
Abstract
Aims:
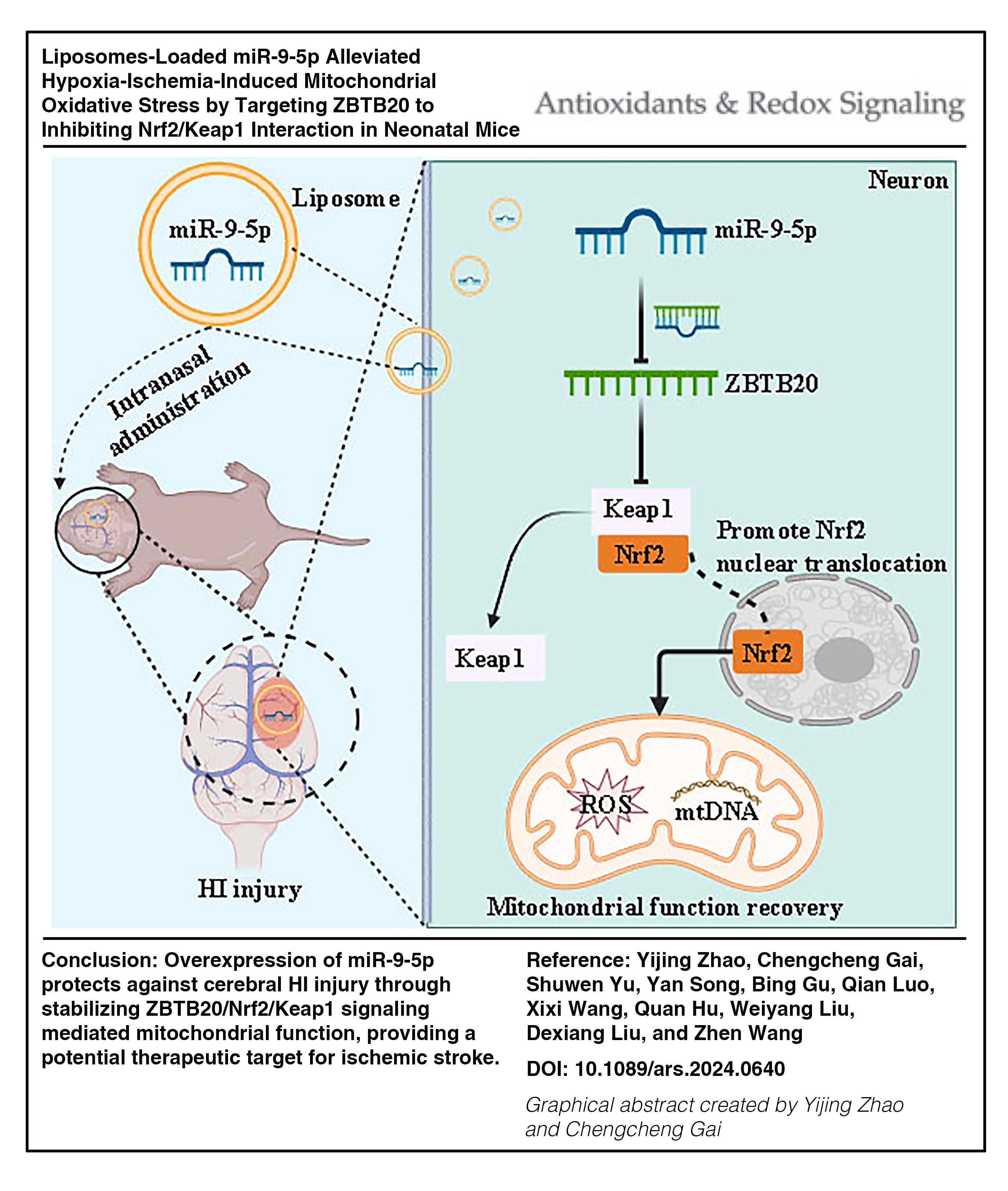
Hypoxia ischemia (HI) is a leading cause of cerebral palsy and long-term neurological sequelae in infants. Given that mitochondrial dysfunction in neurons contributes to HI brain damage, this study aimed to investigate the regulatory role of miR-9-5p in mitochondrial function following HI injury.
Results:
Overexpression of miR-9-5p in HI mice or H2O2-exposed PC12 cells suppressed neuronal injury, associated with increased mitochondrial copy number, normalizing mitochondrial membrane potential, improved nuclear factor-erythroid factor 2-related factor 2 (Nrf2) activation, and downregulation of Keap1. This was mediated, in part, through the ability of this miR-9-5p to bind and regulate the transcriptional activity of zinc finger and BTB domain-containing protein 20 (ZBTB20). Further study suggested that the knockdown of ZBTB20 exerts neuroprotection by inhibiting Nrf2/Keap1 interaction to promote the translocation of Nrf2 from the cytoplasm to the nucleus and the consequent expression of antioxidant proteins. Notably, the protective effects of miR-9-5p overexpression against HI-induced mitochondrial damage were reversed by the Nrf2 inhibitor ML385. Finally, the utilization of liposomes for the delivery of miR-9-5p (miR-9-5p@Lip) presents a promising therapeutic strategy for the treatment of HI injury.
Innovation:
miR-9-5p is a potential therapeutic agent for ischemic stroke through its modulation of the ZBTB20/Nrf2/Keap1 signaling pathway, influencing mitochondrial function and antioxidant response. Furthermore, the use of liposomal delivery for miR-9-5p offers a promising therapeutic strategy for HI injury.
Conclusion:
Overexpression of miR-9-5p protects against cerebral HI injury by modulating mitochondrial function through the ZBTB20/Nrf2/Keap1 signaling pathway. Antioxid. Redox Signal. 42, 512–528.
Get full access to this article
View all access options for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.