
Abstract
Significance:
Preclinical and clinical research in the past two decades has redefined the mechanism of action of some chemotherapeutics that are able to activate the immune system against cancer when cell death is perceived by the immune cells. This immunogenic cell death (ICD) activates antigen-presenting cells (APCs) and T cells to induce immune-mediated tumor clearance. One of the key requirements to achieve this effect is the externalization of the damage-associated molecular patterns (DAMPs), molecules released or exposed by cancer cells during ICD that increase the visibility of the cancer cells by the immune system.
Recent Advances:
In this review, we focus on the role of calreticulin (CRT) and other endoplasmic reticulum (ER) chaperones, such as the heat-shock proteins (HSPs) and the protein disulfide isomerases (PDIs), as surface-exposed DAMPs. Once exposed on the cell membrane, these proteins shift their role from that of ER chaperone and regulator of Ca2+ and protein homeostasis to act as an immunogenic signal for APCs, driving dendritic cell (DC)-mediated phagocytosis and T-mediated antitumor response.
Critical Issues:
However, cancer cells exploit several mechanisms of resistance to immune attack, including subverting the exposure of ER chaperones on their surface to avoid immune recognition.
Future Directions:
Overcoming these mechanisms of resistance represents a potential therapeutic opportunity to improve cancer treatment effectiveness and patient outcomes.
Introduction
Over the past two decades, a significant paradigm shift has occurred in our understanding of cancer treatment. Traditionally, chemotherapy was viewed primarily as a means to directly kill cancer cells. However, a large body of preclinical and clinical evidence from multiple cancer types now suggests that the clinical efficacy of conventional chemotherapy also involves activating the immune system against cancer (Green et al., 2009; Zitvogel et al., 2008). This innovative concept of “immunogenic chemotherapy” has redefined the mechanisms of action of old chemotherapeutics, and allowed us to better understand the processes underlying resistance and to better design combination regimens of chemotherapy/immunotherapy in clinical practice (Galluzzi et al., 2017; Zitvogel et al., 2008).
One of the key mechanisms underlying immunogenic chemotherapy is immunogenic cell death (ICD). ICD is a specialized form of cell death that not only leads to the demise of cancer cells but also stimulates an antitumor immune response (Aaes and Vandenabeele, 2021). The process begins with dead or dying cancer cells, which are phagocytosed by professional antigen-presenting cells (APCs), such as dendritic cells (DCs). These APCs process the tumor cells and present the tumor antigens to T cells, which are then primed and ready to kill more cancer cells (Fig. 1) (Garg et al., 2014; Kroemer et al., 2013).
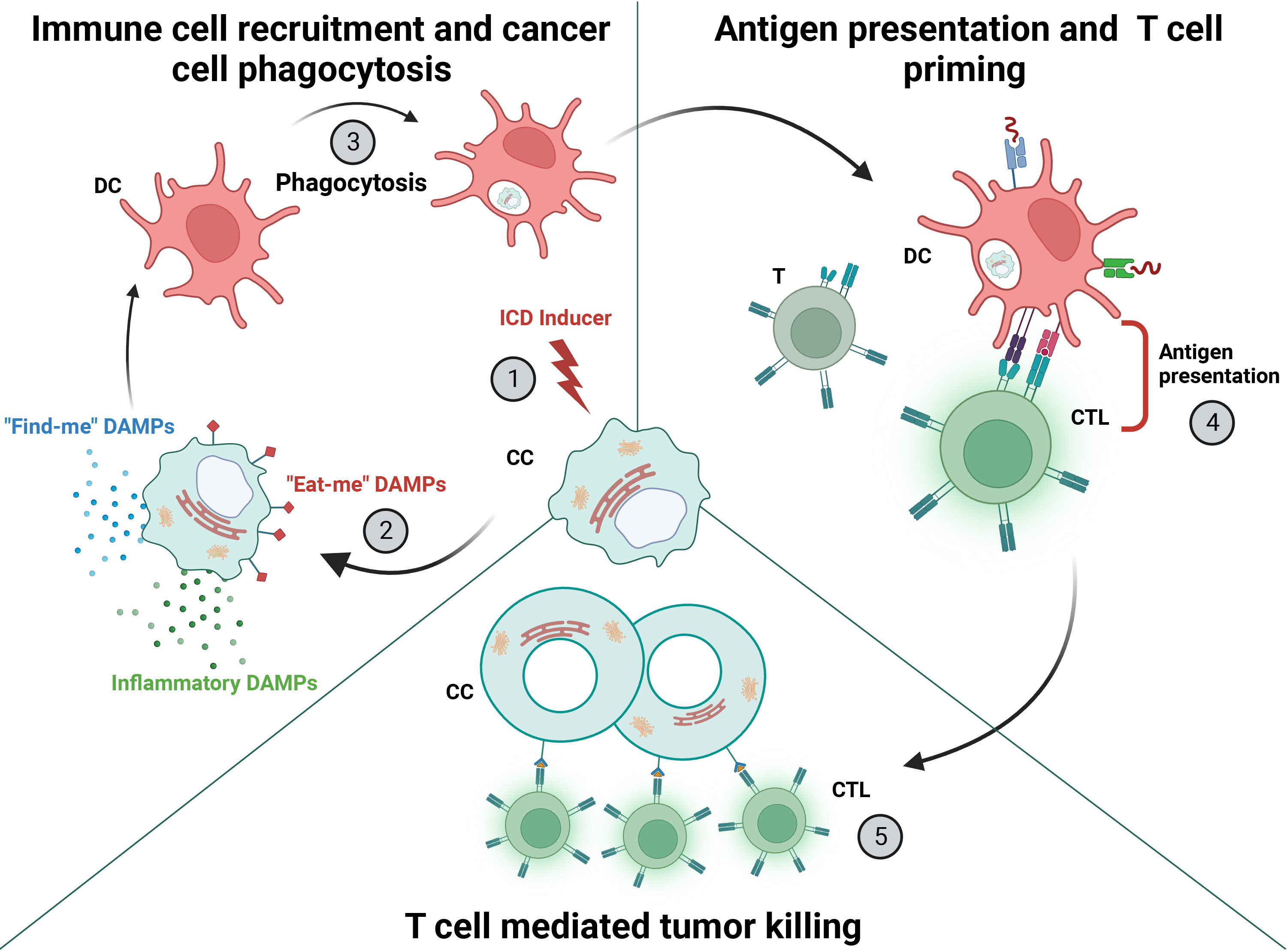
Therapeutic inducers of ICD include chemical and physical agents, such as chemotherapy (Kroemer et al., 2013), irradiation (Obeid et al., 2007a; Perez et al., 2009), photodynamic therapy (PDT) (Garg et al., 2012a; Korbelik et al., 2011), and high hydrostatic pressure (Fucikova et al., 2014; Weiss et al., 2010). Specific examples of medicines include anthracyclines (Fucikova et al., 2011), bleomycin (Bugaut et al., 2013), bortezomib (Gulla et al., 2021; Spisek et al., 2007), cyclophosphamide (Schiavoni et al., 2011), dactinomycin (Humeau et al., 2020), and oxaliplatin (Tesniere et al., 2010). Although ICD inducers have many different modes of action, they all directly or indirectly initiate endoplasmic reticulum (ER) stress pathways, direct the cell to undergo apoptotic death, activate autophagy-related adenosine triphosphate (ATP) secretion, and/or establish an inflammatory tumor state (Johnstone et al., 2022).
Two key requirements for effective ICD are antigenicity and adjuvanticity—processes that, when combined with a permissive microenvironment, drive cancer cell killing (Kroemer et al., 2013; Zitvogel et al., 2008) (Fig. 2). Antigenicity in the context of ICD is primarily mediated by tumor neoantigens (TNAs) and tumor-associated antigens (TAAs). These antigens arise from accumulating mutations, genomic instability, or post-translational modifications that are generally not subject to central tolerance mechanisms (Johnstone et al., 2022; Lawrence et al., 2013; Lee et al., 2018; Tureci et al., 2016). These are the antigens that APCs process from the dying cancer cells and present to T cells during ICD.
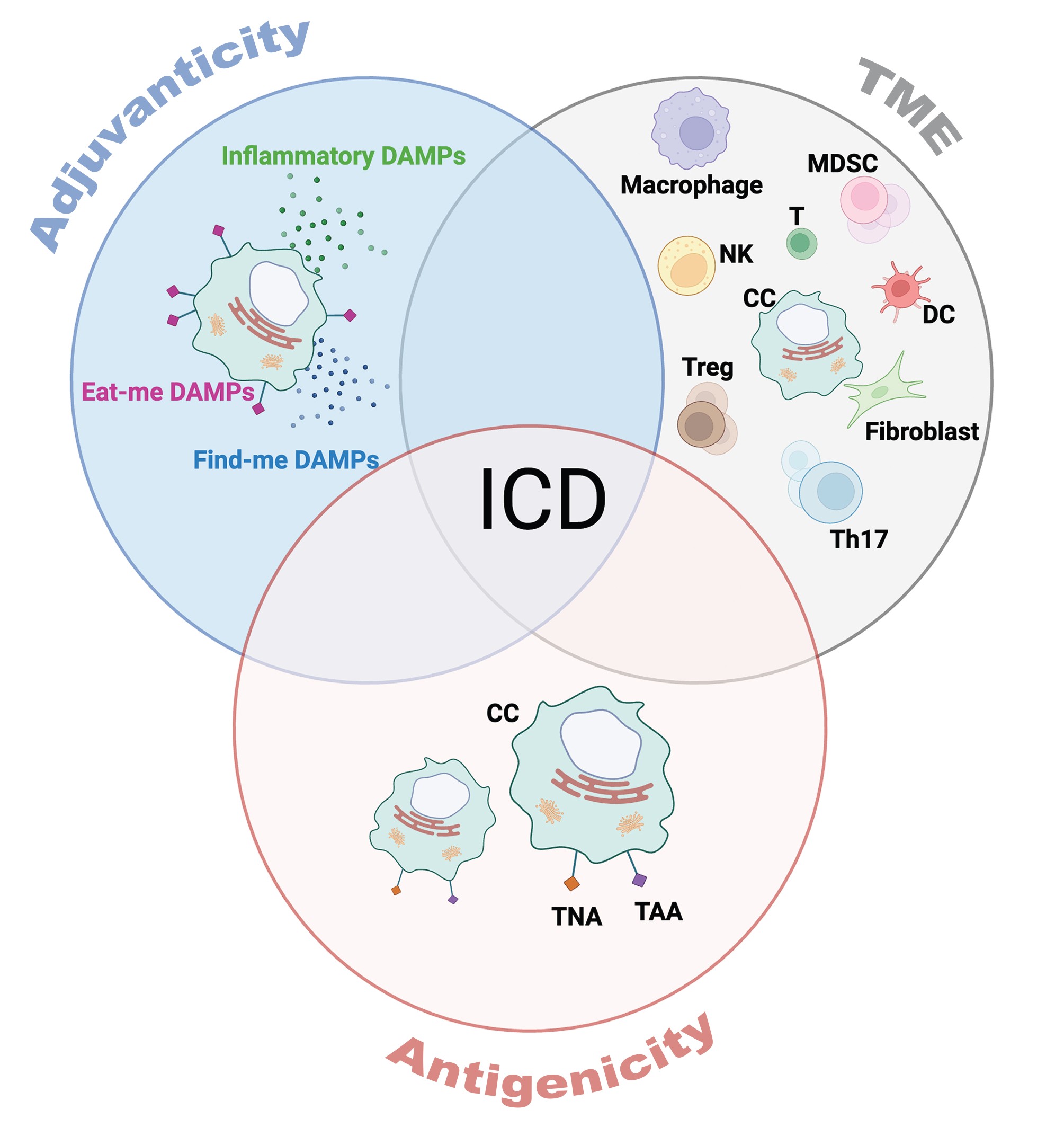
Adjuvanticity refers to the exposure or release of endogenous damage signals, known as damage-associated molecular patterns (DAMPs). These DAMPs include proteins, nucleic acids, and metabolites that are either relocated to the cell surface or released into the microenvironment during ICD (Gong et al., 2020; Kroemer et al., 2022; Krysko et al., 2012). They are sensed by the immune system via pattern recognition receptors (PRRs) and promote an inflammatory environment that supports cancer cell killing (Fuchs and Steller, 2015; Yatim et al., 2017).
In this review, we will delve into the role of certain ER chaperones as DAMPs during the ICD process. Specifically, we will focus on calreticulin (CRT) with a notable mention of other ER chaperones, such as heat-shock proteins (HSPs) and protein disulfide isomerases (PDIs), which have been somewhat better studied. They both relocate to the cell surface during ICD and serve as major “eat-me” signals for tumor cell phagocytosis by APCs. We will also explore the mechanisms by which cancer cells attempt to prevent CRT and other DAMP exposure to subvert ICD, then discuss the open questions and future direction of the field.
DAMPs: Key Drivers of Tumor Immunogenicity
Cell death, or apoptosis as a general term, is not always immunogenic. In fact, apoptotic death can occur with or without the release of DAMPs. If the apoptosis is followed by the release of DAMPs, it is considered immunologically visible, and thus a prerequisite for ICD (Kroemer et al., 2022). “Classical” or tolerogenic apoptosis does not attract the immune system in the way that ICD does (Aaes and Vandenabeele, 2021; Kroemer et al., 2022). In general, four major pathways must happen in parallel to cause DAMP exposure or release during ICD: (1) the ER-stress response via the unfolded protein response (UPR), (2) autophagy, (3) an inflammatory environment similar to a viral infection, and (4) cell death itself (Gulla et al., 2021; Johnstone et al., 2022; Kroemer et al., 2022; Li et al., 2008; Pakos-Zebrucka et al., 2016; Sistigu et al., 2014).
The UPR is associated with the preapoptotic exposure of ER chaperones, such as CRT and HSPs, that act as “eat-me” signals to foster cancer cell phagocytosis by APCs (Panaretakis et al., 2009). Autophagy induction is necessary for the release of ATP, which is the main “find-me” signal that attracts and recruits immune cells, like APCs, to the tumor microenvironment (Michaud et al., 2011). Autophagy-depleted cancer cells lack the preapoptotic secretion of ATP, and thus escape immunosurveillance in comparison with their autophagy-competent counterparts during immunogenic therapy (Martins et al., 2012; Michaud et al., 2011).
ATP secretion does not only pertain to ICD; ATP is secreted in response to inflammation, infection, and hypoxia; however, a robust preapoptotic ATP release during ICD is one of the defining features of ICD as a whole (Dosch et al., 2018; Martins et al., 2014). The inflammatory state within the tumor bed during ICD-inducing treatment mimics that of cells undergoing viral infection.
In short, several mechanisms have been described, including the activation of the cyclic GAS-AMP synthase (cGAS) signaling (Galluzzi et al., 2020a; Gulla et al., 2021), as well as the cancer cell autonomous activation of the endosomal PRR Toll-like receptor 3 (TLR3) (Sistigu et al., 2014), which promote the secretion of type I interferons (IFNs) and inflammatory chemokines. These DAMPs promote T cell priming, T cell-dependent tumor cytotoxicity, DC maturation, and antigen processing (Diamond et al., 2011; Sistigu et al., 2014; Zitvogel et al., 2015).
Cell death itself allows for the release of intracellular proteins, such as the nuclear high mobility group box 1 protein (HGMB1) (Yang et al., 2015) and annexin A1 (ANXA1) (Vacchelli et al., 2015), which stimulate DC maturation and crosspresentation, and facilitate the apoptotic body–DC physical interaction, respectively.
All four pathways are generally required for successful DAMP exposure and ICD induction, and are often referred to as “ICD biomarkers,” ultimately differentiating them from immunogenically silent apoptotic pathways (Galluzzi et al., 2020b). UPR, autophagy, inflammatory state, or cell death may not stimulate efficient adaptive immune response alone. When working in parallel and accompanied by a permissive tumor environment, with antigenic and adjuvant tumor bed properties, these DAMP-related pathways indicate and regulate ICD (Galluzzi et al., 2020b). However, there are some differences between the specific intracellular pathways activated by a variety of immunologically relevant chemotherapies, indicating that there are other unexplored regulators of ICD (Galluzzi et al., 2020b).
Out of the above, the “eat-me” DAMP signals produced by the UPR are especially important. They assist professional phagocytes, such as DCs and macrophages, in docking onto the target cell and engulfing it (Feng et al., 2019). When this intercellular communication is established in a proper immunogenic microenvironment, successful phagocytosis will result in tumor cell antigen processing and subsequent antigen presentation to the T cells (Apetoh et al., 2007; Feng et al., 2019; Garg et al., 2012b; Obeid et al., 2007b), thus causing a T cell response against the tumor. There are many types of “eat-me” signals for different contexts, but the two most well-established ones in the context of ICD of cancer cells are the ER chaperones CRT and HSPs.
ER Stress and Immunogenic ER Chaperones
The ER plays an integral role in the cellular stress response induced by chemotherapy, as it maintains protein homeostasis. This homeostasis can be disturbed by therapies (or the cancer cell's own processes) that alter protein synthesis or degradation (Hetz et al., 2020; Oakes, 2017), thus forcing the ER to induce the UPR when it senses an accumulation of misfolded proteins in its lumen (Cubillos-Ruiz et al., 2017; Hetz et al., 2020; Kroemer et al., 2010; Oakes, 2017; Panaretakis et al., 2009). The UPR activation promotes the restoration of protein homeostasis and cell survival, or may direct the cell to undergo apoptosis when cellular homeostasis cannot be restored (Graner et al., 2014).
Generally, the UPR has three main branches, each initiated by a different ER transmembrane sensor: inositol-requiring enzyme 1 alpha (IRE1a), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) alpha (Cubillos-Ruiz et al., 2017). The initial step of UPR is the dissociation of the binding immunoglobulin protein (BiP) from one or more of these sensors, thus activating them (Hetz et al., 2020; Wang and Kaufman, 2012).
IRE1a ensures the restoration of protein homeostasis by inducing the translation of the XBP1 transcription factor, which upregulates the genes responsible for ER-associated degradation (ERAD) (Ghosh et al., 2014; Han et al., 2009; Wang and Kaufman, 2012). On the contrary, ATF6 translocates from the ER to the Golgi, where it is cleaved by the site-1 (S1P) and site-2 (S2P) proteases, then enters the nucleus as a transcription factor to upregulate the ERAD-related genes (Hetz et al., 2020; Yoshida et al., 1998). It also promotes ER and Golgi biogenesis (Hetz et al., 2020). Similar to these two branches, PERK activation, as will be discussed in depth below, also aims at the restoration of the proteostasis but can similarly lead to apoptosis when the cell cannot adapt to the stress (Chang et al., 2018).
In contrast to the other two branches, PERK activation is the main branch of the UPR involved in ICD (Bezu et al., 2018a). Its activation determines the exposure of ER chaperones on the cell surface, where they dictate immunogenicity.
In health, chaperones within the ER are essential for several processes such as the binding of calcium, protein folding, and the UPR response (Chen and Cubillos-Ruiz, 2021; Graner et al., 2014). During stress, like that induced by immunogenic chemotherapy, ER chaperones can be relocated to the cell surface and become DAMPs (Zitvogel et al., 2010). Several ER chaperones have been associated with such adjuvant properties, but we will focus on CRT and HSPs as the most described and researched ER chaperone DAMPs.
Calreticulin
CRT is an ER-resident protein involved in controlling Ca2+ homeostasis and folding glycoproteins (Migliaccio and Uversky, 2018), but it is also present in the nucleus, Golgi apparatus, and cell surface, perhaps attending to alternative functions there (Migliaccio and Uversky, 2018). The mature form of CRT is generated after the cleavage of an N-terminal signal peptide, which is essential for localization to the ER (Migliaccio and Uversky, 2018).
Structurally, CRT consists of three main domains, each contributing to its diverse functions (Chouquet et al., 2011; Michalak et al., 2009) (Fig. 3). (1) The N-terminal, or globular lectin domain, is highly conserved and specializes in binding to glycoproteins with monoglucosylated oligosaccharides, aiding in their protein folding (Krause and Michalak, 1997; Lum et al., 2016). (2) The proline-rich central domain is similarly involved in chaperone activities and protein folding (Ellgaard et al., 2001). (3) The C-terminal domain primarily serves as a calcium buffer. Its high content of acidic residues allows it to store calcium ions with low affinity but high capacity (Afshar et al., 2005; Varricchio et al., 2017; Villamil Giraldo et al., 2010). The KDEL motif at the end of the C-terminal is an ER-retention signal, favoring CRT accumulation in the ER (Migliaccio and Uversky, 2018).

CRT has been mostly studied in the context of Ca2+ homeostasis and protein folding. However, CRT has a somewhat understudied role as an “eat-me” DAMP that occurs when it translocates to the cell surface during the UPR induced by ICD agents (Kroemer et al., 2013). The process regulating this translocation is complex and must happen in a timely manner with the release of other DAMPs for it to properly operate as a prophagocytic signal (Krysko et al., 2012). The initiating event, as outlined above, is the activation of the PERK pathway by ER stress (Fig. 4), specifically by the dissociation of BiP from PERK's intraluminal domain (Hetz et al., 2020; Wang and Kaufman, 2012).
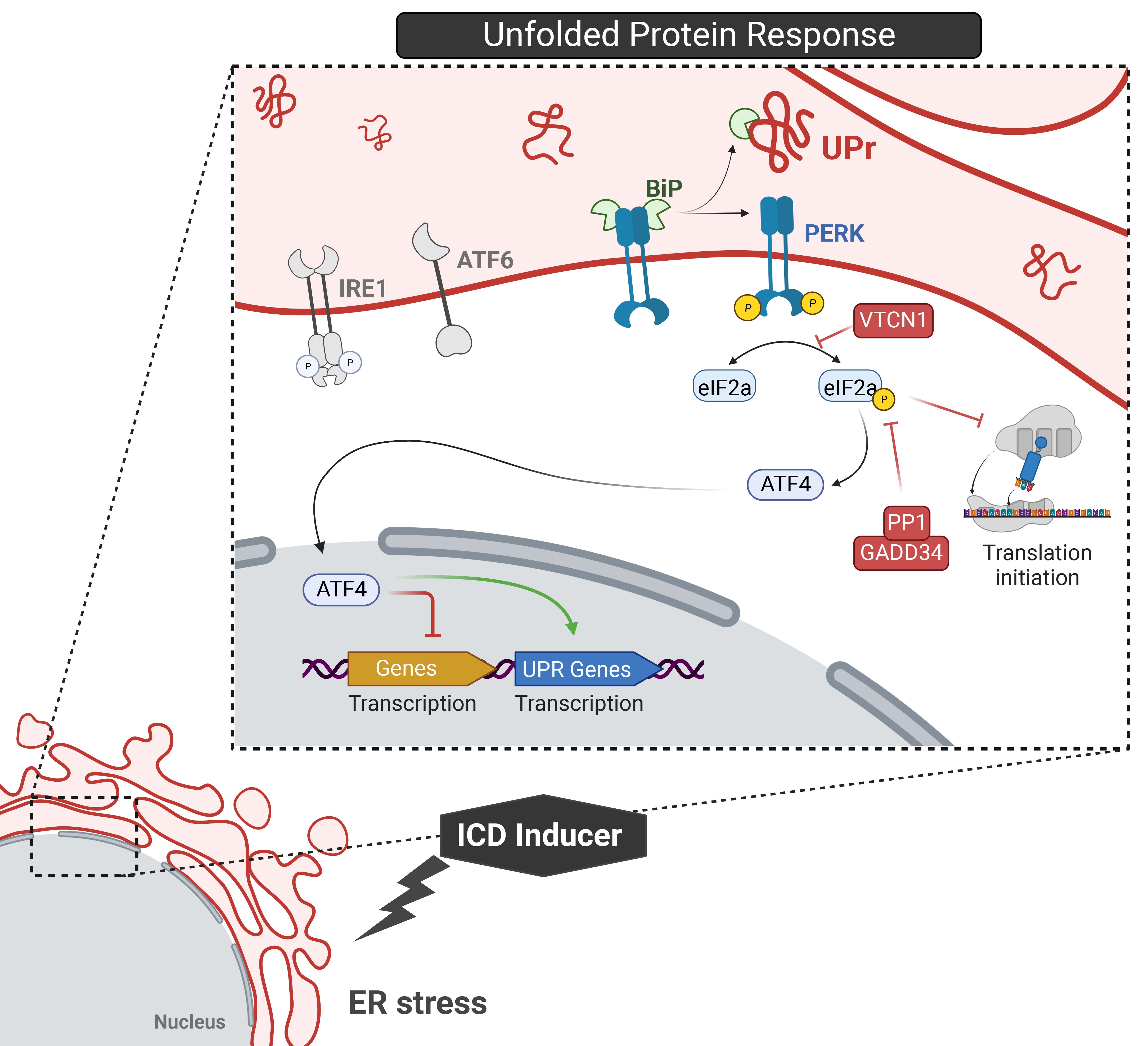
The activated PERK then phosphorylates eukaryotic initiation factor 2 alpha (eIF2a), which arrests global protein synthesis and increases the translation of activating transcription factor 4 (ATF4), which in turn increases the translation of UPR genes (Panaretakis et al., 2009). eIF2a phosphorylation is often referred to as a hallmark of ICD, since depleting PERK or expressing nonphosphorylated forms of eIF2a result in the loss of CRT expression on the cell surface after treatment with ICD inducers (Bezu et al., 2018b; Garg et al., 2012a; Garg et al., 2012b; Obeid et al., 2007b; Panaretakis et al., 2009). CRT and phosphorylation of eIF2a are important in the maintenance of Ca2+ homeostasis and protein translation during ICD-induced ER stress, as they cooperate to help cells cope with stress; however, their coregulation has not been fully elucidated.
Although the PERK branch of the UPR is important for CRT surface expression, the apoptotic cascade also appears important (Rozpedek et al., 2016). Specifically, subapoptotic cleavage of caspase-8 (Casp-8) activates B cell-associated protein 31 (BAP31), which activates the proapoptotic proteins Bcl-2–associated X protein (Bax) and Bcl-2 antagonist killer 1 (Bak) to induce apoptosis (Fig. 5) (Hattori et al., 2024; Kielbik et al., 2021; Lau et al., 2020; Panaretakis et al., 2009; Scorrano et al., 2003; Serrano-Del Valle et al., 2019). This Bax/Bak activation is essential for CRT exposure (Panaretakis et al., 2009; Rozpedek et al., 2016).
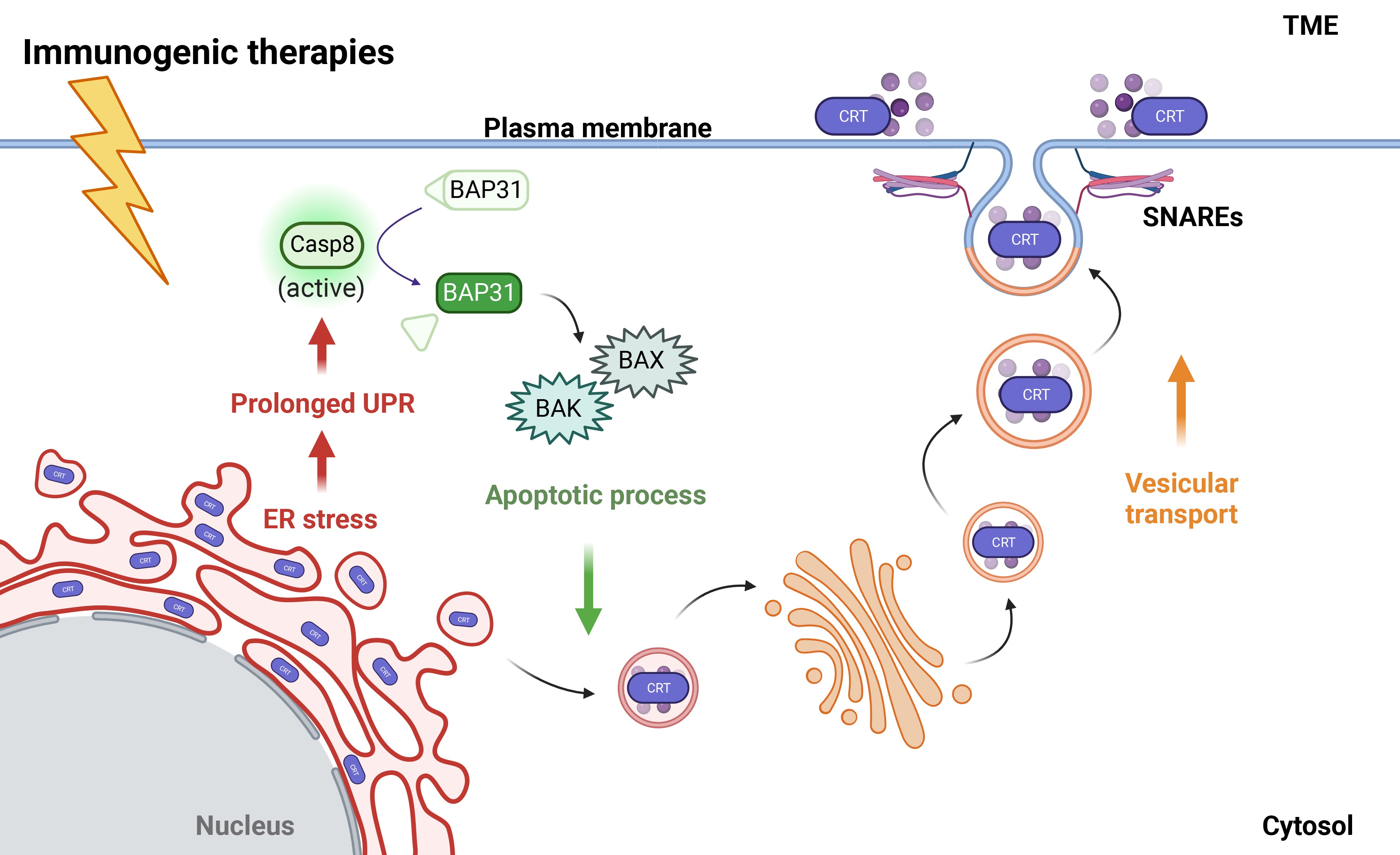
The UPR and Casp-8 activation ultimately trigger CRT translocation from the ER lumen to the plasma membrane although the exact mechanisms for how they are connected are not completely clear (Hattori et al., 2024; Panaretakis et al., 2009). This mechanism is based on actin-dependent ER-to-Golgi anterograde trafficking, whereby endo-CRT is packaged into soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-associated vesicles, and the vesicle buds off from the Golgi apparatus to fuse with the plasma membrane, thus delivering ecto-CRT (Lau et al., 2020; Panaretakis et al., 2009).
We note that certain ICD inducers, such as anthracyclines and oxaliplatin, operate via PERK-dependent ER-stress induction and downstream activation of Casp-8-dependent apoptosis to stimulate CRT translocation to the cell surface, while other ICD inducers, such as hypericin-based PDT, do not (Kepp et al., 2013). These findings suggest that PERK activation and apoptosis work in parallel in some mechanisms of ICD induction, but not all, and this is dependent primarily on the inducing agent (Kepp et al., 2013). Understanding the exact mechanism of this active process of CRT externalization would potentially allow for the identification of therapeutic strategies that would enhance this. Therefore, we have focused on this aspect of the active externalization process of CRT.
Once exposed on the cell surface, CRT acts as an “eat-me” signal for APCs such as DC and macrophages (Gardai et al., 2005). Indeed, CRT binds with CD91 (also known as low-density lipoprotein [LDL] receptor-related protein 1, LRP1) to bring the two cells into proximity, and the activation of CD91 by CRT results in a successful DC-mediated phagocytosis (Gardai et al., 2005). Additional CRT receptors have also been identified in APCs, including the C1q A chain (C1QA) (Eggleton et al., 1994) and mannose-binding 1 lectin (LMAN1) proteins (Eggleton et al., 1994), but their net contribution over CD91 has not been defined yet (Fucikova et al., 2021).
CRT loss during ICD significantly downregulates antitumor immune activation (Garg et al., 2012b; Obeid et al., 2007b; Panaretakis et al., 2009). Surface-exposed CRT upon ICD treatment in vivo was found to act as an adjuvant in immunocompetent syngeneic mice, resulting in effective cytotoxic T lymphocyte (CTL) priming and long-term vaccination against naïve cancer cells (Tatsuno et al., 2019).
Preclinical evidence in several tumor types, including colon (Obeid et al., 2007b), ovarian (Abdullah et al., 2022), and hematological malignancies (such as multiple myeloma) (Gulla et al., 2021) shows that cancer cells engineered to express low amounts of CRT fail to activate phagocytosis in vitro and in vivo when treated with different ICD inducers (such as mitoxantrone and bortezomib) (Gulla et al., 2021; Obeid et al., 2007b); whereby phagocytosis was rescued by re-expression of CRT or by treatment with exogenous recombinant CRT (Gardai et al., 2005). These findings strongly suggest that the externalization of CRT increases cancer cell immunogenicity, and acts as a potent signal for tumor recognition and CTL-mediated killing.
Of note, recent evidence has shown that CRT may also have immunosuppressive properties when mutated at the C-terminal domain (Chachoua et al., 2016; Fucikova et al., 2021; Pecquet et al., 2019). This mutation, which results in the loss of the KDEL retention signal, induces the release of soluble CRT, which binds and saturates the receptors on the phagocytes, thus preventing the phagocytosis of cells exposing CRT on the surface (Liu et al., 2020). Overall, this inhibits phagocytosis and the antitumor response (Liu et al., 2020). CRT mutation frequently occurs in myeloproliferative neoplasms, where truncated CRT promotes malignant transformation (Chachoua et al., 2016; Liu et al., 2020).
Cell surface-exposed CRT is pathognomonic across a spectrum of cancer diagnoses. Interestingly, in some cancer diagnoses, an abundance of CRT signifies a better patient outcome whereas in others it does not. For example, higher surface expression of CRT in patients with acute myeloid leukemia (AML) correlates with a higher number of CTLs with specific TAAs, indicating superior survival (Fucikova et al., 2016). In addition, ovarian carcinoma patient immunohistochemistry samples that express higher levels of ecto-CRT have a higher accumulation of mature DCs and better tumor infiltration by CTLs (Kasikova et al., 2019).
Higher levels of CRT expression also correlate with favorable prognostic factors in neuroblastoma (NB), indicating that CRT can be used to predict survival outcomes at advanced stages of NB (Hsu et al., 2005). On the contrary, robust CRT levels are associated with tumor invasion and poor overall survival in gastric cancer patients (Chen et al., 2009) and pancreatic cancer patients (Matsukuma et al., 2016).
These diverse clinical observations may indicate a multifunctional role for CRT depending on the tumor context. Moreover, they hint at the coexistence of additional mechanisms that counteract its activity. For example, when on the cell surface, CRT can act as a prophagocytic signal; however, it is counteracted by the expression of CD47, which we will discuss later as a mechanism of subversion.
In contrast, the intracellular roles of CRT may explain the negative prognostic scenarios in some disease contexts, in which CRT mediates integrin-dependent signaling and cell adhesion as well as calcium ion homeostasis, which is important in tumor angiogenesis and metastasis (Farfariello et al., 2015; Fucikova et al., 2021; Sheng et al., 2017). Altogether, these findings suggest that more research is needed to address these cancer-intrinsic differences and future directions of ICD induction in these cases.
Other ER chaperones as DAMPs
The induction of ICD can also be achieved by the translocation of several other ER-resident chaperones. In fact, studies exploring the role of HSP families and PDIs as DAMPs report intriguing findings about their immunogenicity as they translocate to the cell surface alone or bound to other DAMPs.
HSPs are a group of highly conserved molecular chaperones that play roles in protein folding, intracellular protein transport, and misfolded protein degradation (Hendrick and Hartl, 1993; Schlesinger, 1990). They are located in several intracellular compartments including the ER, cytosol, and mitochondria (Albakova et al., 2022). In general, HSPs elicit an immunogenic response when externalized. Like CRT, HSP relocation to the cell surface during immunogenic insults promotes the uptake of dying cancer cells by DCs, making them a DAMP (Garg et al., 2010; Obeid et al., 2007b; Spisek et al., 2007). Among the different HSP proteins, the HSP70 and HSP90 family proteins seem to be the most likely to be exposed on the cancer cell surface during treatment with ICD inducers such as bortezomib and anthracycline.
Specifically, bortezomib-induced ER stress stimulates the surface exposure of HSP90, which induces DC activation and CTL-mediated multiple myeloma (MM) cell killing (Spisek et al., 2007), similar to bortezomib-treated mantle cell lymphoma and breast carcinoma cells (Spisek et al., 2007). Likewise, anthracycline-induced ICD in leukemia, ovarian cancer, and prostate cancer induces the translocation of HSP70 and HSP90, along with CRT, to the cell surface, and consequently increases DC-mediated phagocytosis and the CTL antitumor response (Fucikova et al., 2011).
The ability of HSPs to act as DAMPs is closely related to their interactions with the APC receptors CD91/LRP1, LOX-1, and CD40 (Becker et al., 2002; Binder and Srivastava, 2004). These interactions promote phagocytosis and the presentation of TAAs or TNAs by DCs to CTLs to induce an effective antitumor response (Doody et al., 2004; Garg et al., 2010; Schild et al., 1999). In addition, blocking CD91 with an anti-CD91 antibody abolished DC-mediated phagocytosis, antigen crosspresentation, and T-cell activation, suggesting CD91 as an HSP70 binding partner on DCs (Salimu et al., 2015). In line with this evidence, coating tumor cells with recombinant HSP70 increases natural killer (NK) cell proliferation and NK cell-mediated tumor lysis by upregulation of CD94 (Multhoff et al., 1999).
We note that many published studies refer to the HSP70/90 families in general, rather than their specific ER-bound members, as opposed to their cytosolic and nuclear members. However, some studies have successfully identified individual members as DAMPs, such as glucose-related protein 78/GRP78/BiP (also known as HSPA5) from the HSP70 family and GRP94 (also known as gp96) from the HSP90 family (Korbelik et al., 2005; Zheng et al., 2001).
Overexpression of surface-exposed GRP94 plays a role in the activation and maturation of DCs and the reduction of tumor size by recruiting CTL, which is consistent with the notion that surface-exposed ER chaperones can act as DAMPs in activating antitumor response (Zheng et al., 2001). In line with this, GRP94, not GRP78, was found to be externalized in a mouse squamous cell carcinoma cell line upon PDT treatment, which resulted in recognition by macrophages and induced the production of tumor necrosis factor alpha (TNFa) (Korbelik et al., 2005).
Since the 1990s, there has been interest in creating immunotherapies with HSP adjuvants because of their role in chaperoning tumor antigens. Several studies conducted in vitro and in vivo reported promising results with respect to HSP peptide preparations causing immune system engagement and tumor clearance (Li et al., 2005; Sato et al., 2001). These data suggest that HSPs play an important role in the recognition of tumor cells and reducing the tumor load, implying the potential clinical relevance of such therapies in improving the adjuvant properties of anticancer vaccinations.
Moreover, the surface location of gp96 on tumor cells yields antitumor prophylactic effects in vivo (Dai et al., 2003); the GRP78 binding sequence fused to a programmed cell-death–inducing sequence resulted in tumor growth decrease in vivo, suggesting that GRP78 on the cell surface can be used as a DAMP target in eliciting an immune response (Arap et al., 2004). These data suggest that surface-exposed HSPs play an important role in the recognition of tumor cells by APCs and reduction of tumor load, implying the potential clinical relevance of such therapies in improving the adjuvant properties of anticancer vaccinations.
PDIs are described in the context of ICD to a lesser degree, perhaps due to their smaller net contribution to ICD. Protein disulfide isomerase-3 (PDIA3), also known as ERp57, is an ER-bound chaperone that binds luminal CRT and, in some cases, cotranslocates to the cell surface with CRT (Turano et al., 2002). Specifically, ERp57 interacts with CRT in a protein–protein binding manner, and knockdown of ERp57 reduces CRT surface exposure upon anthracycline treatment (Panaretakis et al., 2008).
When ERp57 is knocked down after CRT knockout, there is no significant reduction in the immunogenicity of a mitoxantrone-treated colorectal cancer cell line (Obeid et al., 2007b), implying CRT-dependent ICD. Surface-exposed ERp57 alone is not immunogenic, and its role as a DAMP directly relates to its chaperone activity of translocating CRT to the surface (Liu et al., 2019). However, the precise roles of other members of the HSP family or of other ER-resident proteins, such as the disulfide isomerase (PDI) protein family, in ICD mechanisms have not been clearly defined and are an attractive area of research.
Mechanisms of Subverting ER Chaperone Exposure: Clinical Consequences and Therapeutic Opportunities
Cancer cells constantly adjust to their microenvironment and employ several mechanisms to escape immunosurveillance, which ultimately promotes tumor progression, resistance to therapy, and poor clinical outcome (Zitvogel et al., 2006). In the context of ICD, blocking the externalization of DAMPs from dying cells can maintain an immunosuppressive microenvironment, thus protecting the cancer from the immune system. Specifically, blocking CRT translocation has been observed in tumor cells and is associated with poor long-term control of the disease (Fucikova et al., 2021). Given that there are multiple steps in the translocation of CRT, as well as the general process of ICD, there are multiple resistance mechanisms that can arise (Fig. 6).
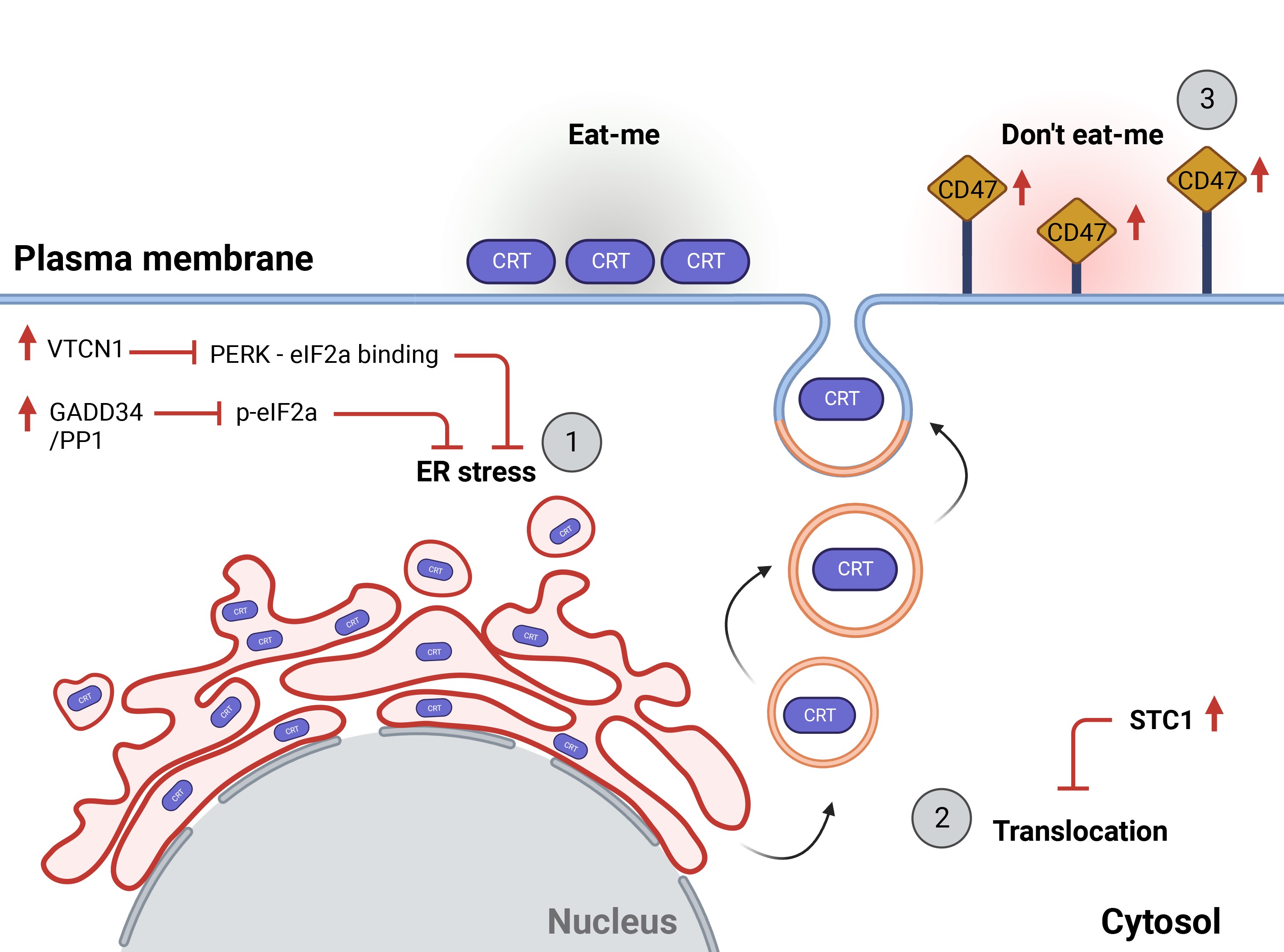
The first step of CRT exposure is ER stress. Therefore, it is of no surprise that the primary mechanism of subversion involves inhibiting ER stress itself, and more specifically, inhibiting eIF2a phosphorylation. Indeed, when tumor cells overexpress proteins that either inhibit eIF2a phosphorylation or increase its dephosphorylation, CRT is not properly exposed on the outer leaflet of the cell membrane during ICD (Kroemer et al., 2022).
Specifically, the B7-H4 or V-set domain containing T cell activation inhibitor 1 (VTCN1) protein is a well-known regulator of eIF2a phosphorylation, and high levels of B7-H4 inhibit PERK binding to eIF2a and are associated with poor immune infiltration and prognosis in patients with triple-negative breast cancer (TNBC) (Song et al., 2020), ovarian tumors (Kryczek et al., 2007), melanoma (Quandt et al., 2011), lung cancer (Sun et al., 2006), and prostate cancer (Podojil and Miller, 2017; Zang et al., 2007).
Indeed, inhibiting B7-H4 protein glycosylation with the NGI-1 compound increases the immunogenicity of TBCN cells in chemotherapy-induced ICD (Song et al., 2020). Similarly, inhibiting eIF2a phosphatase complex, consisting of growth arrest and DNA damage-inducible protein 34 (GADD34) and protein phosphatase 1 (PP1), increases CRT exposure by favoring the persistence of a phosphorylated form of eIF2a (Kepp et al., 2009; Obeid et al., 2007b; Panaretakis et al., 2009). Therefore, therapeutic approaches that maintain proper eIF2a phosphorylation can successfully restore CRT relocation to the cell membrane.
Resistance mechanisms can also target the proteins involved in CRT translocation. For example, low expression of CRT on the cell surface during ICD has been associated with a high level of stanniocalcin 1 (STC1), a cytoplasmic protein whose binding with CRT traps it in the mitochondria (Lin et al., 2021). Cancer patients displaying high STC1 levels have lower immune infiltration, response to immune therapy, and poor clinical outcome (Lin et al., 2021). Therefore, overcoming STC1 overexpression may represent a promising therapeutic approach to improving immune control of the disease.
Even when CRT makes it to the cell surface, resistance mechanisms may still target the phagocytic process. Phagocytosis is tightly regulated by pro- and antiphagocytic signals (Chao et al., 2010b). CRT is an “eat-me” signal, but it can be exposed alongside “don't eat me” signals, which can limit the engulfment of cancer cells (Chao et al., 2010b). A well-studied “don't eat me” signaling pathway is the signal regulatory protein alpha (SIRP1A; expressed on DC) and CD47 (expressed on the tumor cell).
High levels of CD47 in acute lymphocytic leukemia (ALL), AML (Majeti et al., 2009), chronic lymphocytic leukemia (CLL) (Chao et al., 2010b), and human ovarian cancer patients (Brightwell et al., 2016) are correlated with poor clinical outcome (Chao et al., 2010b; Oldenborg et al., 2001). Notably, cell-surface exposed CRT and CD47 expression in physiologically normal cells is significantly lower than that observed in their cancer counterparts (Chao et al., 2010a).
Furthermore, live cancer cells that express higher levels of surface-exposed CRT express higher levels of CD47, suggesting that CD47 expression may counteract CRT-mediated DC phagocytosis (Chao et al., 2010a). These findings also suggest that it is the surface abundance of CRT that renders the cell immunogenic, as the normal cell surface is significantly less abundant in CRT.
Given the balancing act of phagocytic signals, the rate of phagocytosis can in theory be adjusted by targeting either CRT exposure or CD47 exposure. In fact, several therapies to reduce CD47 activity are being tested in the clinical setting, including CD47 monoclonal antibody (NCT03527147), CD47/PD-L1 bispecific antibody (NCT04746131), and SIRPa/Fc fusion protein antibody (NCT03530683) (Yang et al., 2023). Finally, while in most of the described literature, CRT exposure is desirable, it is crucial to address the discrepancy in such generalization in cases where CRT exposure was a negative prognostic marker as we discussed earlier. Therefore, the subversion mechanisms remain to be clearly elucidated with respect to different cancer types and outcomes.
Conclusions and Open Questions
ICD in different cancer types has become a relatively new area of research founded on the idea put simply as “using cancer to beat cancer.” By triggering the release of DAMPs from tumor cells and encouraging APCs to present tumor antigens, we can activate the immune system—once thought to be a passive player during chemotherapy—to recognize and eliminate cancer cells. ER chaperones such as CRT and HSPs, which play homeostatic roles in healthy cells, can become DAMPs because of ER stress induced by therapies promoting ICD. DAMPs label cancer cells with “eat-me” signals to capture the attention of the innate immune system. Once the immune system has been directed to the DAMP location, DC-mediated phagocytosis and antigen crosspresentation to CTLs take place and enhance selective tumor clearance. The ability of DAMPs to initiate this immune machinery is a promising target for immunogenic chemotherapy research, as we must understand its mechanisms to promote ICD in the clinic and overcome the cancer cells' resistance mechanisms to it.
Emerging experimental evidence in support of ICD efficacy in initiating antitumor response serves as an attractive area of research with regard to translational medicine. Results from in vivo and in vitro experiments suggest that ICD induction via cell surface CRT and HSPs exposure, in a favorable environment, enhances and drives DC-mediated phagocytosis and CTL killing of naïve tumor cells. As such, these data show clinical relevance in the administration of chemotherapies such as bortezomib, oxaliplatin, and doxorubicin, indicating both new mechanisms of action and future directions in the improvement and development of ICD agents with adjuvant-like properties.
HSP-derived peptides, in particular, can serve as notable adjuvants for improved vaccination in clinics while surface-exposed CRT and HSP70 and HSP90 induction could initiate adaptive immune machinery, yielding long-term vaccination via the engagement of professional APCs and CTLs.
While the role of ICD in activating the immune system for cancer therapy is promising, several open questions remain. For instance, how do different types of cancer cells vary in their susceptibility to ICD-inducing therapies? What are the specific mechanisms that allow some cancer cells to evade ICD, and can they be targeted to improve therapy outcomes? Are there other cellular components that could serve as DAMPs, and what are the conditions under which they are exposed or released? Understanding the nuances of these questions could provide valuable insights into the limitations and potential improvements of ICD-based therapies.
Given these open questions, future research should focus on identifying the molecular and cellular mechanisms that govern the effectiveness of ICD in different cancer types. This could lead to the identification of patients who are resistant to the immunogenic activation of cell death, and could similarly involve exploring new ICD inducers or combinations of existing ones to overcome resistance mechanisms in cancer cells. These findings would be instrumental for the optimal use of immunogenic chemotherapy as an adjuvant for immunotherapy to improve the proportion of patients who can benefit from therapies.
Furthermore, the development of techniques to measure DAMP exposure and immune system activation in real-time could provide critical data for optimizing treatment protocols. Clinical trials designed to test the efficacy of ICD-based therapies in a more personalized manner, taking into account the specific characteristics of the tumor and the patient's immune system, could be a significant next step in translating these findings into effective treatments.
Footnotes
Acknowledgments
The authors gratefully acknowledge the members of their laboratories for insightful advice and critical discussions. They thank Christina Usher (Dana-Farber Cancer Institute) for editing the article and insightful comments. Figures were created with Biorender.com
Authors' Contributions
A.G., K.C.A., and S.C. designed conceptualization, validation, and writing—review and editing; A.G. and S.C. assisted with methodology, formal analysis, data curation, investigation, and writing—original draft preparation; M.T., P.F., C.C., and S.C. provided software; S.C., T.M., M.T., C.C., and F.B. provided resources; M.T., P.F., and C.C. performed visualization; A.G. provided supervision; A.G. and K.C.A. contributed to project administration and funding acquisition. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
K.C.A. reports personal support from Janssen, Pfizer, Astrazeneca, Amgen, Precision Biosciences, Mana, Starton, NextRNA, Window, and Raqia, and is a Scientific Founder of OncoPep and C4 Therapeutics. The remaining authors declare no competing financial interests.
Funding Information
This work is supported by NIH/NCI grants SPORE-P50CA100707, P01CA155258 (K.C.A.); by the Paula and Roger Riney Foundation grant (K.C.A.); by the Sheldon and Miriam Medical Research Foundation (K.C.A.); the Adelson Medical Research Foundation (K.C.A.). A.G. is a Fellow of The Leukemia & Lymphoma Society and a Scholar of the American Society of Hematology; she received support from the International Myeloma Society (IMS); she is supported by an Individual Start-UP grant from the Italian Association for Cancer Research (AIRC; Project No. 27750); a FPRC “5xmille” 2019 Ministry of Health project (IDEE) and a FPRC “5xmille” 2021 Ministry of Health project (EMAGEN-FaBer). C.C. is supported by NCI grant number 5R25CA174650. K.C.A. is an American Cancer Society Clinical Research Professor.