
Abstract
Aims:
Doxorubicin (Dox) is a potent chemotherapy agent, yet its clinical use is hampered by cardiotoxicity. Although extensive research has focused on Dox-induced cardiotoxicity (DIC), its mechanism remains elusive. Recent evidence implicates ferroptosis as a key contributor to DIC. The 15-lipoxygenase-1 (ALOX15), involved in lipid peroxidation, is known to play an essential role in ischemia-induced myocardial damage and heart failure; however, its function in DIC is undefined. This study seeks to elucidate the role of ALOX15 in DIC and unravel its underlying mechanism.
Results:
Both ALOX15 mRNA and protein levels were elevated in DIC models in vivo and in vitro. Inhibition or silencing of ALOX15 ameliorated lipid peroxidation, ferroptosis, and cardiac dysfunction in Dox-treated mice. Consistently, ALOX15 loss of function protected H9C2 cells against Dox and RSL3-induced toxicity. In addition, we found that linoleic acid increased the susceptibility of H9C2 cells toward Dox-induced damage, which was abolished by ALOX15 inhibition. Furthermore, Alox15 overexpression aggravated Dox-induced cell damage by aggravating reactive oxygen species (ROS)-mediated ferroptosis. Mechanistically, we discovered that the amelioration of Dox-induced ferroptosis by ALOX15 loss of function occurred through inhibiting the ROS-mediated mitogen-activated protein kinase (MAPK) signaling pathway activation.
Innovation and Conclusion:
These results reveal that ALOX15 regulates ferroptosis through ROS-mediated MAPK signaling pathway in DIC, suggesting a potential therapeutic target for DIC intervention. Antioxid. Redox Signal. 43, 363–380.
Introduction
Doxorubicin (Dox) is a commonly used and effective agent for cancer chemotherapy (Mohammadi et al., 2020). However, its clinical application is largely restricted by dosage-dependent cardiotoxicity, including cardiomyocyte loss, progressive cardiac enlargement, and congestive heart failure (Wallace et al., 2020). Despite extensive research on Dox-induced cardiotoxicity (DIC), the underlying mechanism remains incompletely understood, and no effective therapies for DIC are currently available. Therefore, uncovering the underlying mechanism and exploring potential targets for this adverse effect are urgently needed.
Previous studies have elucidated that various factors, including oxidative stress (Kong et al., 2022), impaired mitochondrial function (Tadokoro et al., 2023; Wu et al., 2023b), apoptosis (Wu et al., 2023a; Xu et al., 2022; Zhang et al., 2023b), autophagy (Li et al., 2023; Liu and Zhao, 2022; Sun et al., 2023), and pyroptosis (Dai et al., 2023; Zhang et al., 2023a), are involved in the pathogenesis of DIC. Recently, a novel form of cell death, ferroptosis, has been revealed to play a crucial role in the progression of DIC (Fang et al., 2019; Tadokoro et al., 2020). Ferroptosis is characterized by iron-dependent lipoxygenase (LOX) activation and the consequential cell death induced by excessive reactive oxygen species (ROS) accumulation due to lipid peroxidation of phospholipids having polyunsaturated fatty acid (PUFA) chains (Cao and Dixon, 2016; Dixon et al., 2012).
LOXs, involved in lipid peroxidation, are generally regarded as the main driver of ferroptosis (Shah et al., 2018). Arachidonate 15-lipoxygenase-1 (ALOX15), a member of the LOX family, catalyzes the oxidation of PUFA such as arachidonic acid (AA) and linoleic acid (LA), which are anchored in several phospholipids, including phosphatidylethanolamine (PE) (Kuhn et al., 2005). Several lines of evidence have implied that ALOX15 plays a contributory role in ischemia-induced myocardial damage and heart failure (Cai et al., 2023; Kayama et al., 2009; Ma et al., 2022). Moreover, recent studies have discovered that the ALOX15 inhibitor, PD 146176, can prevent cell death triggered by ferroptosis inducers, indicating a potential role for ALOX15 in ferroptosis (Shintoku et al., 2017; Soriano-Castell et al., 2021). However, whether ALOX15 is involved in DIC remains unclear. Given the pivotal role of ferroptosis in DIC and the lipid peroxidation catalyzed by ALOX15, it is plausible that ALOX15 plays a crucial role in the pathogenesis of DIC. Thus, this study was designed to elucidate the role of cardiac ALOX15 involved in DIC and unravel its underlying mechanism.
Results
Expression of ALOX15 is upregulated in DIC
To elucidate the relationship between ALOX15 and DIC, we analyzed the RNAseq data using mRNA extracted from heart tissue of rats treated with saline (n = 10) or Dox (3 mg/kg/week, n = 6) for 5 weeks, which was available in the Gene Expression Omnibus (GEO) database under the accession number GSE154603. After checking and filtering the data by cluster analysis (Supplementary Fig. S1A and S1B), we conducted differential analysis. We identified 976 differentially expressed genes (DEGs), including 566 upregulated genes and 410 downregulated genes (Fig. 1A, Supplementary Table S1). We then integrated DEGs and ferroptosis-related genes (FRGs) into a Venn diagram analysis, which revealed 28 differentially expressed ferroptosis-related genes (DEFRGs) (Fig. 1B, Supplementary Table S2). Notably, Alox15 was upregulated in the DIC model (Fig. 1A, Supplementary Fig. S1C) and was the most DEG among these 28 DEFRGs. To further understand the biological characteristics of DEFRGs, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. The GO enrichment analysis revealed that DEFRGs were significantly enriched in ROS metabolic process, oxidoreductase complex, and oxidoreductase activity (Supplementary Fig. S1D). Furthermore, these genes were significantly involved in ferroptosis according to the KEGG analysis (Supplementary Fig. S1E). By performing the Gene Set Enrichment Analysis (GSEA), we discovered that the mitogen-activated protein kinase (MAPK) signaling pathway was enriched in the DIC model (Fig. 1C).
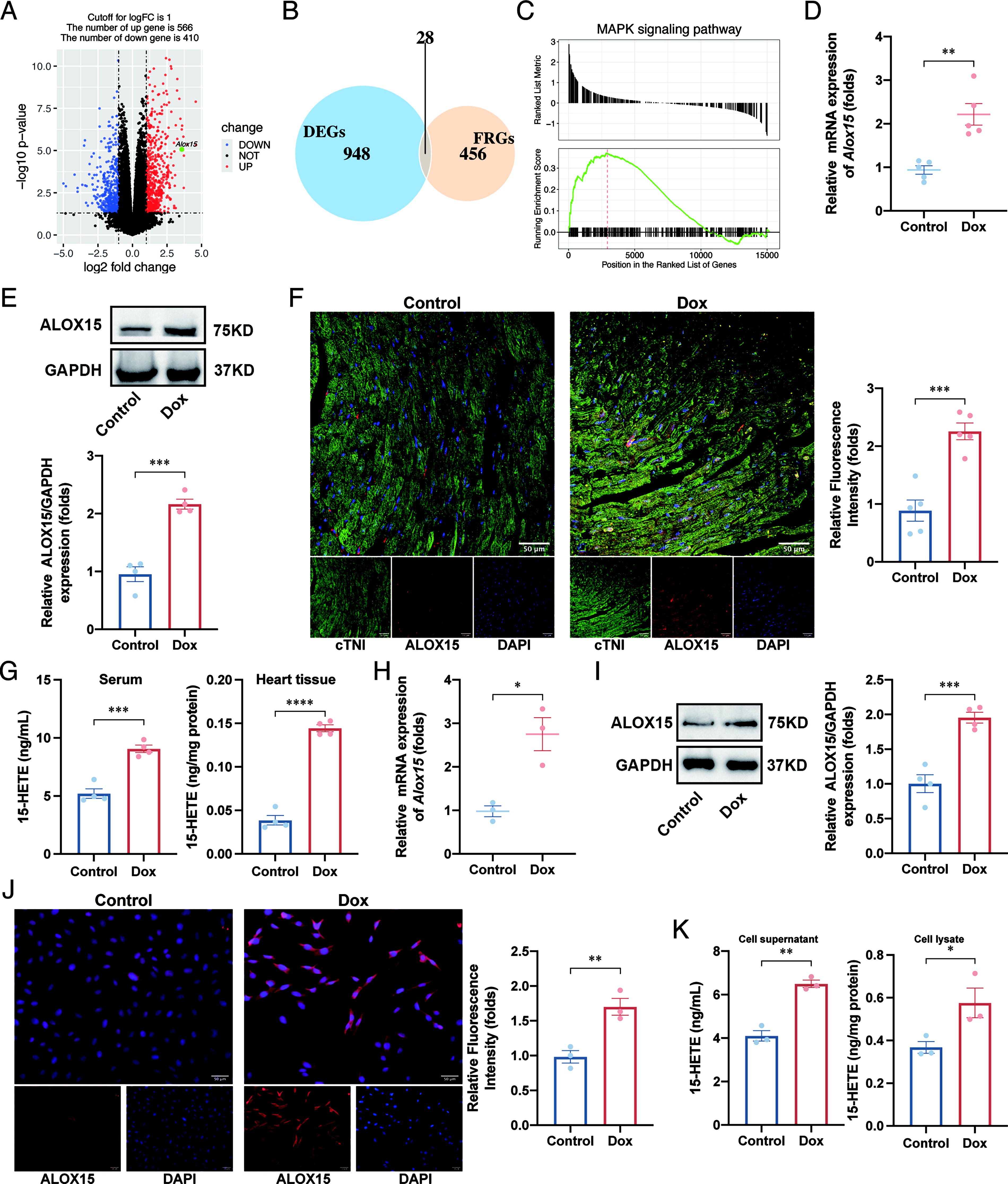
To corroborate these bioinformatic findings, we investigated the expression of ALOX15 in the DIC model in vivo and in vitro. Both the mRNA and protein levels of ALOX15 were increased in the heart tissues of Dox-treated mice, as confirmed by real-time quantitative PCR (RT-qPCR) (Fig. 1D), Western blot (Fig. 1E), and immunofluorescence (IF) analysis (Fig. 1F). In addition, the serum and heart tissue levels of 15-hydroxyeicosatetraenoic acid (15-HETE), a metabolite of ALOX15, were higher in Dox-treated mice, as determined by enzyme-linked immunosorbent assay (ELISA) (Fig. 1G). In line with the results in vivo, the mRNA and protein levels of ALOX15, as well as the 15-HETE levels in cell supernatant and lysate, were increased in Dox-treated H9C2 cells (Fig. 1H–1K).
ALOX15 loss of function attenuates DIC and ferroptosis in vivo
To investigate the role of ALOX15 in DIC, we next employed two complementary loss-of-function approaches: pharmacological inhibition of ALOX15 using PD 146176 and genomic knockdown via adeno-associated virus (AAV) carrying of short hairpin RNA (shRNA) targeting Alox15 (HBAAV2/9-cTNT-miR30-shAlox15). The efficacy of ALOX15 knockdown was confirmed by Western blot analysis (Supplementary Fig. S2A). Concurrently, we observed that the Dox-induced content of ALOX15-derived product 15-HETE was diminished by pharmacological inhibition and genomic knockdown (Fig. 2A, Supplementary Fig. S2B), further confirming the effective loss of function of ALOX15.
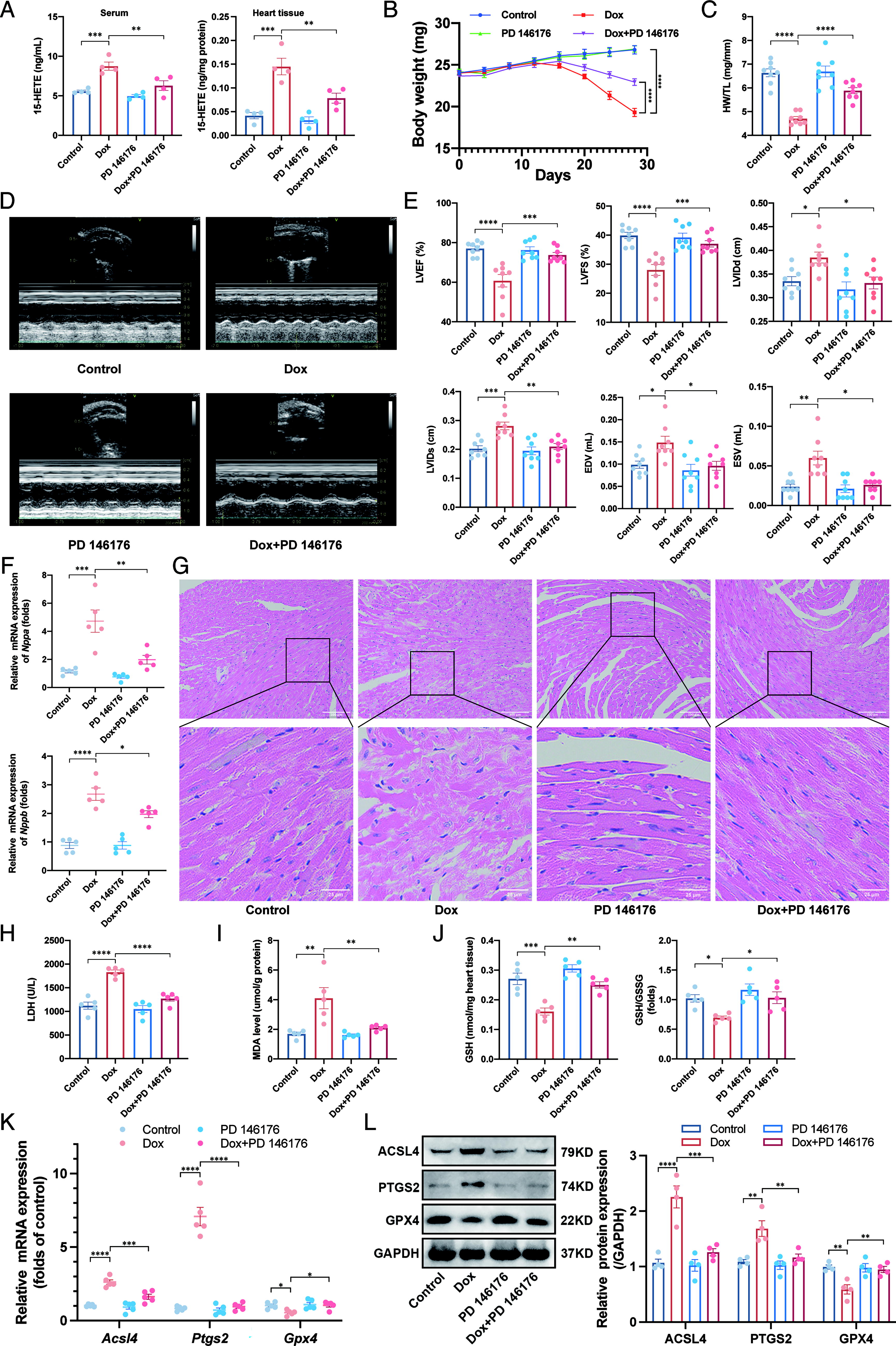
As expected, mice treated with Dox exhibited reduced body weight (Fig. 2B), lower heart weight/tibia length index (Fig. 2C), and impaired cardiac function as assessed by transthoracic echocardiography which was manifested by decreased ejection fraction and fractional shortening, and increased diastolic left ventricular internal diameter (LVIDd), systolic left ventricular internal diameter (LVIDs), end-diastolic volume, and end-systolic volume (Fig. 2D–2E). In addition, Dox treatment elevated markers of heart failure (Nppa and Nppb, Fig. 2F) and induced histopathological changes, including myofibrillar disarrangement and cytoplasmic vacuolization, as observed in hematoxylin and eosin (H&E) staining (Fig. 2G). Furthermore, Dox administration led to increased myocardial cell death, as indicated by elevated serum lactate dehydrogenase (LDH) level (Fig. 2H). Notably, both pharmacological inhibition and genomic knockdown of ALOX15 significantly attenuated Dox-induced cardiac dysfunction (Fig. 2 and Supplementary Fig. S2).
We further explored whether the cardioprotective effects of loss of function ALOX15 were mediated through inhibiting ferroptosis. Results showed that Dox elevated malondialdehyde (MDA) (Fig. 2I) and reduced glutathione (GSH) level and GSH/oxidized glutathione (GSSG) ratio (Fig. 2J). In addition, Dox upregulated the mRNA and protein levels of acyl-CoA synthetase long chain family member 4 (ACSL4) and prostaglandin-endoperoxide synthase 2 (PTGS2), and downregulated the mRNA and protein levels of glutathione peroxidase 4 (GPX4) (Fig. 2K and 2L). However, ALOX15 loss of function significantly attenuated the ferroptosis induced by Dox (Fig. 2 and Supplementary Fig. S2).
ALOX15 loss of function mitigates Dox- and RSL3-induced cytotoxicity and ferroptosis in vitro
To elucidate the role of ALOX15 in cardiomyocyte injury, we conducted in vitro experiments using H9C2 cells and neonatal rat cardiomyocytes (NRCMs). Pharmacological inhibition of ALOX15 with PD 146176 pretreatment and genetic knockdown using small interfering RNA (siRNA) were employed to establish loss-of-function models. Initially, we evaluated the cytotoxicity of PD 146176. Results indicated that concentrations below 5 μM were nontoxic to H9C2 cells, whereas higher concentrations caused slight reductions in cell viability (Fig. 3A). Therefore, 5 μM PD 146176 was selected for subsequent experiments. The efficiency of siRNA-mediated Alox15 knockdown was confirmed by RT-qPCR and Western blot analysis, which demonstrated significant suppression of ALOX15 expression (Supplementary Fig. S3A and S3B).
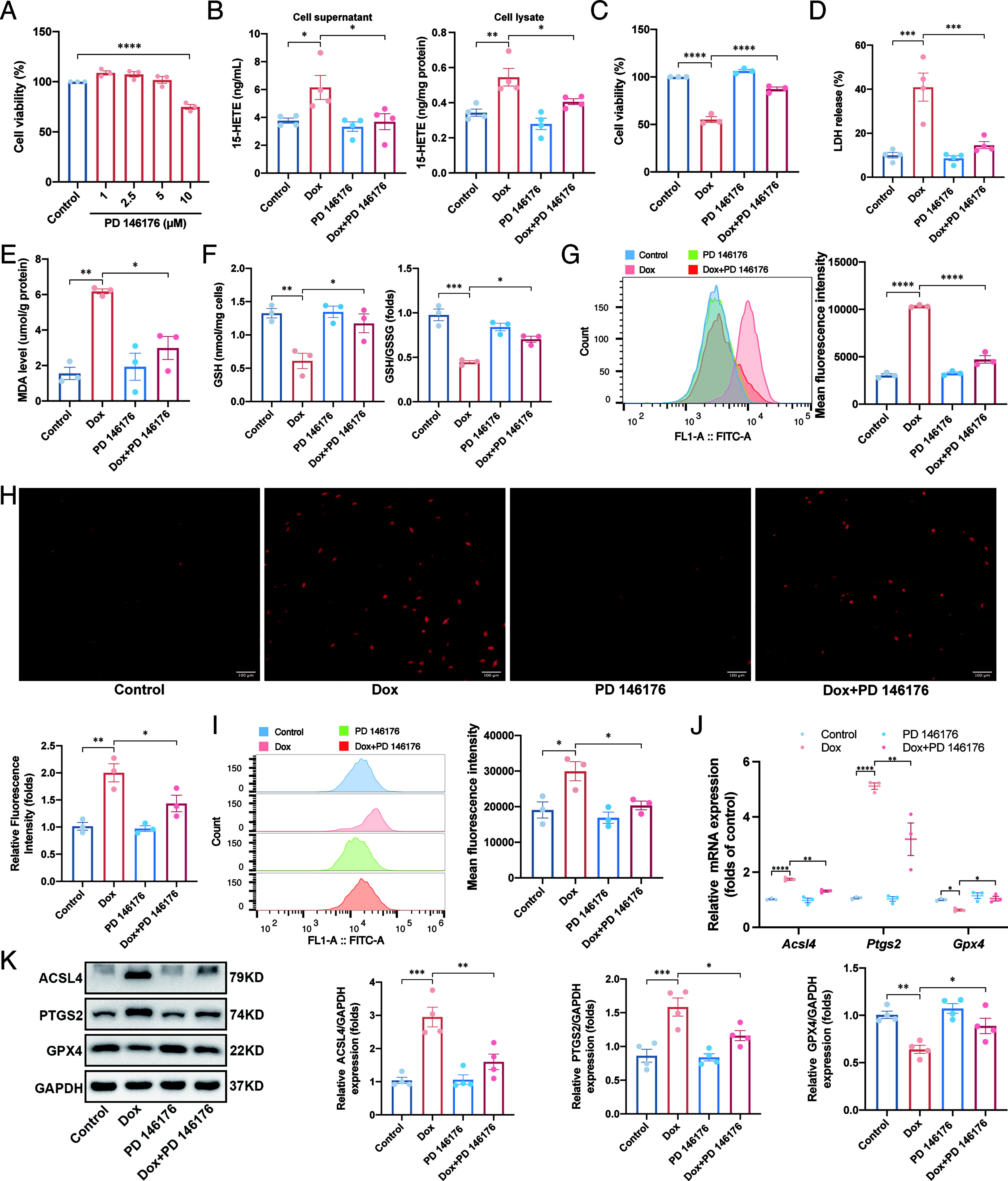
Then we treated H9C2 cells with 1 μM Dox for 24 h to mimic the DIC model. The ELISA results indicated that both pharmacological inhibition and genetic knockdown of ALOX15 reduced the levels of 15-HETE in both the supernatant and lysate of Dox-treated H9C2 cells (Fig. 3B and Supplementary Fig. S3C), confirming the effective loss-of-function approach. ALOX15 loss of function also significantly reduced Dox-induced cytotoxicity, as evidenced by improved cell viability (Fig. 3C and Supplementary Fig. S3D), reduced the LDH release (Fig. 3D and Supplementary Fig. S3E) and MDA level (Fig. 3E and Supplementary Fig. S3F), and elevated the GSH level and GSH/GSSG ratio (Fig. 3F and Supplementary Fig. S3G). Lipid peroxidation, assessed by C11-Bodipy staining, was robustly induced by Dox but was effectively reduced by ALOX15 loss of function (Fig. 3G and Supplementary Fig. S3H). We further investigated the effects of PD 146176 on intracellular ROS by using dihydroethidium (DHE) and dichlorodihydrofluorescein diacetate (DCFHDA) fluorescent probes. Results demonstrated that the promoting effects of Dox on ROS production were alleviated by ALOX15 loss of function (Fig. 3H–3I and Supplementary Fig. S3I–S3J). Next, RT-qPCR and Western blot analyses revealed that Dox treatment in H9C2 cells upregulated the mRNA and protein levels of ACSL4 and PTGS2 while downregulating GPX4, whereas these changes were markedly attenuated by ALOX15 loss of function (Fig. 3J–3K and Supplementary Fig. S3K–S3L).
We further examined the effects of PD 146176 in NRCMs. Similarly, PD 146176 markedly ameliorated Dox-induced LDH release, MDA elevation, and reduction in GSH level and GSH/GSSG ratio (Supplementary Fig. S4A–S4C). In addition, PD 146176 alleviated Dox-induced lipid peroxidation and intracellular ROS generation in NRCMs (Supplementary Fig. S4D and S4E). Western blot analysis further validated that PD 146176 suppressed Dox-induced ferroptosis in NRCMs (Supplementary Fig. S4F).
To determine if the cardioprotective effects of ALOX15 inhibition or silencing were mediated through inhibiting ferroptosis, we employed RSL3, a GPX4 inhibitor known to induce lethal accumulation of lipid hydroperoxides, ROS production, and ferroptotic cell death (Yang et al., 2014), to establish the ferroptotic model in H9C2 cells. The results of the CCK8 assay indicated that treating H9C2 cells with 0.5 μM or higher concentrations of RSL3 for 6 h significantly decreased the cell viability (Supplementary Fig. S5A). Therefore, 0.5 μM RSL3 was selected for subsequent experiments. In addition, we used Ferrostatin-1 (Fer-1), a well-established ferroptosis inhibitor, to validate whether RSL3 indeed induced ferroptosis. As expected, RSL3 treatment caused significant cell death (Supplementary Fig. S5B), increased LDH release (Supplementary Fig. S5C), reduced GSH level and GSH/GSSG ratio (Supplementary Fig. S5D), increased intracellular ROS (Supplementary Fig. S5E), and upregulated the expression of ACSL4 and PTGS2 while downregulating GPX4 (Supplementary Fig. S5F). However, Fer-1 effectively protected against RSL3-induced ferroptosis in H9C2 cells (Supplementary Fig. S5B–S5F). In the RSL3-induced model, we also found that the mRNA and protein levels of ALOX15 were elevated (Supplementary Fig. S5G–S5I).
Next, we explored whether ALOX15 loss of function could attenuate RSL3-induced ferroptosis. Consistent with the effect of Fer-1, ALOX15 loss of function also prevented RSL3-induced cell death (Supplementary Figs. S6A and S7A), LDH release (Supplementary Figs. S6B and S7B), increased MDA level (Supplementary Figs. S6C and S7C), and decreased GSH level and GSH/GSSG ratio (Supplementary Figs. S6D and S7D). In addition, RSL3-induced lipid peroxidation and intracellular ROS accumulation were significantly reduced by ALOX15 inhibition or silencing (Supplementary Figs. S6E–S6G and S7E–S7G). Furthermore, ALOX15 inhibition or silencing suppressed RSL3-induced ferroptosis, as evidenced by the expression of ferroptosis-related markers (Supplementary Figs. S6H–S6I and S7H–S7I).
Collectively, these results demonstrate that both pharmacological inhibition and genetic knockdown of ALOX15 effectively attenuate cytotoxicity induced by Dox through inhibiting ferroptosis.
PD 146176 alleviates the PUFA-triggered increased susceptibility toward Dox-induced cell damage
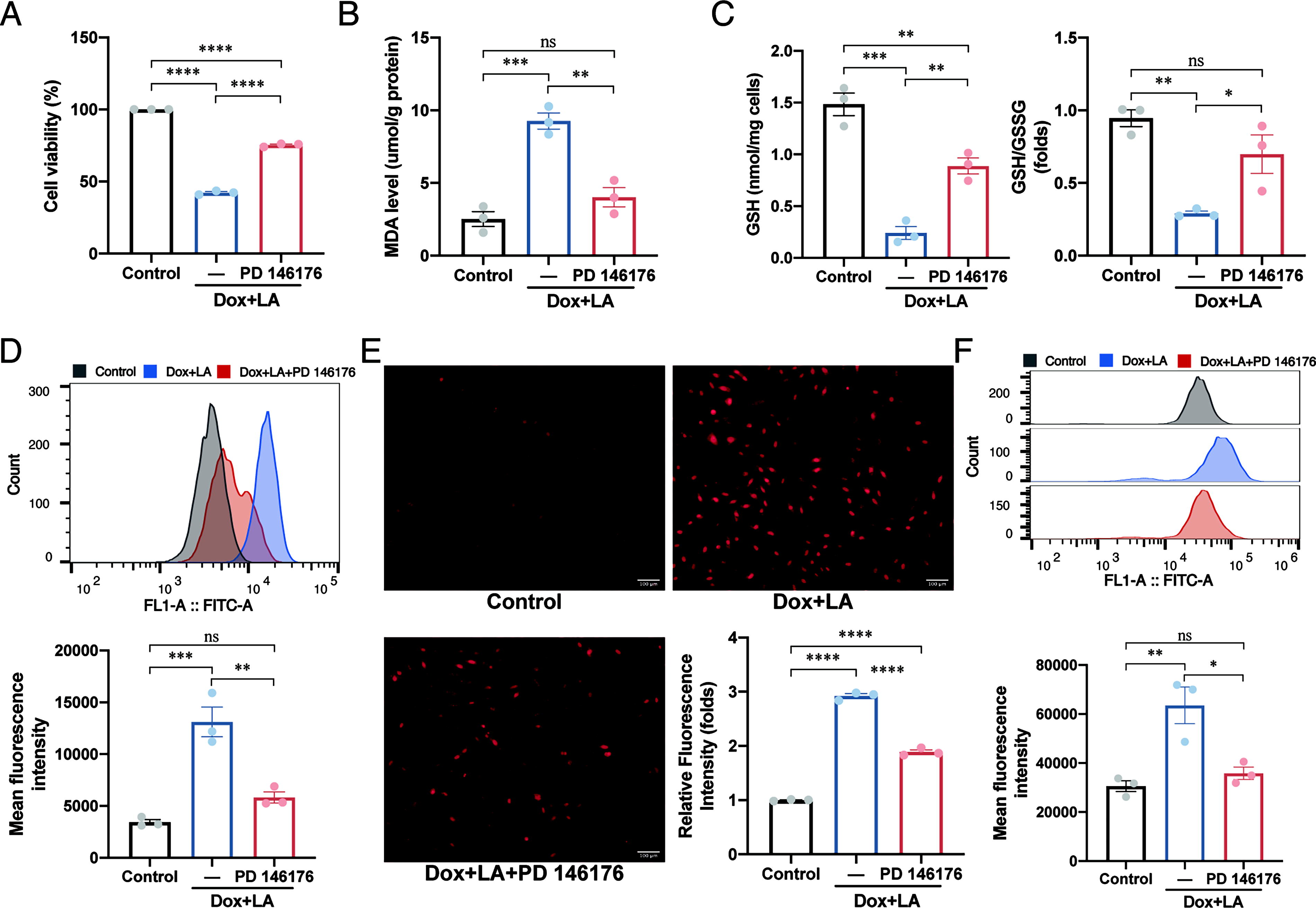
A previous study demonstrated that PUFA, rather than monounsaturated fatty acid (MUFA), induced phospholipids peroxidation, thereby increasing the susceptibility to ischemia-induced myocardial ferroptosis (Ma et al., 2022). In this study, we sought to determine whether PUFA or MUFA would increase susceptibility toward DIC. We incubated H9C2 cells with LA to establish a PUFA-enriched cell model. Meanwhile, the MUFA-enriched cell model was established by incubating H9C2 cells with oleic acid (OA). Cell viability analysis was performed to ascertain the treatment conditions for LA and OA, leading to the selection of incubating H9C2 cells with 160 μM LA and OA for 24 h for subsequent experiments (Supplementary Fig. S8A). Our findings revealed that the LA exposure increased the susceptibility of H9C2 cells to Dox-induced cytotoxicity (Supplementary Fig. S8B), lipid peroxidation (Supplementary Fig. S8C), and ROS accumulation (Supplementary Fig. S8D and S8E). In contrast, OA enrichment had no significant impact on H9C2 cells (Supplementary Fig. S8B–S8E). Furthermore, we found that the metabolism product of ALOX15, 15-HETE, significantly increased the levels of MDA (Supplementary Fig. S9A), lipid peroxidation (Supplementary Fig. S9B), and intracellular ROS (Supplementary Fig. S9C). To elucidate the direct correlation between ALOX15-mediated phospholipid peroxidation and Dox-induced cytotoxicity, we employed PD 146176 to inhibit ALOX15. PD 146176 alleviated the LA-triggered increased susceptibility toward Dox-induced cell damage, as evidenced by cell viability (Fig. 4A), MDA level (Fig. 4B), GSH level and GSH/GSSG ratio (Fig. 4C), lipid peroxidation (Fig. 4D), and intracellular ROS accumulation (Fig. 4E and 4F). These results illustrate that PUFA exposure exacerbates the Dox-induced damage in H9C2 cells, whereas PD 146176 mitigates such damage in the PUFA-enriched DIC model, highlighting the pivotal role of ALOX15 in phospholipid peroxidation and DIC.
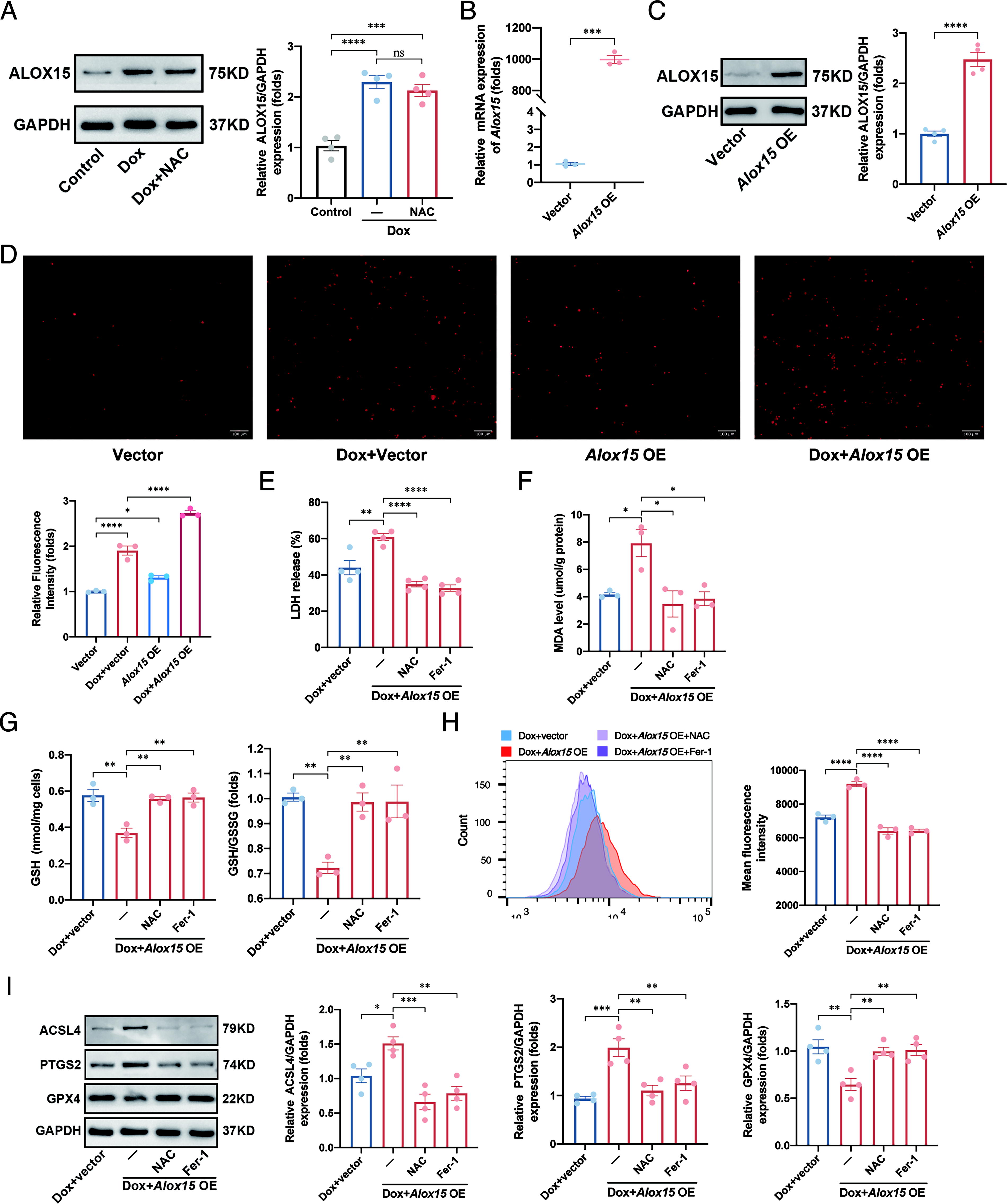
Alox15 overexpression promotes Dox-induced cell damage by aggravating ROS-mediated ferroptosis
To investigate whether the elevated expression of ALOX15 was directly induced by Dox or was a result of intracellular ROS accumulation, cells were pretreated with the ROS scavenger N-acetylcysteine (NAC) 24 h prior to Dox administration. Western blot results revealed that the ALOX15 protein level remained unaffected by NAC treatment (Fig. 5A), indicating that ALOX15 upregulation was ROS-independent. Subsequently, Alox15 overexpression plasmid was transferred into H9C2 cells to elucidate the relationship between ALOX15 and ROS. The significant overexpression effect was confirmed by RT-qPCR and Western blot analysis (Fig. 5B and 5C). Notably, Alox15 overexpression enhanced intracellular ROS accumulation (Fig. 5D), underscoring ALOX15′s pivotal role in ROS generation. Moreover, Alox15 overexpression exacerbated the Dox-induced increase in LDH release and MDA level, reduction in GSH level and the GSH/GSSG ratio, and escalation in lipid peroxidation (Fig. 5E–5H). These effects were mitigated by NAC or Fer-1 pretreatment. In addition, Alox15 overexpression exacerbated the Dox-induced elevation in PTGS2 and ACSL4 protein levels, and the reduction in GPX4 protein level, which were effectively counteracted by NAC or Fer-1 pretreatment (Fig. 5I). Collectively, these findings suggest that ALOX15 promotes ROS generation, thereby aggravating ferroptosis.
ALOX15 inhibition or silencing ameliorates Dox-induced ferroptosis via inhibiting the ROS-mediated MAPK signaling pathway activation
Studies have demonstrated a correlation between ALOX15 and the MAPK signaling pathway, which is closely associated with ferroptosis (Chen et al., 2023; Jiang et al., 2023; Zhao et al., 2011). In addition, bioinformatic analysis revealed an enrichment of the MAPK signaling pathway in DIC. Therefore, we investigated the effect of ALOX15 inhibition or silencing on the MAPK signaling pathway in DIC. We observed that Dox administration significantly upregulated phosphorylation levels of the extracellular signal-regulated kinase (ERK), c-JUN N-terminal kinase (JNK), and p38 MAPK. However, ALOX15 inhibition or silencing mitigated the effects of Dox on the activation of the MAPK pathway both in vivo (Supplementary Fig. S10A and S10B) and in vitro (Fig. 6A and 6B). These findings indicated that the protective effect of ALOX15 inhibition or silencing on ferroptosis may be mediated by inhibiting the activation of the MAPK signaling pathway.
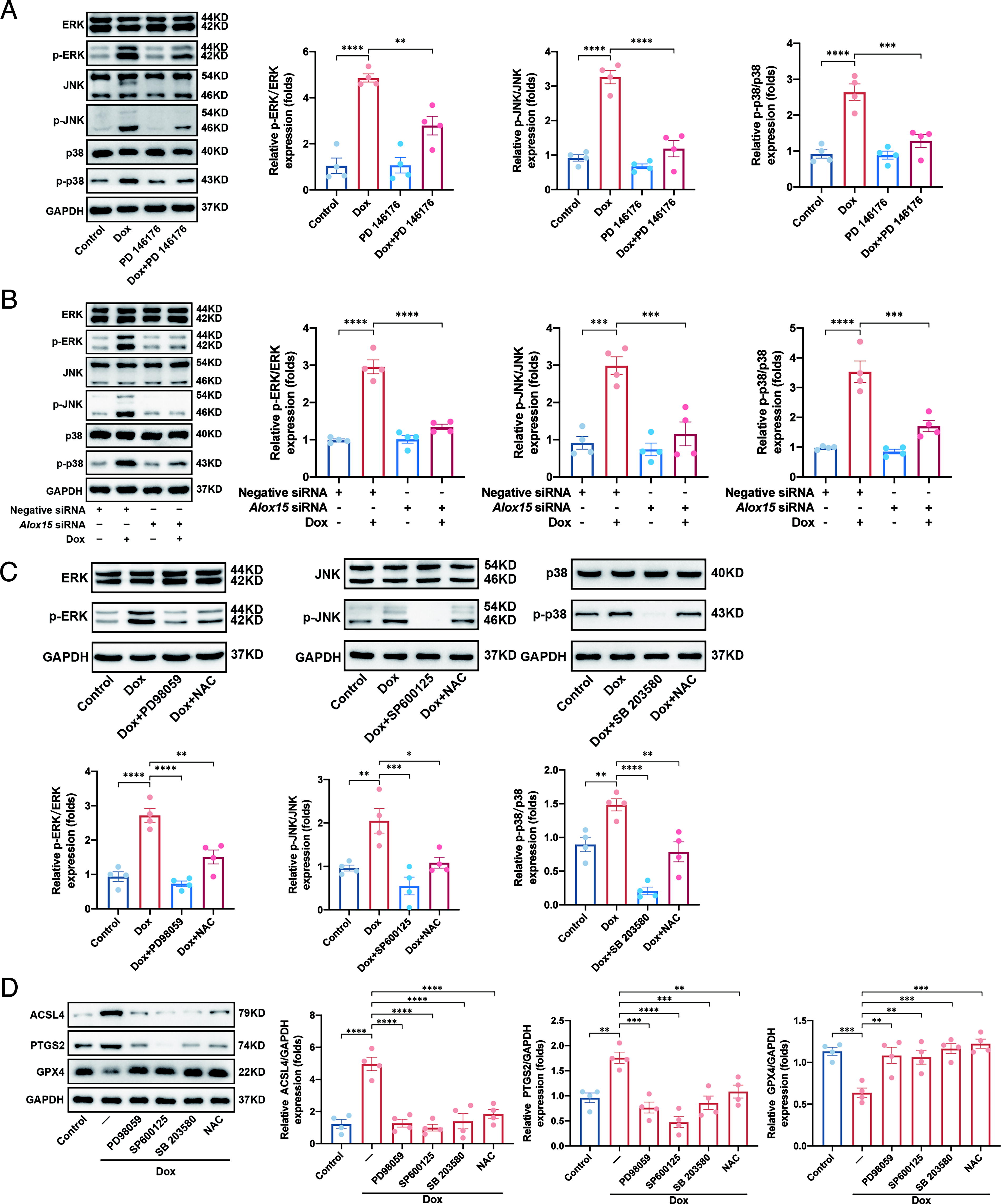
It has been shown that ROS can activate the MAPK signaling pathway (Lee et al., 2024; Qin et al., 2023; Zheng et al., 2024). In this study, we further explored whether the accumulation of lipid ROS mediated by ALOX15 would activate the MAPK signaling pathway. H9C2 cells were pretreated with NAC to determine whether ROS accumulation was an upstream signal for Dox-induced MAPK signaling pathway activation. Our results showed that the Dox-induced activation of ERK, JNK, and p38 MAPK phosphorylation was inhibited by pretreatment with NAC (Fig. 6C), suggesting that the lipid ROS generated by ALOX15 were essential for the MAPK signaling pathway activation. We also used PD98059, SP600125, and SB 203580 to inhibit ERK, JNK, and p38 MAPK, respectively, and observed significant inhibitory effects on the MAPK signaling pathway (Fig. 6C). Western blot revealed that the ferroptosis induced by Dox was significantly inhibited when H9C2 cells were pretreated with PD98059, SP600125, SB 203580, and NAC (Fig. 6D). These results indicate that inhibition or silencing of ALOX15 alleviates Dox-induced ferroptosis by suppressing ROS-mediated MAPK signaling pathway activation.
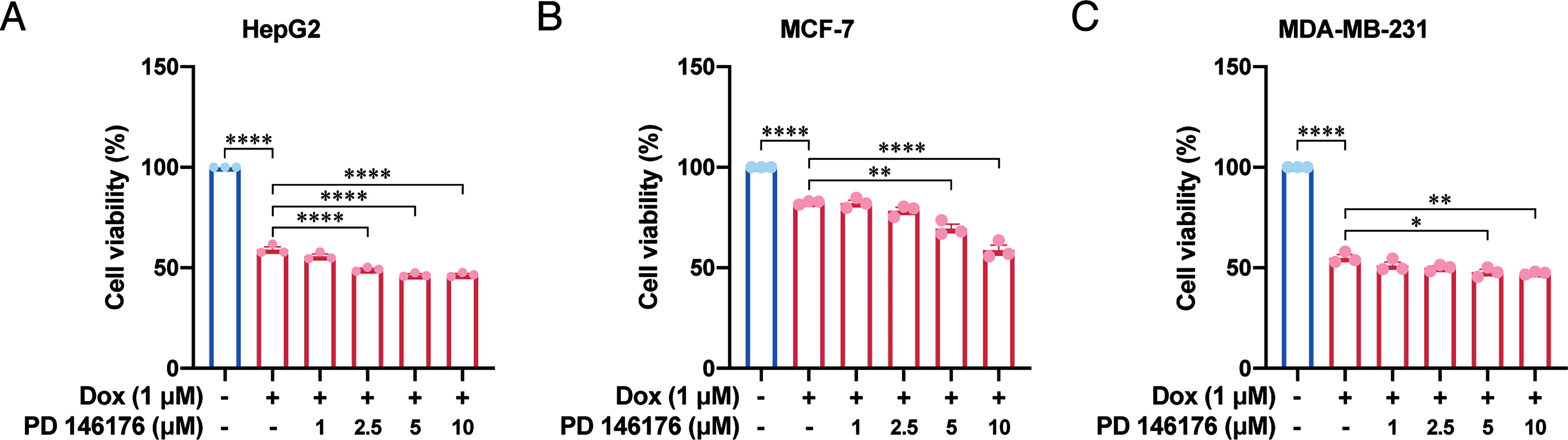
PD 146176 mitigates DIC while enhancing the antitumor effect of Dox in vitro
It is essential to evaluate whether PD 146176 would affect the antitumor effect of Dox. Our results showed that compared with Dox group, the proliferation activity of HepG2 cells was decreased after cotreatment with 2.5–10 μM PD 146176 and Dox (Fig. 7A). Compared with Dox group, although the proliferation activity of MCF-7 and MDA-MB-231 cancer cells did not change after cotreatment with 1 or 2.5 μM PD 146176 and Dox, the antitumor effect was enhanced after cotreatment with 5 or 10 μM PD 146176 and Dox (Fig. 7B and 7C). These findings indicate that 5 μM PD 146176 used in this research enhances the antitumor activity of Dox in vitro.
Discussion
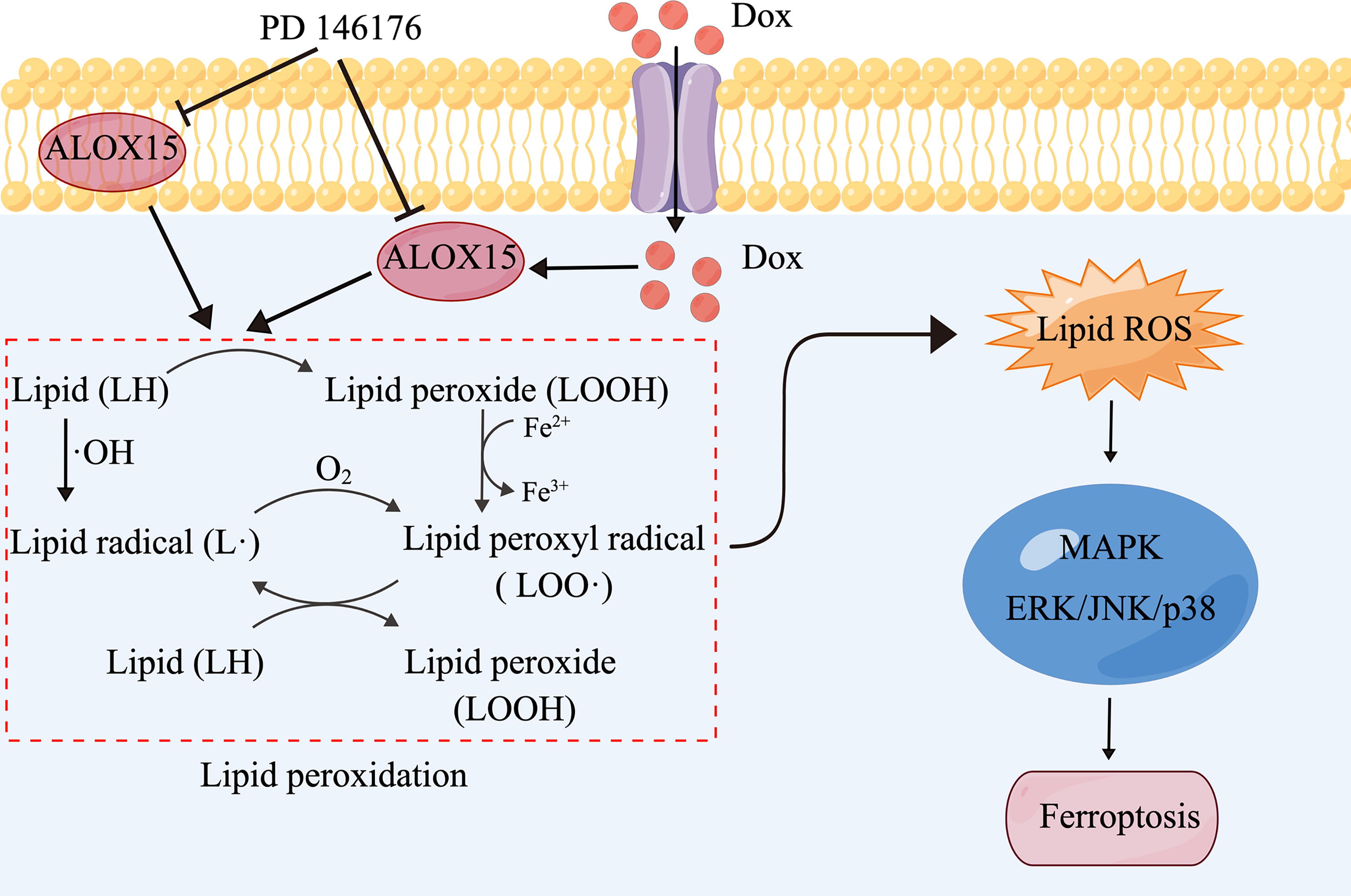
In the present study, we uncovered the essential role of ALOX15 in DIC. We found that ALOX15 expression was markedly increased in Dox-treated cardiomyocytes and hearts. In addition, we obtained evidence that inhibiting or silencing ALOX15 effectively ameliorated Dox-induced lipid peroxidation, ROS accumulation, and ferroptosis, thereby improving DIC. Furthermore, we found that the upregulated expression of ALOX15 catalyzed PUFA, which led to lipid peroxidation and induced ROS-mediated activation of the MAPK signaling pathway, ultimately triggering ferroptosis. These findings are summarized in Figure 8.
Dox is a widely used chemotherapeutic agent, well known for its ability to control cell growth and induce cell death. However, it can also cause dosage-dependent cardiac toxicity (Wallace et al., 2020). Although various types of cell death mechanisms, such as apoptosis, necroptosis, autophagy, and pyroptosis, have been identified to be involved in DIC, the exact molecular mechanism remains unclear. Previous studies have shown that targeting one of these cell death mechanisms failed to protect against DIC completely (Fang et al., 2019; Hou et al., 2021), indicating the existence of other more essential mechanisms. In recent years, a novel form of cell death named ferroptosis has been discovered, which is caused by iron-dependent lipid peroxide accumulation, along with ROS overproduction (Dixon et al., 2012). Since then, emerging evidence has highlighted the significant role of ferroptosis in DIC. For instance, Fang et al. initially reported that the ferroptosis inhibitor could significantly reduce Dox-induced mortality in mice (Fang et al., 2019). Tadokoro et al. also validated the critical role of mitochondria-dependent ferroptosis in the progression of DIC (Tadokoro et al., 2020). Furthermore, an increasing amount of evidence indicated that ferroptosis, rather than other forms of cell death, primarily contributed to the development of DIC, suggesting that targeting ferroptosis may be a potential strategy for treating DIC (Fang et al., 2019; Hou et al., 2021).
The accumulation of lipid ROS is a crucial characteristic of ferroptosis. Through the Fenton reaction, hydrogen peroxide (H2O2) could be catalyzed by Fe2+ into the toxic hydroxyl radical (•OH), which subsequently initiates a chain reaction with PUFAs in cellular membranes. This process leads to the production of substantial amounts of lipid ROS, ultimately resulting in cell death (Shen et al., 2018). In addition, LOXs can directly oxygenate PUFAs, leading to lipid peroxidation and subsequent damage to cellular functions (Kuhn et al., 2018; Kuhn et al., 2005). ALOX15, an enzyme capable of degrading PUFAs, has been reported to be involved in ferroptosis in many diseases, such as renal failure (Friedmann Angeli et al., 2014), neurological diseases (Bouchaoui et al., 2023; Zhao et al., 2022), and asthma (Nagasaki et al., 2022). Notably, recent research has revealed the role of ALOX15 in cardiac ischemia-reperfusion injury, which provided new insights and targets for the treatment of myocardial infarction. Ma et al. found that ALOX15 increases the sensitivity of ferroptosis during ischemia-reperfusion myocardial injury by initiating PUFA-phospholipids peroxidation (Ma et al., 2022). Besides, Cai et al. found that ALOX15 and its intermediate metabolite 15-hydroperoxyeicosatetraenoic acid (15-HPETE) exacerbate cardiac ischemia-reperfusion injury (Cai et al., 2023).
In the present study, we discovered that ALOX15 was upregulated in both the DIC model and RSL3-induced ferroptotic model. This observation is consistent with previous studies demonstrating elevated expression of ALOX15 in the models of myocardial infarction (Cai et al., 2023; Ma et al., 2022), heart failure (Kayama et al., 2009), and diabetic cardiomyopathy (Suzuki et al., 2015). Although one study indicated that RSL3 does not increase ALOX15 protein level in HT1080 cells, the authors suggested that this might be due to nonspecific degradation following cell membrane disruption (Shintoku et al., 2017). However, it is noteworthy that the expression and regulation of ALOX15 can vary across different cell types. Our study further revealed that genetic or pharmacological inhibition of ALOX15 effectively ameliorated Dox-induced lipid peroxidation, ROS accumulation, and ferroptosis, thereby improving Dox-induced cardiac function impairment, which raises the possibility that ALOX15-mediated peroxidation of PUFAs causes ferroptosis in DIC and indicates that ALOX15 is a potential target for DIC intervention.
PUFA, the source of lipid peroxidation, was once deemed beneficial for cardiovascular diseases (CVD) (Kagawa et al., 1982; Mensink et al., 2003). However, the cardioprotective role of PUFAs has been challenged recently. A series of systematic reviews of randomized clinical trials indicated that increasing PUFA intake has a modest to no effect on CVD events or mortality (Abdelhamid et al., 2018a; Abdelhamid et al., 2018b; Hooper et al., 2018). LA has been reported to increase collagen I/III ratio and cardiac “stiffening” with early diastolic dysfunction (Beam et al., 2015). A previous study also found that PUFA-enriched animals/cells were more susceptible to ischemia-hypoxia injury (Ma et al., 2022). LA and AA could be catalyzed by ALOX15 to 13-hydroperoxyoctadecadienoic acid and 15-HPETE, which have been validated to induce loss of cardiomyocytes membrane integrity and aggravate myocardial ischemia-reperfusion injury (Cai et al., 2023; Thollon et al., 1995). The present study also provided evidence that LA increased the susceptibility of H9C2 cells to Dox-induced cell damage due to ALOX15-catalyzed phospholipid peroxidation. In contrast, treatment with MUFA did not have a significant impact on Dox-induced cell damage, which is consistent with the previous study (Ma et al., 2022). This may be attributed to the ability of the less oxidizable MUFA to substitute more oxidizable PUFA from the membrane phospholipids, consequently limiting lipid peroxidation (Pope and Dixon, 2023). Moreover, this study found that overexpressing Alox15 exacerbated Dox-induced cell damage by augmenting intracellular ROS accumulation, while scavenging ROS or inhibiting ferroptosis was found to alleviate this effect. These results further confirm that Dox-induced upregulation of ALOX15 expression facilitates lipid peroxidation, thereby aggravating ferroptosis.
The MAPK family, comprising JNK, ERK, and p38 MAPK, plays a vital role in various cell death pathways. Notably, the MAPK signaling pathway has been reported to be intimately linked to ferroptosis across a spectrum of diseases, including acute respiratory distress syndrome (Wang et al., 2022), osteoarthritis (Miao et al., 2022), hypoxic-ischemic brain damage (Zhu et al., 2021), and myocardial ischemia/reperfusion injury (Chen et al., 2023; Jiang et al., 2023). Chen et al. discovered that Dapagliflozin treatment protected against myocardial ischemia-reperfusion injury by downregulating MAPK phosphorylation in vitro and in vivo (Chen et al., 2023). Jiang et al. showed that adaptor protein HIP-55 alleviated ferroptosis by negatively regulating the MAP4K1-dependent JNK pathway in myocardial infarction (Jiang et al., 2023). In addition, ALOX15 is closely related to the MAPK signaling pathway. One study demonstrated that ALOX15 and its product, 15-HETE conjugated to PE, function as signaling molecules that intersect with PEBP1 to activate Raf-1/mitogen-activated protein kinase kinase (MEK)/ERK (Zhao et al., 2011). In line with these results, our data revealed that ALOX15 inhibition and silencing suppressed the activated phosphorylation of JNK, ERK, and p38 induced by Dox. On the other hand, an extensive body of studies indicated that oxidative stress was the primary reason for MAPK pathway activation (He et al., 2022; Lee et al., 2024; Qin et al., 2023; Zheng et al., 2024). Our study further substantiated that the activation of the MAPK pathway was mediated by ROS accumulation, which subsequently triggered ferroptosis.
The present study is not without limitations. While we employed the putative ALOX15 inhibitor PD 146176 in this study, a study has demonstrated that 20 μM PD 146176 inhibits CYP epoxygenase rather than functions as an ALOX15 inhibitor in endothelial cells (Du et al., 2022). This raises the possibility that the protective effect of PD 146176 on the heart might be attributed to its inhibition of the CYP family. However, this likelihood appears to be low in our study, as our findings revealed a significant reduction in the metabolite of ALOX15, 15-HETE, after using 5 μM PD 146176. Moreover, we must acknowledge that ALOX15 may engage in DIC through other pathways. A study demonstrated that MITOL/MARCH5 determines cardiomyocyte fate via the ferroptosis process (Kitakata et al., 2021). As an E3 ubiquitin ligase, MITOL/MARCH5 plays a critical role in regulating mitochondrial quality and function, potentially being involved in ALOX15-mediated ROS generation. Furthermore, the protective effect of ALOX15 inhibition or silencing against RSL3-induced toxicity appears to be more pronounced than against DIC in our study, suggesting that while ferroptosis predominates in the pathological process of DIC, other forms of cell death might also be involved.
Materials and Methods
Bioinformatic tools
The expression profiling data used in this study were obtained from GEO (https://www.ncbi.nlm.nih.gov/geo/). The “GEOquery” package in R (Davis and Meltzer, 2007) was used to get the raw datasets (GSE154603). All data were converted into a log2 scale to facilitate further analysis. The R package “limma” (Ritchie et al., 2015) was utilized to obtain DEGs, which were identified based on the criteria of p < 0.05 and |log2 (fold change)| >1. The resulting DEGs were visualized using volcano plots. Furthermore, we obtained 484 FRGs from the FerrDb database (http://www.zhounan.org/ferrdb/current/, accessed: January 2023) and intersected them with DEGs to identify DEFRGs. GO enrichment analysis and KEGG pathway analysis were performed in R using the “clusterProfiler” package (Yu et al., 2012) to investigate the biological functions of these DEFRGs. In addition, we performed the GSEA analysis utilizing the “clusterProfiler” package (Yu et al., 2012). A threshold of adjusted p < 0.05 was considered significant.
Animals
All animal experiments were approved by the Animal Experimental Ethical Inspection of the First Affiliated Hospital, Zhejiang University, School of Medicine (Reference Number: 2022-1124), in strict accordance with relevant ethical guidelines. Male C57BL/6J mice (8–10 weeks, 20–25 g) sourced from the Academy of Medical Science (Zhejiang, China) were maintained in a temperature-controlled room (22 ± 2°C) under a 12-h light–dark cycle. Mice were initially randomly divided into four groups: the Control group, the Dox group, the PD 146176 group, and the Dox+PD 146176 group (n = 8 for each group). Mice in the Control group received intraperitoneal injection of an equal volume of sterile saline daily. Mice in the PD 146176 group and Dox + PD 146176 group received PD 146176 (10 mg/kg/day; GlpBio, USA) for 4 weeks. The DIC model was established by intraperitoneal injections of Dox (GlpBio, USA) with a 24 mg/kg cumulative dose (three times/week for the last 2 weeks). In a separate cohort, mice were randomly divided into four groups: AAV9-shCtrl group, Dox+AAV9-shCtrl group, AAV9-shAlox15 group, and Dox+AAV9-shAlox15 group (n = 6 for each group). These mice initially received an injection of either AAV9-shCtrl or AAV9-shAlox15 via the tail veins. Following a 2-week interval, the mice received intraperitoneal injections of an equivalent volume of sterile saline solution or a cumulative dose of 24 mg/kg of Dox (same method as described above). Twenty-four hours after the last injection of Dox, transthoracic echocardiography examination was performed under anesthesia (3% pentobarbital sodium). After that, all mice were sacrificed, and heart tissues were collected, frozen in liquid nitrogen, and stored at −80°C for further experiments or fixed with 4% paraformaldehyde for the histological examination.
H&E staining
Myocardial tissue samples from the mice were fixed with 4% paraformaldehyde for 24 h and subsequently embedded with paraffin. Embedded tissues were cut into 5 μm sections, and then H&E staining (Servicebio, China) was performed to investigate the histological changes. The images were captured using a light microscope (Olympus, BX53, Japan).
Cell culture and treatment
Rat cardiac myoblast cell line H9C2 was purchased from the Cell Bank of the Chinese Academy of Sciences. The cells were routinely cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA), supplemented with 10% fetal bovine serum (FBS, ExCell Bio, Uruguay) and 1% penicillin/streptomycin, and then incubated at 37°C with 5% CO2. To construct DIC and ferroptotic model in vitro, H9C2 cells were treated with 1 μM Dox (GlpBio, USA) for 24 h, 0.5 μM RSL3 (MCE, USA) for 6 h, or 5 μM 15-HETE (GlpBio, USA) for 24 h. For the rescue experiments in vitro, the cells were treated with PD 146176 (5 μM) or Fer-1 (5 μM; MCE, USA) for 24 h before Dox or RSL3 administration. A consistent concentration of DMSO (0.1%) was applied across all groups.
Primary NRCMs were isolated with the cold trypsin digestion method, following a standardized laboratory protocol. Briefly, myocardial tissues from Sprague–Dawley rats aged 0 to 1 day old were incubated with trypsin at 4°C for 10 h. Subsequently, these myocardial tissues were subjected to digestion with warm trypsin every 5 min, a process that was repeated five times. The isolated cells were then allowed to differentially adhere for 1.5 h. Following this, the primary NRCMs were cultured in high-glucose DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 0.1 mmol/L BrdU for 24 h. NRCMs were pretreated with PD 146176 (5 μM) for 24 h and then stimulated with Dox (2 µM) for an additional 24 h to establish the DIC model.
In the present study, H9C2 cells were pretreated with the ROS scavenger NAC at a concentration of 500 μM for 24 h before the administration of Dox. Furthermore, to inhibit the MAPK signaling pathway, the following MAPK pathway inhibitors were employed: 10 μM PD98059 (ERK inhibitor) (Gutiérrez-Venegas et al., 2017), 10 μM SP600125 (JNK inhibitor) (Gutiérrez-Venegas et al., 2017), 3 μM SB 203580 (p38 MAPK inhibitor) (Guo et al., 2013), which were used to pretreat H9C2 cells for 1 h before Dox stimulation.
Dox-sensitive cancer cells, liver hepatocellular carcinoma cells (HepG2), and human breast cancer cell lines (MCF-7 and MDA-MB-231) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were cotreated with 1 µM Dox and PD 146176 for 24 h to evaluate the impact of PD 146176 on the antitumor effect of Dox in tumor cells.
After the treatments, the cells were subsequently stained or collected for a variety of assays.
IF staining
IF staining was used to measure the expression of ALOX15. In the case of H9C2 cells, the cells were washed three times with phosphate-buffered saline. Subsequently, the cells were fixed with 4% paraformaldehyde (Servicebio, China) for 15 min and then permeabilized with 0.5% Triton X-100 for 20 min at room temperature. After being covered with 5% bovine serum albumin (Sigma-Aldrich, USA) for 30 min, the cells were incubated with the primary antibody against ALOX15 (ab244205, Abcam, USA, diluted with 5% bovine serum albumin) overnight at 4°C. The following day, the cells were covered with secondary antibody (Cy3 conjugated, AS007, ABclonal, China), and the nuclei were stained with DAPI (Servicebio, China). The procedure for IF staining of mouse heart tissues was analogous to that of the cells. After deparaffinization, rehydration, and serum blocking, the slides were incubated with primary antibodies against ALOX15 and Troponin-I (GB122408, Servicebio, China). The following day, the slides were covered with secondary antibodies [Fluorescein isothiocyanate (FITC) or Cy3 conjugated], and the nuclei were stained with 4′,6-DiAmidino-2-PhenylIndole (DAPI). Three random visual fields were captured using a confocal laser scanning microscope (Olympus FV3000, Japan).
Enzyme-linked immunosorbent assay
H9C2 cells were seeded in 6 cm dishes, and before treatments, the median was changed to 2 mL per dish to avoid too low concentration of 15-HETE in cell supernatant. After treatments, the cell supernatant and lysate of H9C2 cells were collected. In addition, serum from mice blood samples and heart tissues was collected. The concentration of 15-HETE was measured using the 15-HETE ELISA kit (EU2612, Fine Biotech, China), in accordance with the manufacturer’s instructions.
Cell viability
Cells were seeded in 96-well plates. After treatments, cell viability was determined by using the CCK-8 assay (GlpBio, USA). In brief, 10 μL CCK8 reagent was added to each well and incubated for 1 h. Subsequently, a microplate reader (SpectraMax i3x, USA) was used to measure the absorbance values at the wavelength of 450 nm.
LDH release detection
The release of LDH was regarded as an indicator of cytotoxicity. After the indicated treatments, a commercially available kit (A020-2-2, Nanjing Jiancheng Bioengineering Institute, China) was utilized to determine the concentration of LDH in the serum obtained from mice blood samples. Furthermore, another kit (C0017, Beyotime, China) was employed to measure the LDH level in the cell supernatant, following the manufacturer’s instructions. The absorbance of samples was measured at wavelengths of 490 nm and 600 nm (reference wavelength) using a microplate reader (SpectraMax i3x, USA).
MDA detection
MDA measurement was performed by a commercial kit (S0131S, Beyotime, China) per the manufacturer’s instructions. Protein concentrations were detected by the enhanced BCA protein assay kit (P0010, Beyotime, China).
GSH content detection
H9C2 cells (10 mg) or heart lysate (30 mg) were used to detect the level of GSH in vitro and in vivo by the kit (S0053, Beyotime, China) per the manufacturer’s instructions.
Lipid peroxidation and intracellular ROS detection
For lipid peroxidation measurements, H9C2 cells were harvested with Hank’s balanced salt solution (HBSS) containing 5 μmol/L Bodipy (BODIPY™ 581/591 C11, Invitrogen, USA) and incubated at 37°C in the dark for 30 min. Following incubation, the cells were washed twice with HBSS to remove excess C11-BODIPY. The labeled cells were then trypsinized and resuspended in HBSS. Subsequently, fluorescence was measured using a flow cytometer (BECKMAN CytoFLEX, USA). Intracellular ROS was assessed using the fluorescent dye DHE (S0063, Beyotime, China). H9C2 cells were incubated with 10 μmol/L DHE at 37°C in the dark for 30 min. For each well, three random visual fields were selected under a microscope (Olympus IX 73, Japan), and the mean fluorescence intensity was assessed for analysis using Image J. In addition, 10 μmol/L DCFHDA (S0033S, Beyotime, China) was used to stain the H9C2 cells for 30 min to detect ROS level, and fluorescent signals were collected using flow cytometry (BECKMAN CytoFLEX, USA).
RNA extraction and RT-qPCR
Total RNA was extracted from H9C2 cells and mice heart samples by Trizol reagent per the manufacturer’s instructions. After extraction, the cDNA Synthesis kit (G3330, Servicebio, China) was used to synthesize cDNA by using 1 μg of total RNA in a 20 μL reaction. RT-qPCR amplification of the generated cDNA was performed using the SYBR Green qPCR Master Mix (Low ROX, G3321, Servicebio, China). The primers used are listed as follows:
Alox15 (rat): forward, 5′-AGGCTTGCTACTTCATCAC-3′, reverse, 5′-CTTCTCCATTGTTGCTTCC-3′; Gapdh (rat): forward, 5′-GGTGGACCTCATGGCCTACA-3′, reverse, 5′-CTCTCTTGCTCTCAGTATCCTTGCT-3′; Acsl4 (rat): forward, 5′-CTCAAGCATTCCTCCAAGT-3′, reverse, 5′-GGTGACGAATATCCAATCCT-3′; Ptgs2 (rat): forward, 5′-ATGTTCGCATTCTTTGCCCAG-3′, reverse, 5′-TACACCTCTCCACCGATGAC-3′; Gpx4 (rat): forward, 5′-GGAGGCAGGAGCCAGGAAGTAA-3′, reverse, 5′-AGCCGTTCTTATCAATGAGAAACTTGG-3′; Alox15 (mouse): forward, 5′-GCTGTTTGTGAGAGTGCAGAA-3′, reverse, 5′-AGGGGAACGTGTACTCCGAT-3′; Gapdh (mouse): forward, 5′-GGTTGTCTCCTGCGACTTCA-3′, reverse, 5′-GGTGGTCCAGGGTTTCTTACTC-3′; Acsl4 (mouse): forward, 5′-CCACACTTATGGCCGCTGTT-3′, reverse, 5′-GGGCGTCATAGCCTTTCTTG-3′; Ptgs2 (mouse): forward, 5′-TTCCAATCCATGTCAAAACCGT-3′, reverse, 5′-AGTCCGGGTACAGTCACACTT-3′; Gpx4 (mouse): forward, 5′-GATGGAGCCCATTCCTGAACC-3′, reverse, 5′-CCCTGTACTTATCCAGGCAGA-3′; Nppa (mouse): forward, 5′-GTGCGGTGTCCAACACAGAT-3′, reverse, 5′-TCCAATCCTGTCAATCCTACCC-3′; Nppb (mouse) forward, 5′-GAGGTCACTCCTATCCTCTGG-3′, reverse, 5′-GCCATTTCCTCCGACTTTTCTC-3′.
Western blot
Total proteins were isolated from cells or mice heart samples on ice using Radio Immunoprecipitation Assay (RIPA) buffer (Beyotime, China) containing protease and phosphatase inhibitors (MCE, USA). Equal amounts of protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred to 0.22 μm polyvinylidene fluoride (PVDF) membrane (Merck Millipore, Billerica, MA, USA). The membranes were then blocked with 5% skim milk (Beyotime, China) in Tris-buffered saline for 1 h at room temperature. Following this, the blots were incubated overnight at 4°C with corresponding primary antibodies. The primary antibodies used were ALOX15 (ab244205, Abcam, USA), GPX4 (A11243, ABclonal, China), PTGS2 (12282, CST, USA), ACSL4 (GB113871, Servicebio, China), ERK (A16686, ABclonal, China), p-ERK (AP0974, ABclonal, China), JNK (9252, CST, USA), p-JNK (4668, CST, USA), p38 MAPK (8690, CST, USA), and p-p38 MAPK (4511, CST, USA). Afterward, the blots were incubated with secondary antibodies for 1 h at room temperature and then quantified using the Clinx Image Analysis System (CLiNX, China) with the enhanced chemiluminescence reagent (Millipore, USA).
siRNA and plasmid transfection
Rat siRNA targeting Alox15 and control siRNA were synthesized by RiboBio (China). The sequences for si-Alox15 were CCAGAAAGGCACUCUGUUUTT (sense) and AAACAGAGUGCCUUUCUGGTT (antisense), which were identical to the siRNA used in previous study (Ma et al., 2022). The sequences for the negative control siRNA were UUCUCCGAACGUGUCACGUTT (sense) and ACGUGACACGUUCGGAGAATT (antisense). H9C2 cells were initially seeded into 6-well plates and cultured in 2 mL of antibiotic-free DMEM supplemented with 10% FBS. Subsequently, cells at a 50% confluence were transfected with either control or target siRNAs duplex, which was incubated with Lipofectamine 3000 (Invitrogen). After 48 h of transfection, the cells were harvested for further analysis. The silencing of ALOX15 was then confirmed by RT-qPCR and Western blot analysis.
For ALOX15 overexpression, H9C2 cells were seeded in 6-well plates and cultured in 2 mL of antibiotic-free DMEM supplemented with 10% FBS. Upon reaching 50% confluence, the cells were transfected with either the pCMV-Alox15(rat)-3×Flag-Neo plasmid or empty vector (Miaoling, China) utilizing Lipofectamine 3000 (Invitrogen). After 48 h of transfection, the cells were harvested for further analysis. The overexpression of ALOX15 was subsequently confirmed by RT-qPCR and Western blot analysis.
Statistical analysis
All experimental results are presented as mean ± standard error of the mean (SEM). Statistical analysis was conducted using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). Initially, the Shapiro–Wilk test was employed to determine if the data were normally distributed. For comparing two groups with normally distributed data, the Student’s t test was used for similar variances; if equal standard deviations are not assumed based on an F test, Welch’s correction was applied. When comparing more than two groups, the Brown–Forsythe test was performed to assess similar variances, and then ordinary analysis of variance (ANOVA) or Welch ANOVA analysis was conducted depending on whether the assumption of similar variances was met. For nonnormally distributed data, the Kruskal–Wallis with Dunn’s multiple comparisons test was utilized. Values of p < 0.05 were considered statistically significant. Electronic laboratory notebook was not used in this study.
Footnotes
Acknowledgments
The authors would like to acknowledge Home for Researchers (www.home-for-researchers.com) and Figdraw (https://www.figdraw.com/static/index.html) for their assistance in creating the ![]() .
.
Authors’ Contributions
X.S., L.C., L.W., and L.Z.: Conceived and designed the study. X.S., L.C., J.H., W.C., S.L., T.C., M.C., H.Z., and Y.H.: Performed all the experiments. X.S. and L.C.: Collected and analyzed the data, and prepared the initial version of the article. J.H., W.C., S.L., and T.C.: Helped revise the article. L.W. and L.Z.: Initiated and supervised the project, and revised the article. All authors approved the final version of the article.
Data Availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the grants from the National Key Research and Development Program of China (No. 2023YFC3606500), the National Natural Science Foundation of China (No. 82470428), the Zhejiang Provincial Natural Science Foundation of China (LHDMD25H020001 and LY23H020003), and the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2023KY679).
Supplementary Material
Supplementary Figures
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.