which these stressors influence the cardiovascular system. Noise primarily causes annoyance and sleep disturbance, promoting the release of stress hormones. Air pollution primarily damages the lung, where it causes local inflammation and an increase in oxidative stress, which can propagate to the circulation and remote organs.
Critical Issues:
Both noise and air pollution converge at the vascular level, where the inflammatory state and oxidative stress cause dysfunction in vascular signaling and promote atherosclerotic plaque formation and thrombosis. Both inflammation and oxidative stress are key aspects of traditional cardiovascular risk factors, such as arterial hypertension. The similarities among the mechanisms of environmental risk factor-induced CVD and hypertension indicate that a complex interplay between them can drive the onset and progression of CVDs, leading to synergistic health impacts.
Future Directions:
Our present overview of the negative effects of noise and air pollution on the cardiovascular system provides a mechanistic link to the traditional CVD risk factor, hypertension, which could be used to protect patients with preexisting CVD better. Antioxid. Redox Signal. 42, 827–847.
Introduction
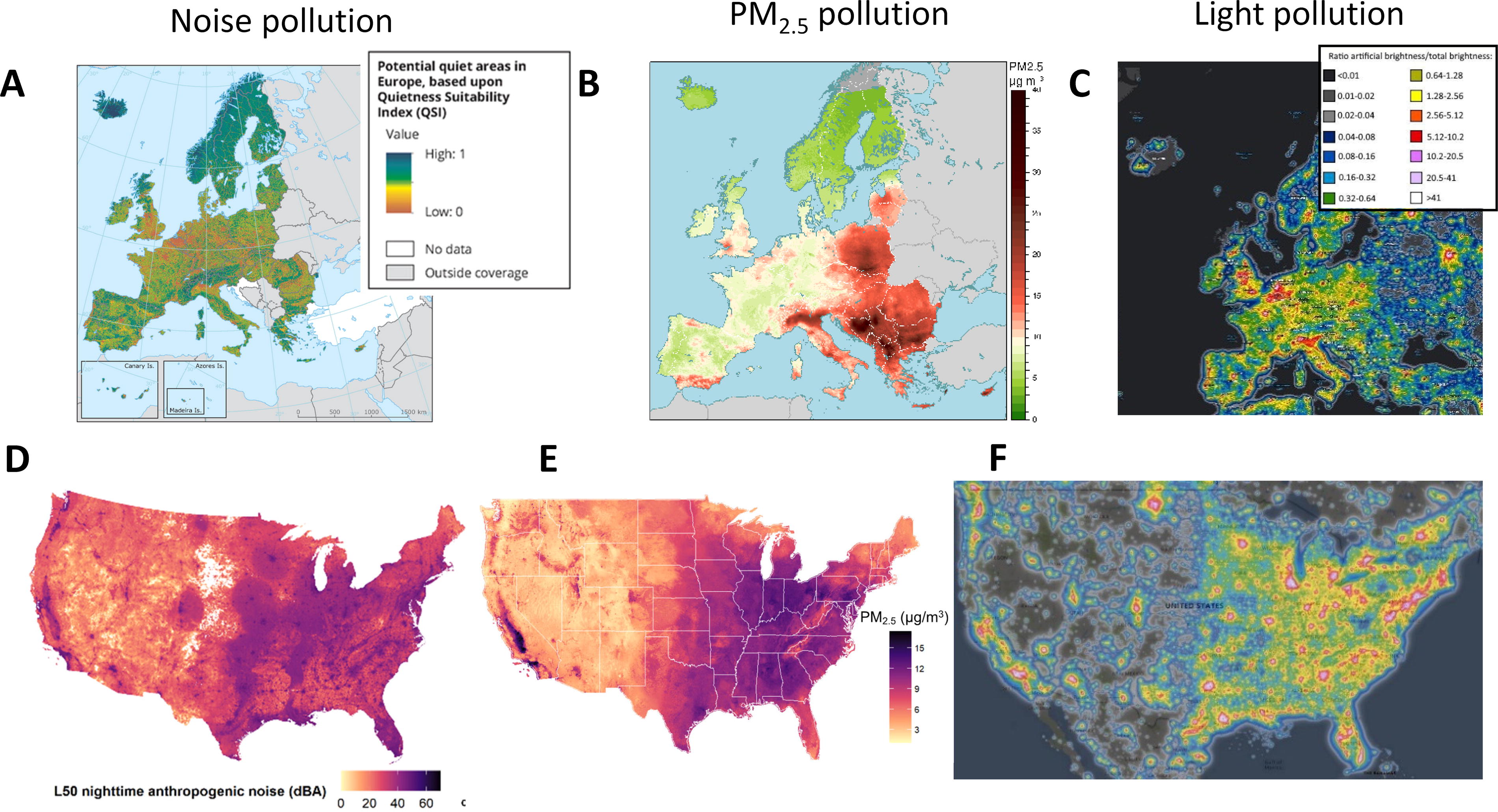
Cardiovascular diseases (CVDs) like ischemic heart disease and stroke continue to be the leading causes of global disability-adjusted life years (GBD 2019 Risk Factors Collaborators, 2020), and hypertension is the leading risk factor for global disability and mortality (GBD 2019 Risk Factors Collaborators, 2020). There is significant interest in understanding environmental and behavioral stressors contributing to hypertension and CVD development. In recent years substantial evidence has emerged, pointing to environmental risk factors, such as noise and air pollution, as major impediments to human health (EEA, 2020 and EEA, 2021). Both noise and air pollution are known to lead to cardiovascular events and increase blood pressure independently (Hahad et al., 2023b), posing the question as to whether these stressors share the same pathophysiologic mechanisms and whether they would be responsive to the same pharmacological interventions used for classical hypertension. Studying the mutual effects of noise and air pollution is essential as they are highly co-localized in urban environments in the USA, and noise/light also in Europe (see Fig. 1). This figure shows how challenging it is for epidemiological studies to discriminate between different environmental risk factors and also highlights the potential for additive health effects in multi-exposure scenarios. Accordingly, studies focusing on only one risk factor are prone to bias toward individual impacts on the disease burden.
This review focuses on the environmental stressors of noise and air pollution (specifically fine particulate matter) and the combination of both. We also emphasize the pathophysiology and incidence of arterial hypertension caused by these stressors. We also highlight the pathophysiologic similarities of developing classical hypertension or noise/air pollution-induced blood pressure increase.
Clinical and epidemiological evidence for cardiovascular damage and induction of hypertension by traffic noise
Environmental noise, derived chiefly from traffic, is a primarily neglected risk factor for CVD (WHO, Munzel et al., 2018b; Munzel et al., 2021; Krittanawong et al., 2023c; Munzel et al., 2024). The World Health Organization (WHO) provides guidelines for the average sound pressure levels that different types of transportation noise (road traffic, rail, and aircraft) should not exceed during daytime and nighttime (WHO, 2018). The 2018 WHO report suggests that over 100 million people in Europe are exposed to noise levels exceeding the recommended values and that traffic noise is associated with an increased risk for ischemic heart disease (Relative Risk (RR) 1.08 95% Confidence Interval [CI]: 1.01–1.15) per 10 dB(A) increase in noise exposure, starting at 53 dB(A) (Kempen et al., 2018). Traffic noise is especially detrimental during nighttime, increasing the risk for acute cardiovascular morbidity and mortality (Munzel et al., 2020b). Arterial stiffness, a subclinical marker of atherosclerosis and a prognostic marker for the development of CVD, was observed to be increased in subjects exposed to railway and road noise (Foraster et al., 2017). Other cardiovascular events, such as stroke, heart failure, and arrhythmias, were also associated with road, railway, and aircraft noise (Munzel et al., 2014a; Munzel et al., 2018b), together with cardiometabolic disorders, such as diabetes mellitus (Munzel et al., 2021). Estimates about the global disease burden due to noise pollution are still unavailable. In the EU, about 95 million people are affected by road traffic noise levels of at least 55 dB(A), which are considered harmful to health. In total, estimates indicate about 11,000 premature deaths and 40,000 new cases of ischemic heart disease due to chronic exposure to transport noise per year (EEA, 2023). Despite the public health burden of noise, the EU is behind on efforts to reduce noise levels substantially. The Zero Pollution Action Plan issued by The European Commission aims to reduce the proportion of individuals chronically disturbed by transportation noise by 30% by 2030 compared with 2017, which is likely to fail to be achieved (EEA, 2022b). Under a so-called optimistic scenario, a decline of about 19% of the number of people highly annoyed by transport noise can be expected by 2030. Under a more conservative scenario, there might be even an increase of 3% rather than a decrease. Effective measures that could contribute to achieving the 2030 target include the reduction of traffic speed limits, redesigning roadways, creating low-noise emission zones, and retrofitting trains with quiet brakes and pads (EEA, 2022a). Reducing transportation noise exposure will demand obligatory collective action and long-term policies at the national and local levels. Given the magnitude of noise exposure, there is great potential for implementing effective measures. Although the data on the socioeconomic status and noise effects is not yet clear, noise exposure was shown to disproportionately impact materially deprived people (Dreger et al., 2019), showing that vulnerable groups have the most need for implementation of noise reduction solutions.
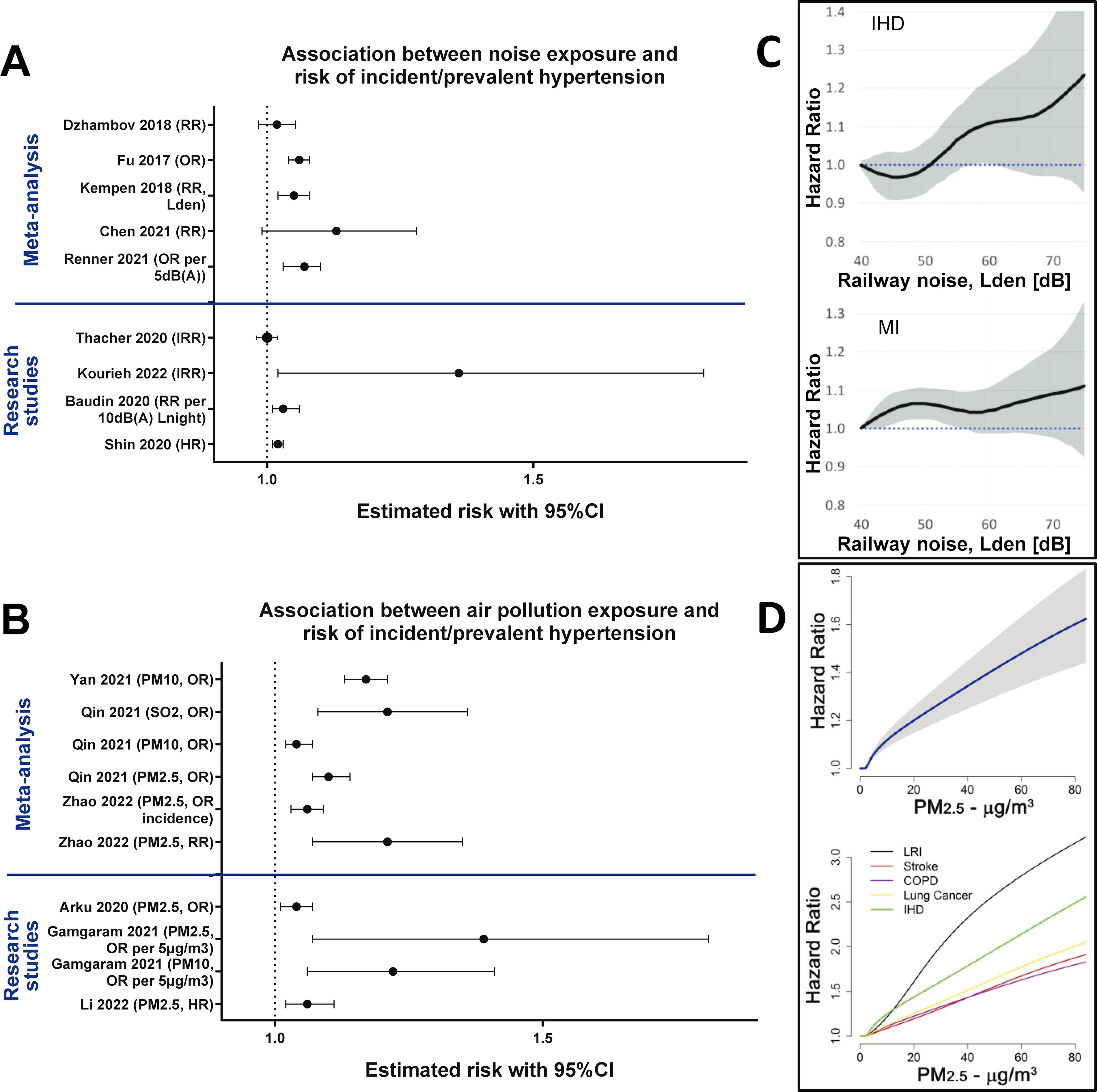
Importantly, noise was also associated with an increased incidence of hypertension (Hahad et al., 2023b), and recently, the European Society of Hypertension has recognized traffic noise as an important risk factor for the development of hypertension by including it together with air pollution in their 2023 report (Mancia et al., 2023). Early data associating noise with hypertension came from the HYENA (Hypertension and Exposure to Noise near Airports) study, where hypertension was significantly associated with nighttime noise (Odds ratio [OR] = 1.14, 95% CI: 1.01–1.29, per 10 dB(A)) (Jarup et al., 2008). Another more recent study, DEBATS (Discussion on the health effects of aircraft noise), also correlated the nighttime aircraft noise exposure to hypertension [OR = 1.34 95% CI: 1.00–1.97, per 10 dB(A)] (Evrard et al., 2017). On the other hand, results of the European study of cohorts for air pollution effects (ESCAPE) found that although road traffic noise was correlated to an increase in self-reported hypertension, the effects were no longer significant when the results were adjusted for air pollution (PM2.5) as a confounder (Fuks et al., 2017). In Europe alone, 1.7 million cases of hypertension and 18,000 excess deaths (from coronary heart disease and stroke) can be attributed to noise pollution (ETC/ACM, 2014). It is estimated that a reduction in noise levels by only 5 dB(A) could help reduce the incidence of hypertension by 1.4% and the incidence of ischemic heart disease by 1.8%, providing a cost saving of up to 3.9 billion USD in health expenses (Swinburn et al., 2015). Interventional studies of nighttime aircraft noise exposure also demonstrated a dose-dependent impairment of endothelial function visible by a reduction in flow-mediated dilation (FMD) (Schmidt et al., 2013). A summary of the association between noise and the risk of development/incidence of hypertension or cardiovascular sequelae (ischemic heart disease, myocardial infarction) is shown in Figure 2A and C.
Clinical and epidemiological evidence for PM-induced CVD and hypertension
Air pollution, in general, has been widely recognized as one of the leading risk factors for cardiovascular and total mortality and morbidity (Krittanawong et al., 2023a). Studies have estimated excess deaths from air pollution, ranging from 4.2 million to 10.2 million (WHO, 2019; Lelieveld et al., 2019; Vohra et al., 2021). The 2019 Global Burden of Disease study ranked air pollution 4th among all risk factors for all-cause morbidity and mortality (GBD 2019 Risk Factors Collaborators, 2020). PM exposure is often estimated from ground monitoring stations or multi-modal computational models, linked with health outcomes in large cohorts (Krittanawong et al., 2023b). Data from the United States Medicare program show that every increment of 10 µg/m3 of PM2.5 exposure is significantly associated with increased risk of all-cause mortality (RR = 1.050; 95% CI: 1.044, 1.056), CVD mortality (RR = 1.088 95% CI: 1.078, 1.098), and respiratory mortality (RR = 1.056; 95% CI: 1.038, 1.074) (Wang et al., 2020). The European ELAPSE (Effects of Low-Level Air Pollution: A Study in Europe) project determined the hazard ratio (HR) for all-cause mortality with smaller increments of PM2.5 concentration of 5 µg/m3 (HR = 1.13, 95% CI: 1.11, 116) (Strak et al., 2021). The ESCAPE study also examined all-cause mortality using multiple European cohorts (HR = 1.14, 95% CI: 1.04, 1.26, for every 10 µg/m3 increase in PM2.5) (Lipfert, 2017). National Institutes of Health-American Association of Retired Persons (AARP) cohort found increased CVD mortality associated with PM2.5 (HR = 1.10; 95% CI: 1.05, 1.15, for every 10 µg/m3) (Thurston et al., 2016). A smaller cohort found a 12% decrease in reactive hyperaemia index, a measurement of endothelial function, for every 10 µg/m3 increase in PM2.5 concentration (95% CI: 21.0, −2.7) (Riggs et al., 2020). Air pollution was also observed to disproportionately affect people of lower socio-economic status, as the communities preferentially inhabited by low-income individuals experienced higher total exposure to PM2.5 levels in comparison to high-income communities (Jbaily et al., 2022), and reduction in PM2.5 exposure was shown to disproportionately benefit the lower-income communities, lowering the all-cause mortality HR (Josey et al., 2023).
In addition to the cohort studies, interventional studies have shed light on the causal effects of PM in humans. In a study on 20 healthy volunteers, exposure to a low concentration of ambient PM2.5 was associated with an increase in serum amyloid A, C-reactive protein (CRP), soluble intercellular adhesion molecule-1 (sICAM-1), and soluble vascular cell adhesion protein-1 (sVCAM-1), pointing to a pro-inflammatory state (Wyatt et al., 2020). Another controlled study of 60 volunteers showed that exposure to low concentrations of PM2.5 worsened nitroglycerine-induced vasorelaxation, but changes in redox or inflammatory markers were not observed (Hemmingsen et al., 2015). Higher PM exposure at a mass concentration of 150 µg/m3 was shown to cause significant brachial artery vasoconstriction in a study on 25 healthy subjects (Brook et al., 2002). Studies on exposure to diesel PM were also performed in humans. Acute exposure to 300 µg/m3 of diesel PM reduced acetylcholine and bradykinin-induced forearm vasodilatation (Tornqvist et al., 2007). Other studies showed that diesel PM exposure causes elevated heart rate, endothelial dysfunction, and thrombus formation (Lucking et al., 2008; Langrish et al., 2009; Mills et al., 2011). Interventional studies featuring mitigation strategies like air filtration systems showed a reduction in both systolic and diastolic blood pressure (Faridi et al., 2023; Morishita et al., 2018), and physical activity showed highly varied results depending on the air pollution levels and exercise intensity (Hahad et al., 2023a; Krittanawong et al., 2023d; Park et al., 2023; Sun et al., 2023). A summary of the association between air pollution and the risk of development/incidence of hypertension or cardiovascular mortality (ischemic heart disease, stroke) is shown in Figure 2B and D.
Pathophysiological Mechanisms of Environmental Stressors
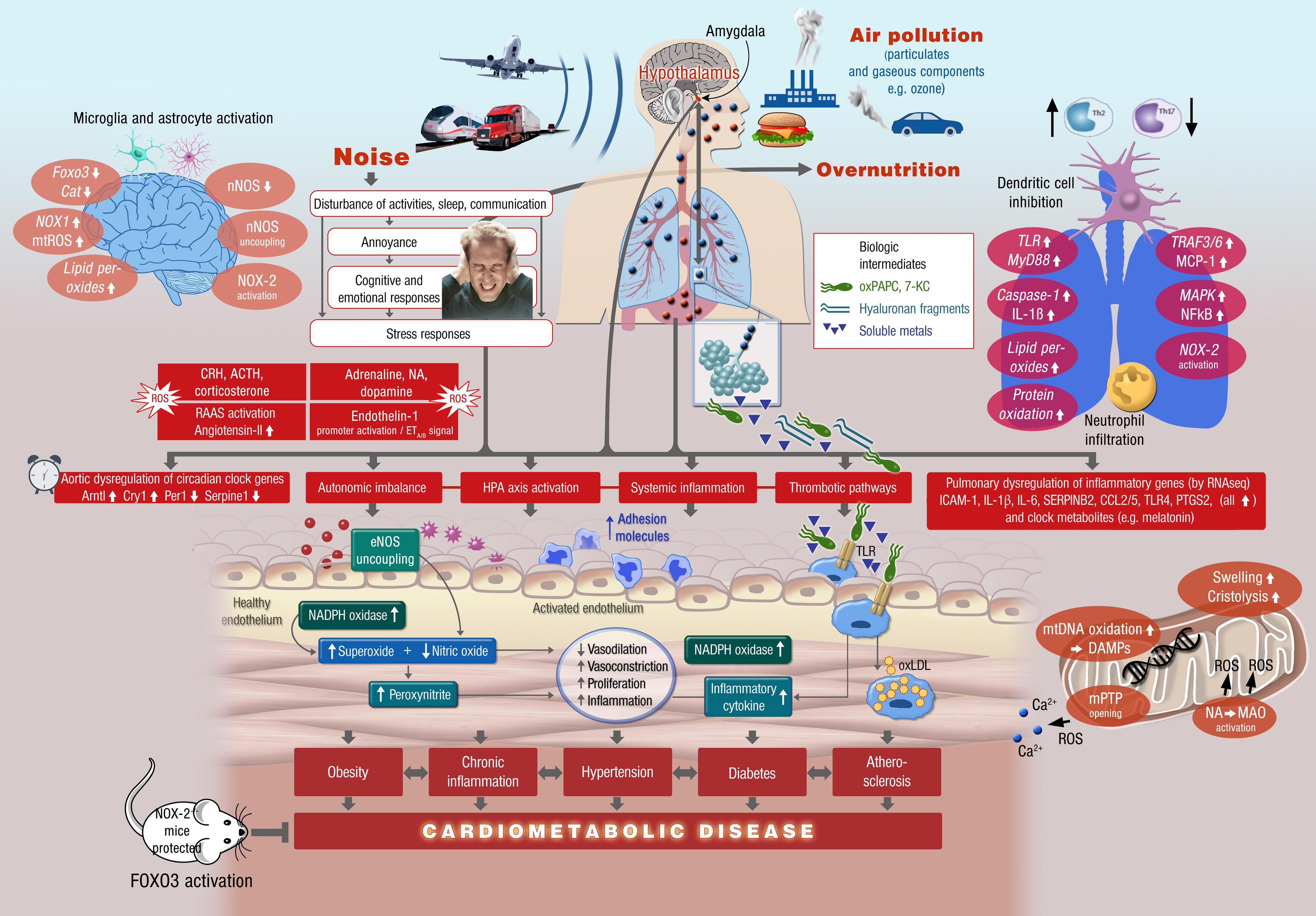
Noise and PM have different “points of entry” into the human body and thus have different initiating mechanisms. Noise is primarily a psychological stressor, that exerts its effects predominantly through the nervous system (Babisch, 2002; Hahad et al., 2022; Munzel et al., 2021; van Kamp et al., 2020). All downstream complications arising from noise exposure start as neuronal arousals that can be categorized into annoyance, sleep disturbance, and general psychological stress. PM and air pollution in general exert their negative effects when internalized into the human body through inhalation or direct uptake of (ultra)fine particles into the brain or circulation (Al-Kindi et al., 2020; Munzel et al., 2018a; Miller and Newby, 2020; Rajagopalan et al., 2018). However, skin contact can be important as well (Fussell and Kelly, 2020; Magnani et al., 2016). Inhaled PM can elicit local to systemic inflammatory reactions (Arias-Perez et al., 2020). Although these different stress mechanisms of noise and PM pollution converge at the cardiovascular system level, they should first be considered individually. Individual and combined effects of aircraft noise and particulate matter (mostly fine PM) exposure were recently characterized, highlighting the brain as the primary target organ of noise and the lung for PM, with substantial additive damage in the vasculature by co-exposure (Kuntic et al., 2023).
Mechanism of noise-induced cardiovascular and cerebral damage
Conceptually, the effects of noise can be separated into the “direct” and the “indirect” pathway (Babisch, 2003). The direct pathway deals with intense sound pressure exposure that results in inner ear (cochlear) damage and hearing loss, whereas the indirect pathway is more nuanced and manifests itself as annoyance, sleep disruption, and general stress. The indirect pathway is characterized by a release of stress hormones that follow from the activation of the pituitary-adrenal-cortical axis (e.g., cortisol) and the sympathetic-adrenal-medullary axis (e.g., catecholamines) (Babisch, 2002). Confirmation of this pathway came from the observation of the activation of the amygdala and the glutamatergic signaling in the brains of rats acutely exposed to noise (Singewald et al., 2000) and by observing a general cognitive decline in the chronically exposed rats (Jafari et al., 2018). Stress hormones, such as epinephrine, norepinephrine, and cortisol (catecholamines and steroid hormones in general), are often found to be upregulated in noise exposure studies (Daiber et al., 2019). Prolonged persistence of this phenotype can increase immune system activation, disrupt the circadian clock, induce vascular dysfunction, and lead to CVD (Babisch et al., 2013; Babisch et al., 2003; Kroller-Schon et al., 2018; Munzel et al., 2017a; Recio et al., 2016; Babisch, 2014). Field studies of human exposure to aircraft and train noise showed that nighttime exposure worsens sleep quality and increases levels of stress hormones and blood pressure (Herzog et al., 2019; Schmidt et al., 2013). This was also associated with decreased FMD, a surrogate measurement for endothelial function. The patients with preexisting coronary artery disease had a more pronounced endothelial dysfunction after nighttime noise exposure (Munzel et al., 2018b; Schmidt et al., 2015). Acute administration of vitamin C partially improved endothelial function, suggesting oxidative stress’s involvement in noise-induced endothelial dysfunction (Herzog et al., 2019; Schmidt et al., 2013).
A recent study established a mechanistic link between transportation noise exposure and vascular inflammation (Osborne et al., 2020). Studies have shown that noise leads to increased amygdala activity, resulting in the increase of major adverse cardiovascular events (MACE) [HR = 1.341, 95% CI: 1.147–1.567, per 5 dB(A)]. Activation of the amygdala by noise is an important finding, as it is a part of the limbic system and is involved in perception and response to stress (Muscatell et al., 2015). In addition, higher stress resilience is associated with a lower amygdala activation and a lower rate of vascular inflammation and MACE (Dar et al., 2020). Transportation noise, especially nighttime noise, is known to fragment sleep, disrupt the circadian rhythm, and increase the release of stress hormones (Cappuccio et al., 2011). Stress hormone release can be directly linked to vascular dysfunction, as activation of the sympathetic nervous system can further activate the renin-angiotensin-aldosterone system and initiate catecholamine release (Lob et al., 2010; Ye et al., 2006). Catecholamines can promote the monoamine oxidase activity or activate microglia, generating more reactive oxygen species (ROS 1 ) through NADPH oxidase (Lee et al., 2017; Neri et al., 2007). Angiotensin-II and endothelin-1, both potent vasoconstrictors, can directly mediate the activation of NADPH oxidase, feeding the oxidative stress/vascular dysfunction loop (Chen et al., 2012; Kroller-Schon et al., 2018; Munzel et al., 2017a; Wright et al., 1985).
Mechanisms of PM-induced cardiovascular damage
PM enters the human body mainly through inhalation, making the lung the entry point. The severity of the health effects of PM may be attributed to their size, as smaller particles like PM2.5 and ultrafine particles (< 0.1 µm) can penetrate deeper into the respiratory tract and even translocate into circulation (Miller et al., 2017). Ultrafine particles were also observed to be present in the brain where they came either by direct uptake through the olfactory nerve (Hahad et al., 2020), or through circulation after penetrating the blood-brain-barrier (Daiber et al., 2020b). Ultrafine PM can even enter individual cells where they can cause damage to the mitochondria, increasing mitochondrial ROS production (Daiber et al., 2020b). Although ultrafine PM can cause detrimental effects by directly damaging even subcellular compartments, many toxic PM effects are caused by larger particles that remain in the alveolar space (Pinkerton et al., 2000). Chronic exposure can result in the accumulation of PM in the lungs, promoting inflammatory reactions. This local inflammation in the lung then causes systemic stress through activated macrophages and granulocytes that either enter circulation directly or release pro-inflammatory cytokines or damage-associated molecular patterns (Munzel et al., 2018a; Rao et al., 2018). PM in the lungs can also stimulate mechano-receptors, causing the lung arc reflex and the activation of the sympathetic nervous system, further releasing catecholamines (Hajat et al., 2019). Sensory neurons in the airway can be triggered by environmental toxicants, specifically aerogenic oxidants, resulting in the onset of neurogenic inflammation (Simon and Liedtke, 2008).
Although PM can carry ROS on its surface (such as semiquinone radicals and •O2 −) and directly introduce it to the organism (Dellinger et al., 2001), oxidative stress in response to PM is primarily due to the respiratory defense mechanisms (Munzel et al., 2018a; Rao et al., 2018). The whole array of possible toxic chemicals carried by the PM, such as transition metals, primary and secondary organic compounds, endotoxins, and reactive quinones/aldehydes, can lead to oxidative stress induction in the lung and circulation (Poschl and Shiraiwa, 2015). These secondary toxicants can also deplete antioxidants in the lung lining fluid, such as ascorbate, glutathione, and tocopherol (Poschl and Shiraiwa, 2015). Oxidative stress derived in this manner is a potent mechanism for vascular dysfunction in the pulmonary and other blood vessels.
Overlap of the noise and PM mechanisms of cardiovascular damage
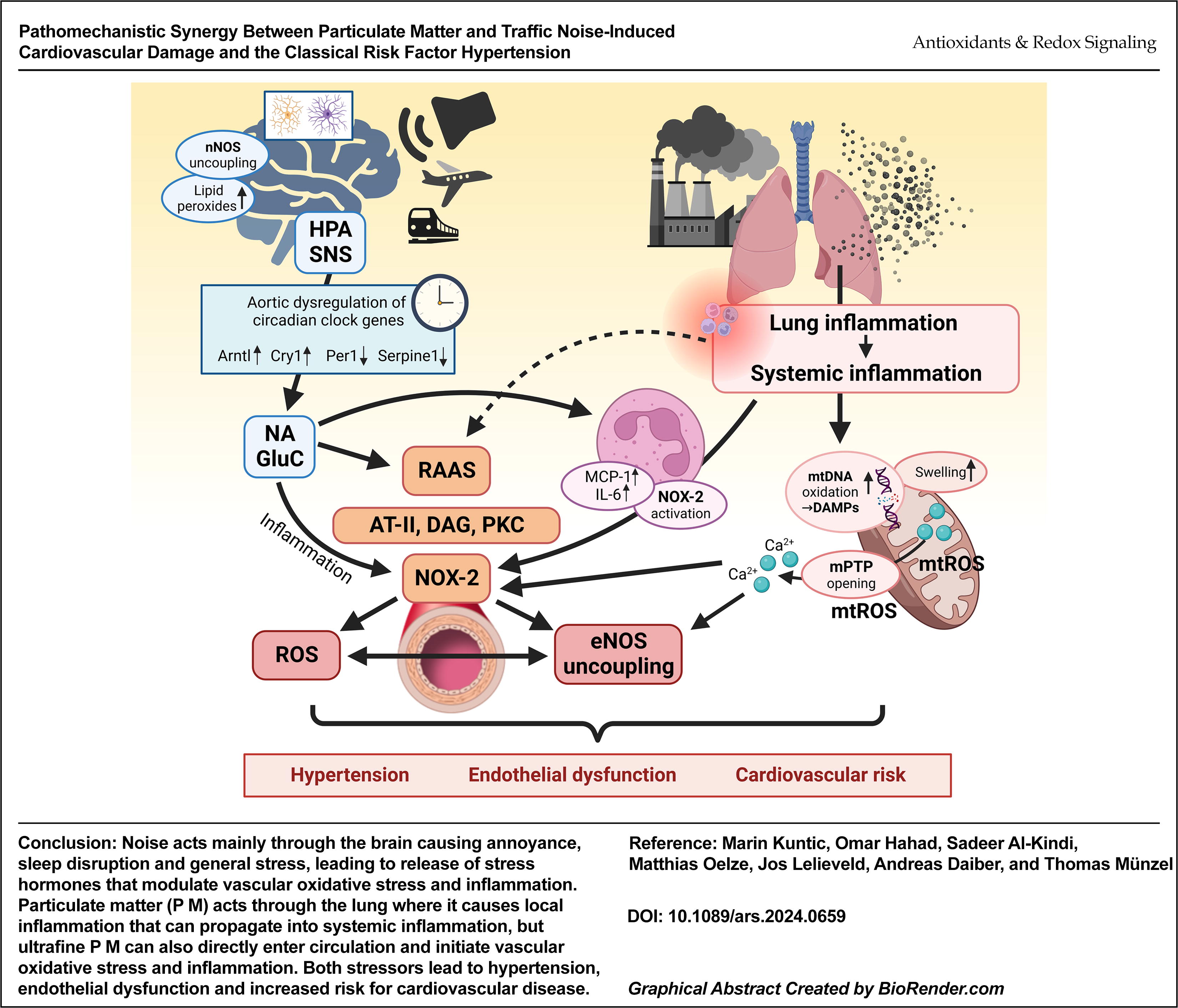
The overlap of the noise- and PM-induced mechanisms of cardiovascular damage are shown in Figure 3, and explained here in brief. Noise causes primary damage in the brain by neuronal activation (e.g., of the amygdala), cerebral oxidative stress, and neuroinflammation, which is then further propagated via stress response pathways by activation of the hypothalamic–pituitary–adrenal (HPA) axis and cortisol (human) or corticosterone (mouse) synthesis as well as sympathetic nervous system (SNS) activation and catecholamine release (e.g., adrenaline or noradrenaline [NA]) (Munzel et al., 2021). Stress responses may subsequently activate secondary endocrinal stress hormones such as angiotensin-II and endothelin-1 (Munzel et al., 2017a). Particulate matter causes primary damage in the lung by pulmonary inflammation via Toll-like receptor (TLR) activation and enhanced NFkB, TRAF, and MyD88/MCP-1 signaling (Kampfrath et al., 2011), leading to impaired dendritic cell function, neutrophil activation and infiltration as well as adverse regulation of T helper cells. Lung inflammation and oxidative stress will lead to the release of cytokines such as interleukin 1β (IL-1β) or MCP-1 as well as damage-associated molecular patterns (DAMPs) to the circulation, e.g., 7-ketocholesterol (7-KC) and oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (oxPAPC) (Kampfrath et al., 2011; Rao et al., 2014). In addition, ultrafine particles (nanometer scale) and soluble toxins from the particles such as heavy metals can directly penetrate the lung and reach the circulation (Al-Kindi et al., 2020; Munzel et al., 2018a). Ultrafine particles are also known to cause mitochondrial damage by mtDNA oxidation, swelling, and cristolysis, mitochondrial ROS formation by, e.g., monoamine oxidase (MAO) activation by noradrenaline (NA) and mitochondrial DAMPs, ROS, and calcium are released to the cytosol upon mitochondrial permeability transition pore (mPTP) opening (Daiber et al., 2020b). All of the adverse effects of noise and particulate matter will lead to dysregulation of circadian rhythms, autonomic imbalance, systemic inflammation, and activation of thrombotic pathways (Al-Kindi et al., 2020; Daiber et al., 2022; Munzel et al., 2018a). These pathophysiological pathways converge at the vascular level and cause vascular oxidative stress, immune cell adhesion and infiltration, endothelial nitric oxide synthase (eNOS) uncoupling, and NADPH oxidase activation, leading to enhanced vasoconstriction, impaired endothelial function, and increased proliferation. This will promote cardiometabolic risk factors such as obesity, diabetes, chronic inflammation, atherosclerosis and hypertension, leading to cardiometabolic disease. As a protective measure against noise and particulate matter genetic deletion of the phagocytic NADPH oxidase (NOX-2) was identified (Kroller-Schon et al., 2018; Kampfrath et al., 2011). Pharmacological Forkhead box proteins O3 (FOXO3) activation proved effective against noise-induced damage (Kroller-Schon et al., 2018), and genetic interference with inflammatory signaling (e.g., ablation of lysozyme M (LysM)-positive cells or deletion of TLR4) proved effective against most of the noise (Frenis et al., 2021a) and particulate matter-mediated complications (Kampfrath et al., 2011). Importantly, in a mouse study the effects of combined noise and particulate matter exposure seemed to be additive at the level of the circulation and the vasculature (Kuntic et al., 2023).
Mechanistic Similarities between Classical Arterial Hypertension and Noise and/or Particulate Matter-Induced Cardiovascular Complications
Oxidative stress in the cardiovascular pathology
Oxidative stress is a state of imbalance between the production of reactive oxygen and nitrogen species (RONS) and their scavenging. This shift in redox balance is a hallmark of many cardiovascular and cerebrovascular diseases (Sies, 1991; Sies, 2015), a side effect of many drug therapies (Munzel et al., 2005; Munzel et al., 2014b), and is associated with environmental risk factors, such as transportation noise and air pollution (Araujo et al., 2008; Munzel et al., 2017b, Munzel et al., 2017c; Munzel et al., 2014a). The essential signaling cascades in vascular tissue is nitric oxide (•NO)-mediated signaling, as •NO plays a role in the regulation of vascular tone and suppression of thrombosis through the control of platelet activation (Sies, 2017; Stamler, 1994; Rhee, 1999; Ullrich and Kissner, 2006). Vascular •NO signaling is susceptible to interference from the superoxide radical (•O2 −). The reaction between •O2 − and •NO is kinetically favorable and therefore competes with the detoxification of •O2 − by superoxide dismutase (Beckman and Koppenol, 1996; Kissner et al., 1997) and leads to the formation of peroxynitrite (ONOO-) (Beckman and Koppenol, 1996). ONOO- can nitrate protein tyrosine residues (3-nitrotyrosine), which leads to inactivation of certain enzymes, like prostacyclin synthase (Zou et al., 1999; Hink et al., 2003; Zou and Bachschmid, 1999), and was previously observed in atherosclerotic plaques and in other pathophysiological settings (Beckmann et al., 1994; Crow and Beckman, 1995; Daiber et al., 2021; Ischiropoulos and Beckman, 2003).
Improvement of vascular function after administration of vitamin C demonstrated the importance of oxidative stress in CVD (Daiber et al., 2017b; Herzog et al., 2019; Heitzer et al., 1996, Heitzer et al., 2001; Schmidt et al., 2013), but a more comprehensive insight came from the studies in genetically modified animals where genes coding for •O2 −-generating enzymes were deleted (Daiber and Chlopicki, 2020; Daiber et al., 2017a; Daiber et al., 2020c). In mice lacking the NADPH oxidase subunits p47phox or gp91phox vascular oxidative stress and endothelial function were improved after noise-, angiotensin II, or nitroglycerin-induced vascular oxidative stress (Landmesser et al., 2002; Matsuno et al., 2005; Kroller-Schon et al., 2018; Wenzel et al., 2008). The cardiovascular system is also susceptible to the concept of “kindling radicals” (“bonfire” hypothesis), where any formation of ROS leads to further generation of ROS through different mechanisms by activating “redox switches” (Daiber et al., 2017a; Frenis et al., 2021b). For example, mitochondrial H2O2 can drive the activation of NADPH oxidase complex through activation of the protein kinase C (Lin and Takemoto, 2005), and uncoupling of eNOS by ROS results in the production of •O2 − rather than •NO (Kuhn et al., 2017; Leo et al., 2021), fueling the cycle of ROS formation (Kroller-Schon et al., 2014; Dikalov, 2011). Xanthine oxidase is another example of a secondary source of ROS triggered by the activation of NADPH oxidase (Daiber et al., 2017a; Schulz et al., 2014).
Role of oxidative stress and inflammation in models of classical hypertension
Traditional risk factors that are associated with vascular dysfunction, such as hypertension (Mollnau et al., 2002), diabetes mellitus (Hink et al., 2001), hypercholesterolemia (Oelze et al., 2000), and smoking (Munzel et al., 2020a), are accompanied by a similar increase in inflammation and oxidative stress as observed in noise and air pollution exposure models, raising the question of whether these pollutants can have an additive effect on the existing “traditional” risk factors or if they can contribute to their onset. Indeed, both aircraft noise and air pollution exacerbated the cardiovascular damage by hypertension in mouse models (Nemmar et al., 2011; Steven et al., 2020). The common mechanisms of cardiovascular injury caused by noise and air pollution and established hypertension, derived from animal studies, are presented in Figure 4.
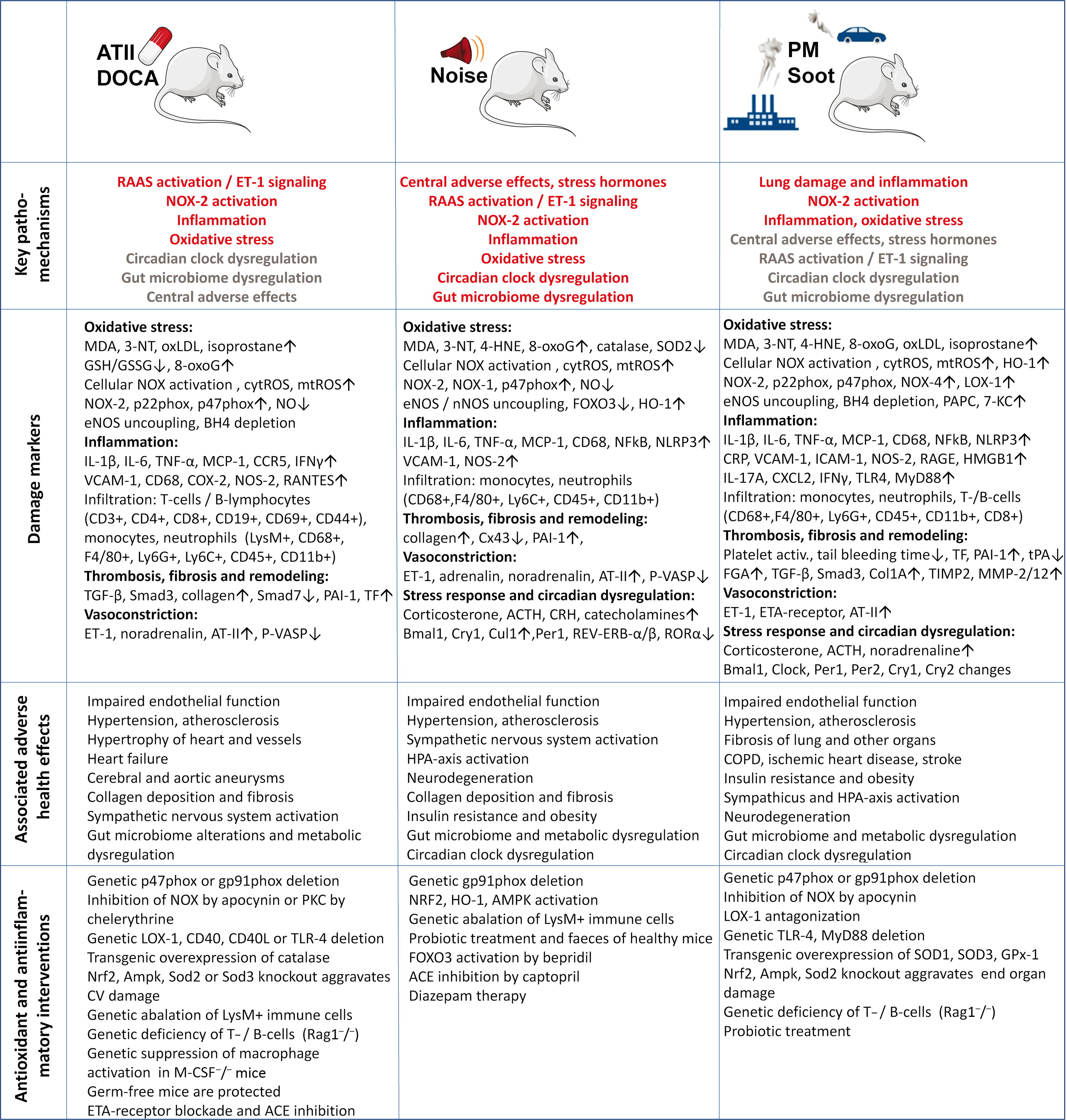
There is substantial animal experimental evidence that oxidative stress triggers and promotes arterial hypertension, as shown in mouse models of angiotensin-II or deoxycorticosterone acetate (DOCA) salt-induced hypertension (Landmesser et al., 2003; Rajagopalan et al., 1996). NOX-2 plays a central role in the underlying mechanisms since the significant cardiovascular complications of arterial hypertension, can be prevented by genetic deletion of the p47phox or gp91phox subunit of NOX-2 (Landmesser et al., 2003; Landmesser et al., 2002). There is also clear evidence for a crosstalk between mitochondrial ROS formation and NADPH oxidases (NOX-1 and NOX-2) in arterial hypertension (Kroller-Schon et al., 2014, Dikalov, 2011). This crosstalk also explains the observation that the inhibition of only one enzymatic source of ROS is sufficient to normalize vascular function in various CVD settings completely (Daiber et al., 2017a; Daiber and Chlopicki, 2020). Vasoconstrictors, such as endothelin-1, are directly linked to the NADPH oxidase redox regulatory system, which increases vascular •O2 − formation (Li et al., 2003b; Chen et al., 2012; Duerrschmidt et al., 2000) and inflammatory processes (Li et al., 2003a; Javeshghani et al., 2013) in different models of hypertension. Conversely, NADPH oxidase-derived •O2 − formation amplifies the endothelin-1-dependent signaling and vasoconstriction (Li et al., 2003c; Kahler et al., 2001; Kahler et al., 2000). Endothelin-1 also shares several cross-activation mechanisms with the renin-angiotensin-aldosterone system (Rajagopalan et al., 1997; Tran et al., 2009) and the mitochondria (Wenzel et al., 2017).
Previous reports established a crucial role of inflammatory cells, mainly monocytes/macrophages (Wenzel et al., 2011; De Ciuceis et al., 2005) as well as T-cell/lymphocyte dyad (Guzik et al., 2007; Barhoumi et al., 2011), for angiotensin-II-induced arterial hypertension and associated cardiovascular complications such as endothelial dysfunction, oxidative stress and increased NOX-2 activity, since immune cell deficient mice, e.g., LysMCreiDTR plus diphtheria toxin (Wenzel et al., 2011) and recombination activating gene 1 (Rag1)−/− (Guzik et al., 2007) genetic models of ablation, were protected. Therefore, it is tempting to speculate that, e.g., the beneficial effects of pharmacological inhibition of endothelin-1 receptors (Rajagopalan et al., 1997) were successful because of the anti-inflammatory effects. Moreover, redox activation of immune cells by mitochondrial •O2 −/hydrogen peroxide formation with subsequent activation of NOX-2 was proposed as a central mechanism for the development and progression of inflammation (Dikalov et al., 2012; Kroller-Schon et al., 2014). NOX-2 activation seems to be indispensable for the activation, recruitment, and infiltration of myelomonocytic cells (De Ciuceis et al., 2005; Wenzel et al., 2011) and T-cells/lymphocytes (Guzik et al., 2007; Barhoumi et al., 2011), since adoptive cell transfer of immune cells from gp91phox −/− or p47phox −/− mice to the LysM-iDTR or RAG1−/− mice did not lead to the typical symptoms of hypertension, whereas adoptive cell transfer of immune cells from wildtype mice restored all adverse effects of angiotensin-II infusion (Guzik et al., 2007; Wenzel et al., 2011).
The impact of NOX-2 derived •O2 − can be best explained by the fact that the migration of monocytes/recruitment of macrophages is controlled by the cellular redox potential (Qiao et al., 2009), involving redox modifications of mitogen-activated protein kinase (MAPK) phosphatase 1 (Kim et al., 2012) and changes in the S-glutathionylation pattern (Ullevig et al., 2013). Angiotensin-II also activates thrombo-inflammation by enhanced platelet reactivity and impaired coagulation via tissue factor signaling (Kossmann et al., 2017). Importantly, peripheral and central mechanisms act synergistically on T-lymphocyte activation and the development of angiotensin-II-induced arterial hypertension (Lob et al., 2010; Lob et al., 2013; Marvar et al., 2010).
Induction of oxidative stress and inflammation by noise exposure
Few studies on animals have examined nonauditory effects of noise on the cardiovascular and cerebral system. Although chronic noise exposure studies in rats (Altura et al., 1992) and monkeys (Peterson et al., 1981) have observed an increase in blood pressure, the sound pressure level used in these preclinical studies is often high, exceeding 100 dB(A). White noise at the sound pressure levels of ≥100 dB(A) was demonstrated to increase oxidative DNA damage in the cardiovascular system and adrenal glands, pointing to the possibility of direct auditory damage (Frenzilli et al., 2004; Lenzi et al., 2003). Our own study in mice showed that even exposure to lower sound pressure levels [aircraft noise with a mean sound pressure level of 72 and peak level of 85 dB(A)] can have detrimental effects on the cardiovascular system (Munzel et al., 2017a). After acute noise exposure, mice developed endothelial dysfunction and increased blood pressure, accompanied by increased vascular and cerebral oxidative stress and inflammation. Notably, the detrimental effects of aircraft noise were not observed in mice exposed to the same sound pressure levels of white noise. Transcriptome analysis pointed to changes in the regulation of vascular function, circadian rhythm, vascular remodeling, and cell death. In a subsequent study, NADPH oxidase was identified as the primary source of vascular ROS in response to noise, as Nox2 knockout mice showed protection against aircraft noise-induced vascular dysfunction and oxidative stress (Kroller-Schon et al., 2018). As NOX-2 is the phagocytic isoform of NADPH oxidase, it is clear that inflammation plays a significant role in the detrimental effects of aircraft noise on the vasculature.
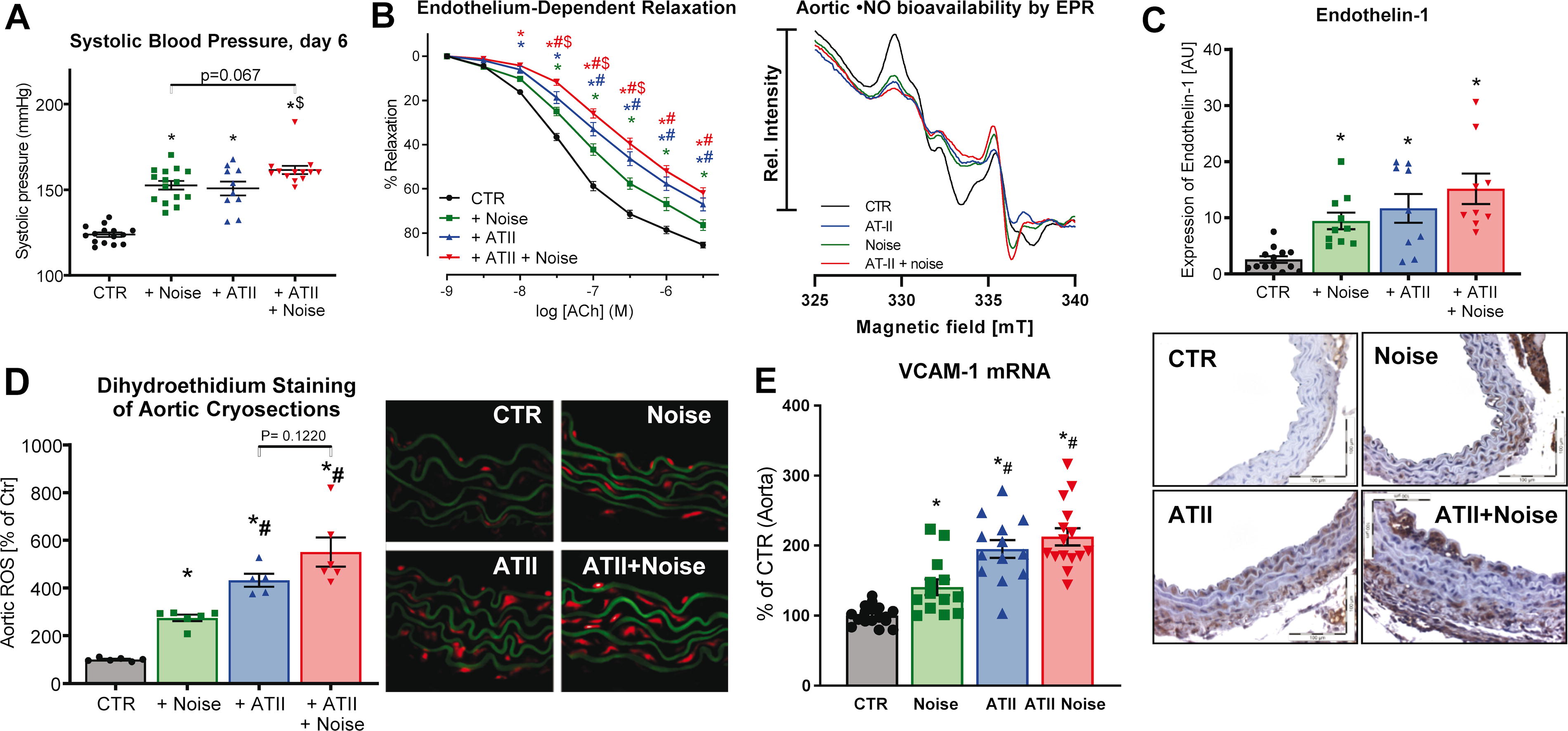
As discussed above, the adverse health effects of noise in animals can be assigned to the activation of the HPA axis and the sympathetic nervous system as measured by increased levels of corticosterone, adrenaline, and noradrenaline in noise-exposed mice (Munzel et al., 2017a) and rats (Gannouni et al., 2013). Activation of these stress response systems accounts for the adverse cardiovascular outcomes detected in noise-exposed animals, including increases in blood pressure in monkeys and endothelial dysfunction, oxidative stress, and inflammation in mice (Peterson et al., 1981; Munzel et al., 2017a; Kroller-Schon et al., 2018). We have even shown that noise exposure exacerbates these cardiovascular complications in a mouse model of preexisting angiotensin-II-induced hypertension, e.g., as shown by the additive increase of blood pressure and vasoconstrictors, impairment of endothelial function and •NO bioavailability as well as markers of inflammation and oxidative stress (Fig. 5; Steven et al., 2020). Like in arterial hypertension, a central role of oxidative stress for noise-triggered pathophysiology is also supported by oxidative stress markers [e.g., 3-nitrotyrosine-, malondialdehyde (MDA)- or 4-hydroxynonenal-positive proteins, uncoupled eNOS] as well as higher formation rates of ROS in noise-exposed animals (Kroller-Schon et al., 2018; Munzel et al., 2017a; Steven et al., 2020). The central role of NOX-2 became evident by increased protein and gene expression in noise-exposed mice (Munzel et al., 2017a). Also, more pronounced activation mechanisms of NOX-2 by noise, such as angiotensin-II dependent diacylglycerol-mediated protein kinase C activation with subsequent phosphorylation of the significant cytosolic regulator of NOX-2, p47phox, at serine 328, support a key mechanism centered around NOX-2 (Kroller-Schon et al., 2018). In addition, NOX-2 inhibition by GSK2795039 suppressed ROS signals in cerebral cryosections of noise-exposed mice (Kroller-Schon et al., 2018).
A pro-oxidative phenotype was also revealed by RNA sequencing data indicating down-regulation of genes encoding for antioxidant defense proteins such as superoxide dismutase 1 and glutathione peroxidase 1 (GPx1) as well as antioxidant transcription factors such as forkhead box proteins O3 (FOXO) (Munzel et al., 2017a). The noise-induced oxidative stress leads to secondary damage, such as the uncoupling of eNOS and neuronal NOS (nNOS) (Daiber et al., 2020a). Direct scavenging of •NO by the diffusion-controlled reaction with •O2 − to form ONOO- in noise-exposed mice leads to impaired endothelial function as well as anti-aggregatory and anti-adhesive properties of the endothelium, also envisaged by enhanced tyrosine nitration (Munzel et al., 2017a). We also found induction of endothelin-1 expression in the aorta of noise-exposed mice and exacerbation of endothelin-receptor signaling as envisaged by more pronounced endothelin-1 dependent vasoconstriction (Kroller-Schon et al., 2018; Munzel et al., 2017a). In support of a critical link between oxidative stress and the onset of adverse cardiovascular effects of noise, induction of the antioxidant principle nuclear factor E2-related factor-2 (Nrf2)/heme oxygenase 1 (HO-1) axis was highly protective in noise-exposed mice (Bayo Jimenez et al., 2021).
The important mechanistic role of NOX-2 was further supported by noise-triggered infiltration of macrophages/monocytes into the vascular wall (Munzel et al., 2017a; Steven et al., 2020), which is per se promoted by oxidative stress conditions as explained for the setting of arterial hypertension in the preceding section. In general, it may be assumed that noise-induced ROS formation promotes an inflammatory phenotype in the heart, vessels, and the brain as central mediators of inflammatory reactions (e.g., by redox activation of NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome, high-mobility group box 1 protein (HMGB1), and transcription factors such as nuclear factor kappa B (NF-κB) (Wenzel et al., 2017). Our group also provided molecular proof that the phagocytic NOX-2 in LysM-positive inflammatory cells (most probably monocytes and macrophages) is responsible for the adverse cardiovascular effects of noise. Genetic ablation of these LysM-positive cells by diphtheria toxin treatment of mice with transgenic LysM-specific diphtheria toxin receptor expression (LysMCreiDTR), prevented noise-induced vascular oxidative stress, inflammation, endothelial dysfunction and increase in blood pressure (Frenis et al., 2021a). Untargeted plasma proteome analysis supported a pro-inflammatory phenotype in noise-exposed mice that was associated with enhanced interaction of leukocytes with the endothelium and overall microvascular dysfunction, which was all corrected by genetic deletion of Nox2 (Eckrich et al., 2021). Neutrophils, monocytes, and macrophages are constitutive expressors of NOX-2, explaining why genetic Nox2 deletion alters the function of these cells.
The overall pro-inflammatory phenotype and increased infiltration of immune cells into tissues by noise are also reflected by numerous markers of inflammation that are upregulated in noise-exposed mice, e.g., circulating cytokines such as IL-6, IL-1β monocyte chemoattractant protein-1 (MCP-1, CCL2), transcription factor NF-κB, adhesion molecule VCAM-1, aortic inducible nitric oxide synthase (iNOS), and cerebral cluster of differentiation (CD) 68 (CD68 or macrosialin) (Frenis et al., 2021a; Kroller-Schon et al., 2018; Munzel et al., 2017a; Steven et al., 2020). Likewise, experimental myocardial infarction ischemic heart and vascular damage were also aggravated by preceding noise exposure by a “priming” of heart and vessel toward an inflammatory phenotype (Molitor et al., 2023). These animal data were also in accordance with the observed shift to a pro-atherothrombotic phenotype of the plasma proteome of train noise-exposed healthy human subjects (Herzog et al., 2019), epigenetic changes that promote immune cell activation and expression of CRP in a large population (Eze et al., 2020; Cai et al., 2017) and amygdala activation-driven coronary atherosclerosis (Osborne et al., 2020). Noise-mediated inflammation in mice was also prevented by genetic Nox2 deletion, as shown by two independent preclinical studies (Eckrich et al., 2021; Kroller-Schon et al., 2018) and antioxidant pharmacological activation/induction of the NRF-2-HO-1-axis (Bayo Jimenez et al., 2021), further supporting the link between oxidative stress and inflammation. With the LysMCreiDTR model, we were also able to show that ablation of peripheral LysM+ immune cells is sufficient to prevent the noise-induced blood pressure increase and peripheral endothelial dysfunction, inflammation, and oxidative stress. In contrast, neuroinflammation and stress response were unaltered in this model because microglia, the major inflammatory cells in the brain, were not ablated due to low LysM expression (Frenis et al., 2021a). Significantly, inflammation and oxidative stress in noise-exposed mice were suppressed by non-pharmacological interventions, physical exercise and intermittent fasting, or pharmacological activation of the AMP-activated kinase (AMPK) (Kvandova et al., 2023). A summary of the mechanistic features of noise-induced cardiovascular damage and hypertension in animal models is provided in Figures 3 and 4. The role of oxidative stress in noise stress was reviewed in full detail, also highlighting the involved ROS sources and inflicted oxidative damage (Sorensen et al., 2024).
Induction of oxidative stress and inflammation by particulate matter exposure
Numerous studies have demonstrated the presence of oxidative stress and modification of RONS pathways in animal models of PM exposure. In general, it was shown that antioxidant therapies or inhibition of ROS sources in PM-exposed animals have beneficial effects on blood pressure, endothelial function, bioavailability of •NO, and inflammation (Rao et al., 2018, Munzel et al., 2018a). For example, PM2.5 exposure amplified vasoconstrictor responses and impaired endothelial function of the aorta of apolipoprotein E (ApoE)−/− mice in association with more pronounced macrophage infiltration, iNOS expression, ROS generation, and higher 3-nitrotyrosine levels (Sun et al., 2005). Likewise, oxidative stress was dependent on p22phox and p47phox in PM2.5-exposed rats (Sun et al., 2008). Mice deficient in phagocytic NADPH oxidase activity (p47phox−/−) were mostly protected from insulin resistance, adipose inflammation, and visceral fat •O2 − formation upon PM exposure (Xu et al., 2010). Also, endothelial dysfunction, adhesion, and infiltration of immune cells (Ly6Chigh monocytes) into the vasculature, induction of cytokines (tumor necrosis factor alpha-TNF-α, MCP-1), formation of oxidized phospholipid derivatives (e.g., PAPC) in lungs and ROS formation in aorta triggered by PM2.5 exposure were largely prevented in mice with a genetic deficiency in Nox2 and Tlr4 (Kampfrath et al., 2011). CD36-dependent 7-KC accumulation in macrophages promoted atherosclerosis upon exposure to PM2.5 (Rao et al., 2014). These processes may participate in endothelial barrier dysfunction and inflammatory cell recruitment and enable facile penetration of air pollutants, chemokines, and other secondary mediator signals as well as immune cells into the systemic circulation, feeding into TLR4-mediated oxidative stress pathways. Alveolar macrophages play a prominent role in both oxidative stress and inflammation homeostasis, as they are the first line of defense against inhaled pathogens and PM (Tao et al., 2003). It was previously demonstrated that alveolar macrophages exposed to PM2.5 show an increased M1 polarization and release of pro-inflammatory cytokines such as IL-6, IL-1β, and TNFα (Becker et al., 2005). The activation of alveolar macrophages also resulted in the increased activation of NOX-2 and the production of •O2 − and other ROS, propagating oxidative stress (Kuntic et al., 2023).
In addition, exposure of mice to PM2.5 caused suppression of protein kinase B (Akt) and eNOS phosphorylation and decreased aortic expression of the NF-κB inhibitor alpha (IκBα) (Haberzettl et al., 2016). Diesel exhaust is a major source of airborne ultrafine PM, and studies in diesel exhaust-exposed animals are a good model of ultrafine PM exposure. Diesel exhaust particles impaired endothelial function and increased aortic •O2 − formation in exposed rats, all of which was corrected by administration of tetrahydrobiopterin (BH4) (Cherng et al., 2011). Exposure of rats to diesel exhaust particles induced aortic expression of biomarkers of oxidative stress, thrombosis, inflammation, vasoconstriction, and fibrosis (e.g., HO-1, tissue factor, tissue-type plasminogen activator-tPA, plasminogen activator inhibitor-1-PAI-1, von Willebrand factor-vWF, endothelin-1, steroidogenic acute regulatory protein-ETAR, matrix metalloproteinase-2-MMP-2, tissue inhibitor of metalloproteinase 2-TIMP-2, lectin-type oxidized LDL receptor 1-LOX-1, receptor for advanced glycation endproducts-RAGE and HMGB1) (Kodavanti et al., 2011). Exposure of ApoE−/− mice to diesel exhaust particles increased markers of adverse cardiovascular effects such as oxidized low-density lipoprotein (oxLDL), MDA, LOX-1, endothelin-1, and MMP-9 expression and monocyte/macrophage infiltration, all of which were prevented by treatment with a LOX-1 antibody (Lund et al., 2011). •O2 − production in response to chronic PM2.5 exposure was abolished by the NOX-2 or eNOS inhibition (Cherng et al., 2010; Sun et al., 2008), supporting a central mechanistic role of NOX-2 and uncoupled eNOS as proposed for classical arterial hypertension and noise exposure. While there are limited comparative studies of ultrafine particles relative to PM2.5, these generally show equal or in some cases, larger effects for diesel exhaust exposures. Besides the higher systemic penetration, exposure to ultrafine particles may result in inhibition of the anti-inflammatory capacity of high-density lipoprotein (HDL) and greater systemic oxidative stress as evidenced by increased hepatic MDA, up-regulation of NRF-2-regulated antioxidant genes, and lipid peroxidation products in the plasma and liver (Araujo et al., 2008; Yin et al., 2013). Mitochondria-derived •O2 − was also shown to activate the NLRP3 inflammasome complex, which lead to the increased expression of IL-1β, driving inflammation (Caceres et al., 2024; Zhong et al., 2023).
In addition, activation of the renin-angiotensin-aldosterone system, increased levels of angiotensin-II, aldosterone, and increased blood pressure were reported for exposure of mice to PM2.5 (Du et al., 2020). Importantly, thrombotic events caused by diesel exhaust particle were exacerbated in a mouse model of angiotensin-II-induced arterial hypertension (Nemmar et al., 2011). Exposure of spontaneously hypertensive rats with ultrafine carbon particles caused aggravation of blood pressure increase, markers of thrombo-inflammation (e.g., tissue factor and PAI-1), and vasoconstriction pathways (e.g., renin, angiotensin-II, and endothelin-1) (Upadhyay et al., 2008, Upadhyay et al., 2010). The thrombotic phenotype was also aggravated in mice with angiotensin-II-induced arterial hypertension upon diesel PM exposure (Nemmar et al., 2011). A summary of the mechanistic features of PM-induced cardiovascular damage and hypertension in animal models is provided in Figure 3 and 4. Interestingly, gaseous pollutants like ozone (O3) and nitrogen dioxide (•NO2) were also reported to independently induce cardiovascular damage through oxidative stress and inflammation, although to a lower extent than PM (Munzel et al., 2018a; Al-Kindi et al., 2020).
Role of environmental dysregulation of circadian rhythms and the gut microbiome for the development of hypertension
Disturbance of circadian rhythms is a potent trigger of inflammation, oxidative stress, arterial hypertension and other cardiovascular risk factors (Morris et al., 2016; Crnko et al., 2019), and chronotherapy by melatonin was shown to reduce blood pressure in patients with essential hypertension (Scheer et al., 2004). Notably, almost all environmental risk factors, including traffic noise and air pollution, cause dysregulation of the circadian clock, which may contribute to developing hypertension, and other cardiovascular sequelae (Daiber et al., 2022; Li et al., 2020). A salt diet caused a loss of blood pressure rhythmicity and increased renin levels in period circadian regulator (Per)−/− mice in contrast to wildtype mice, and diurnal blood pressure regulation in the knockout mice was restored by treatment with the angiotensin 1-receptor blocker losartan (Pati et al., 2016). The reason for environmental regulation of the circadian rhythm is the impact of oxidative stress on the circadian clock (Putker and O’Neill, 2016). Besides the direct redox regulation of cryptochrome (CRY), PER, and F-box/LRR-repeat protein 3 (FBXL3) via thiol oxidation/reduction, the clock system is redox-regulated by RONS-dependent activation of redox-sensitive kinases, histone deacetylases, stress-response proteins, and transcription factors, all of which are modulated by environmental stressors (Fig. 6A) (Daiber et al., 2022; Li et al., 2020). Exposure of mice to continuous aircraft noise caused changes in the expression pattern of circadian genes in the aorta and kidney (Kroller-Schon et al., 2018). Bioinformatical analyses revealed down-regulation of the transcription factor FoxO3 expression, which may represent a circadian regulator via its binding site in brain and muscle ARNT-Like 1 (BMAL-1). More than 10 different circadian clock genes were dysregulated including Bmal1, Cry1, and Per1.
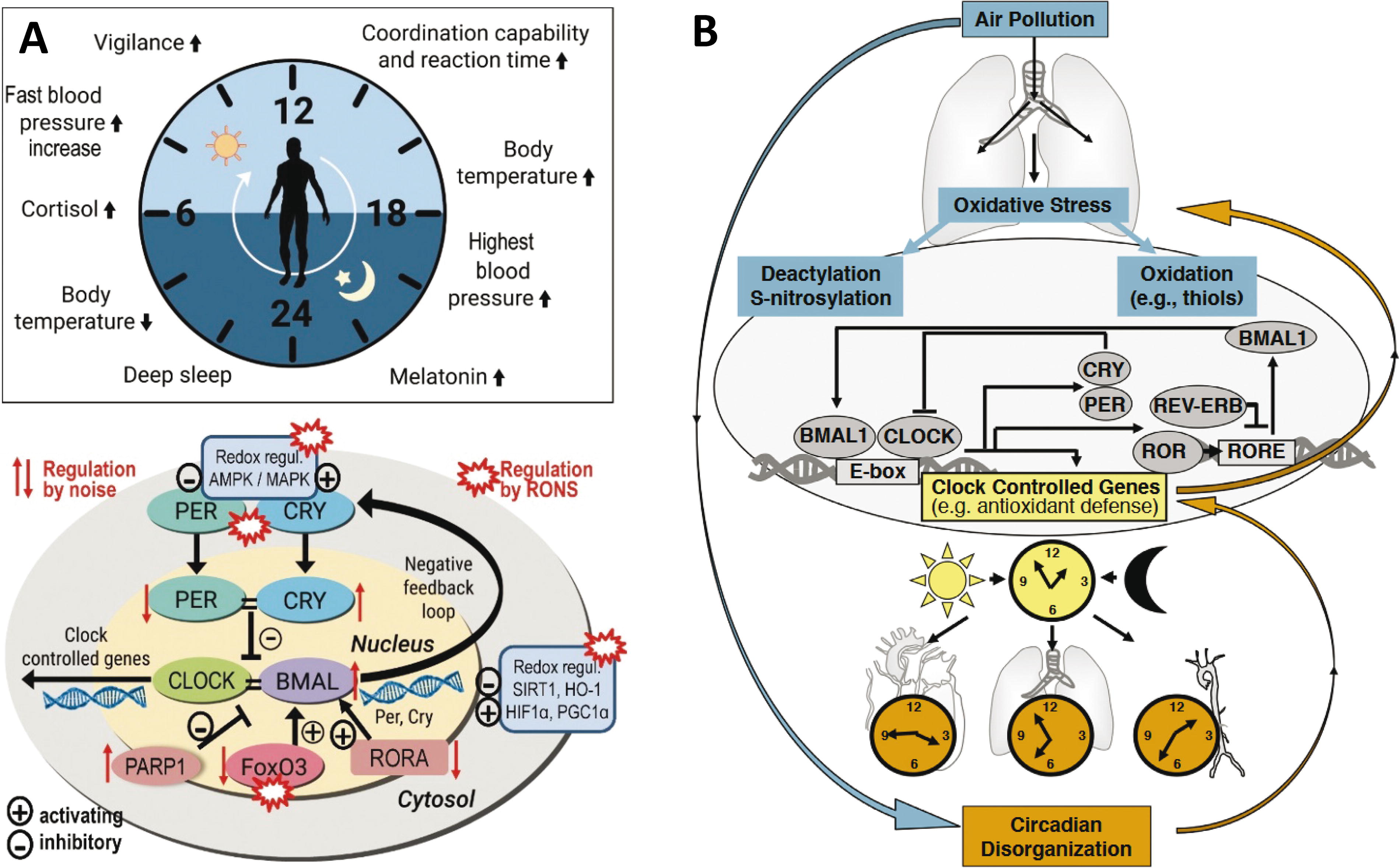
Noise exposure also changed the mRNA profile of clock genes in inferior colliculus neurons, an auditory structure essential for sound processing (Park et al., 2016). Also, exposure of both pregnant and offspring rats to air containing particulate matter affected the circadian clock. It caused the downregulation of key clock genes Per1, Per2, Per3, Rev-erbα, Dbp, and the upregulation of Bmal1 compared to filtered-air-exposed controls (Song et al., 2017). Likewise, exposure of mice to PM2.5 induced adverse metabolic changes (e.g., higher fasting blood glucose, insulin resistance) and more pronounced infiltration of macrophages, all of which were associated with amplitude shifts of circadian gene expression (e.g., Bmal1, Clock, Per1, Per2, Cry1, and Cry2) as well as a phase shift in Cry2 expression (Palanivel et al., 2020). Similarities between noise and air pollution-induced disruptions in the circadian rhythm are shown in Figures 3 and 6.
Also, the gut microbiome may affect CVD risk (Campbell and Colgan, 2019), e.g., by adverse redox and inflammatory signals by unhealthy bacterial populations. The gut microbiota-brain axis has been identified as a central player responsible for developing neuropsychiatric disorders, intestinal inflammatory disease, and regulation of mood and behavior (Cryan and Dinan, 2012, Collins et al., 2012), all linked to increased cardiovascular risk. In a mouse study, the development of arterial hypertension by inflammation in response to angiotensin-II treatment was promoted in the presence of the gut microbiome. In contrast, germ-free mice were protected from hypertensive complications (Karbach et al., 2016), all of which are supported by adverse metabolic changes caused by angiotensin-II treatment in wildtype but not in germ-free mice (Cheema and Pluznick, 2019). Noise exposure induced metabolic changes and increased inflammatory cytokines (TNF-α and IL-1β) in rats, all of which were associated with an increase in health-compromising proteobacteria and a decrease in health-promoting actinobacteria as measured by 16S rRNAseq (Cui et al., 2016). In another study, the microbiome changes in rats upon exposure to construction noise were associated with alterations of body weight, hematological parameters and histopathological changes in the organs (Zymantiene et al., 2017). A noise-triggered signaling pathway along the gut microbiota-brain-axis was associated with an impairment of cognitive function, anxiety-like behavior, and higher serum corticosterone concentrations, all of which were improved by probiotic treatment of rats (Hadizadeh et al., 2019).
In summary, noise may modulate the gut microbiota with a potential impact on the development of CVD (Karl et al., 2018; Munzel et al., 2021). Likewise, air pollutants changed the microbiome in spontaneously hypertensive rats (Chen et al., 2019). Exposure to PM2.5 altered gut microbiota composition in mice (Wang et al., 2018), and these changes in microbiota were associated with an atherogenic lipid metabolite profile (Li et al., 2017). Importantly, noise rather impacts the gut microbiome by dysregulation of central pathway (Karl et al., 2018), whereas changes of microbiota by inhaled PM are most likely triggered by intestinal and circulatory inflammation (Mutlu et al., 2018), as shown by studies using intra-tracheal instillation.
Conclusions
In the age of good sanitation and advanced medical care, where communicable diseases no longer present the most important burden to human health, new risk factors are emerging, all summarized by the exposome concept (Munzel et al., 2023). These new risk factors, either behavioral or environmental, influence noncommunicable diseases over an individual’s lifespan. It is noise and air pollution that are especially hazardous because of their inseparable presence in urbanized and industrialized societies, as reflected by their leading ranks as risk factors for global deaths and disability-adjusted life years. Research in animals and in humans has shown that noise and air pollution are associated with the increased risk of cardiovascular events and the onset of CVD in general (Al-Kindi et al., 2020; Munzel et al., 2021) and hypertension in particular (Hahad et al., 2023b; Hahad et al., 2023c). The advancement of research, especially of the combined exposure to both risk factors, can provide deeper insight into the molecular mechanisms of their action and provide solutions in the form of either therapies or legislative intervention. For now, both of these risk factors should be taken seriously, especially noise pollution, which is still neither included in the global burden of disease studies nor in actual clinical guidelines or global action plans. Of note, animal studies revealed additive vascular damage by combined noise and PM exposure (Kuntic et al., 2023), whereas human studies reported a cumulative risk index for concomitant exposure to noise and PM2.5 or ultrafine particle and higher numbers of diabetes, stroke, and myocardial infarction (Poulsen et al., 2024; Poulsen et al., 2023; Sorensen et al., 2022).
Footnotes
Acknowledgment
The authors thank Margot Neuser for graphical assistance.
Authors’ Contributions
M.K., A.D., and T.M. performed review/editing, conceptualization and writing major parts of the original draft (lead). O.H., S.A.-K., M.O., and J.L. performed review/editing, and writing parts of the original draft.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
A.D. and T.M. were supported by vascular biology research grants from the Boehringer Ingelheim Foundation for the collaborative research group “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutics” and through continuous research support from Foundation Heart of Mainz and the Center for Translational Vascular Biology (CTVB). T.M. is a PI, and M.K., O.H., and A.D. are (young) Scientists of the DZHK (German Center for Cardiovascular Research), partner site Rhein-Main, Mainz, Germany. The work was also supported by the environmental network EXPOHEALTH "Gesundheitliche Risiken von Umweltstressoren" funded by the research initiative of the state Rhineland-Palatinate, Germany.