
Abstract
Aims:
Myocardial ischemia–reperfusion (I/R) injury facilitates cardiomyocyte death and endangers human health. N6-methyladenosine (m6A) methylation plays a critical role in cardiovascular diseases. The m6A reader YTHDF2 identifies m6A-modified RNA and promotes target RNA degradation. Hence, we hypothesized that YTHDF2 affects I/R injury by regulating RNA stability.
Results:
Both messenger RNA (mRNA) and protein levels of YTHDF2 were upregulated in I/R mice and hypoxia-reoxygenation (H/R)-induced cardiomyocytes. Silencing endogenous YTHDF2 abrogated cardiac dysfunction and lowered the infarct size in I/R mice, and the forced expression of YTHDF2 aggravated these adverse pathological processes. Consistently, the protective effect of silencing YTHDF2 occurred in cardiomyocytes exposed to H/R and erastin. Further, RNA-Seq and RNA-binding protein immunoprecipitation (RIP) revealed that YTHDF2 recognized the m6A modification sites of the ferroptosis-related gene solute carrier family 7 member 11 (SLC7A11) mRNA to promote its degradation both in vivo and in vitro. Inhibition of SLC7A11 impaired cardiac function, increased infarct size, and the release of lactate dehydrogenase (LDH) in I/R mice after silencing YTHDF2. The beneficial effects of si-YTHDF2 on H/R injury were reversed by co-transfection with SLC7A11-specific siRNA (si-SLC7A11), which substantially exacerbated ferroptosis and the production of reactive oxygen species.
Innovation and Conclusion:
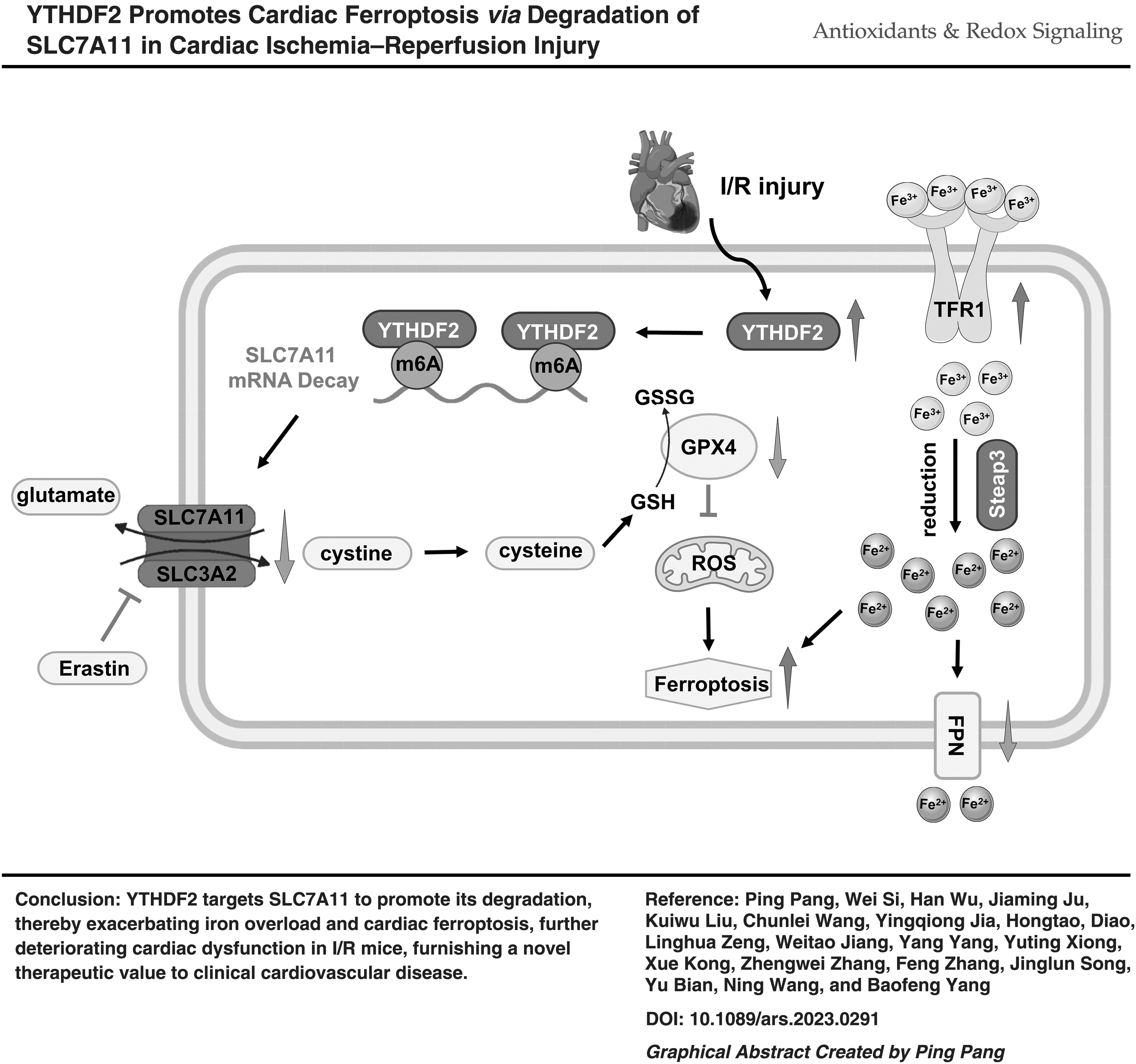
The cardioprotective effects of silencing YTHDF2 are accomplished by increasing SLC7A11 stability and expression, reducing ferroptosis, and providing novel potential therapeutic targets for treating ischemic cardiac diseases.
Introduction
Cardiovascular diseases threaten human life and significantly influence global health load (Chouchani et al., 2014). Myocardial ischemia–reperfusion (I/R) injury is caused by oxidative stress damage to cardiomyocytes following coronary artery recanalization (Chen et al., 2020). During I/R injury, oxidative stress can severely impair cardiac function (Gumpper-Fedus et al., 2022; Guo et al., 2020a).
Excessive mitochondrial reactive oxygen species (ROS) can affect the permeability of mitochondria and cause oxidative damage to specific internal molecules, which is the main driving factor leading to I/R (Cai et al., 2022). Cardiac I/R injury induces various inflammatory responses during cardiomyocyte necrosis and apoptosis, leading to cardiac systolic and diastolic dysfunctions (Heusch, 2015). Cardiomyocytes are characterized by terminal differentiation and non-regeneration; hence, there is an urgent need to mitigate irreversible lesions in cardiomyocytes to cure cardiac diseases.
Emerging evidence has revealed that other processes, in addition to apoptosis and necrosis, participate in myocardial cell death, including pyroptosis, autophagy, and ferroptosis (Del Re et al., 2019). Ferroptosis, an iron-dependent form of regulated programmed cell death that was newly discovered in 2012, is accompanied by an increase in intracellular iron levels and engendering of oxidative hydroxyl radicals, as distinguishing factors from the other cell death modes (Dixon et al., 2012; Wu et al., 2020).
Innovation
Myocardial ischemia–reperfusion (I/R) injury is caused by oxidative stress damage to cardiomyocytes after coronary artery recanalization, which endangers human health (Chen et al., 2020). The main aim of this study was to explore the potential regulatory mechanism of the N6-methyladenosine (m6A) reader YTHDF2 in I/R injury. Our findings showed that the cardioprotective effects of silencing YTHDF2 were accomplished by enhancing the stability and expression of solute carrier family 7 member 11 (SLC7A11). We propose that the downregulation of SLC7A11 affects the transport of glutamate and cysteine and increases the synthesis of reactive oxygen species, which may provide novel potential therapeutic targets for treating ischemic cardiac diseases.
Ferroptosis affects lipid peroxidation and increases ROS levels (Dixon et al., 2012). Moreover, a ruptured mitochondrial outer membrane, increased membrane density, decreased mitochondrial volume, and significant morphological modifications arise owing to ferroptosis (Yao et al., 2021). Cells have a similar array of compounds against ferroptosis; solute carrier family 7 member 11 (SLC7A11) transduces extracellular cystine into glutathione (GSH), and then glutathione peroxidase 4 (GPX4) transforms toxic lipid hydroperoxides to nontoxic lipid alcohols with GSH to synergistically restrain ferroptosis (Mao et al., 2022; Zhang et al., 2021a).
Further, ferroptosis participates in numerous pathological processes, such as those involved in multiple sclerosis and nonalcoholic steatohepatitis (Feng et al., 2022). Ferroptosis is an important factor in cardiac impairment (Fang et al., 2020). Consequently, deciphering the connection between ferroptosis and cardiac I/R injury is crucial for the development of innovative therapies for angiocardiopathy.
Epigenetic regulation is conducive to adjusting and controlling the restored functions of angiogenesis (Shi et al., 2021). N6-methyladenosine (m6A), a dynamic reversible methylation modification, occurs on the sixth nitrogen atom of adenine in RNA (Gilbert et al., 2016). Moreover, m6A requires the joint participation of m6A methyltransferase complexes (writers), m6A demethylases (erasers), and m6A recognition proteins (readers) to modulate RNA metabolic processes associated with alternative splicing, localization, stabilization, and decay (Zhang et al., 2020; Zhao et al., 2021). Particular m6A “readers,” such as the YT521-B homology (YTH) domain family, exploit the functions of the whole (Lu et al., 2021).
YTHDF2, an m6A reader that promotes the degradation of m6A-modified messenger RNA (mRNA) (Wang et al., 2014), is associated with neoplastic diseases, such as prostate cancer, hepatocellular carcinoma, and pancreatic cancer (Guo et al., 2020b; Hou et al., 2019; Li et al., 2020). However, the relationship between YTHDF2 and cardiac I/R injury remains unclear.
Thus, this study was designed to investigate the effect of YTHDF2 on myocardial impairment after I/R injury and clarify the mechanism by which YTHDF2 regulates myocardial I/R injury in mice. These results may provide potential therapeutic targets for the treatment of ischemic cardiac diseases.
Results
Upregulation of YTHDF2 in I/R mouse hearts and hypoxia-reoxygenation cardiomyocytes
m6A methylation modification plays a critical role in cardiovascular diseases. We initially quantified the mRNA levels of the m6A methyltransferases METTL3, METTL14, and WTAP; demethylases FTO and ALKBH5; and m6A-binding proteins YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 in ischemic hearts after I/R injury to determine the pattern of m6A methylation. mRNA and protein expression levels of the m6A reader YTHDF2 were seven- and two-fold higher than in the sham control, respectively (Fig. 1A, B).
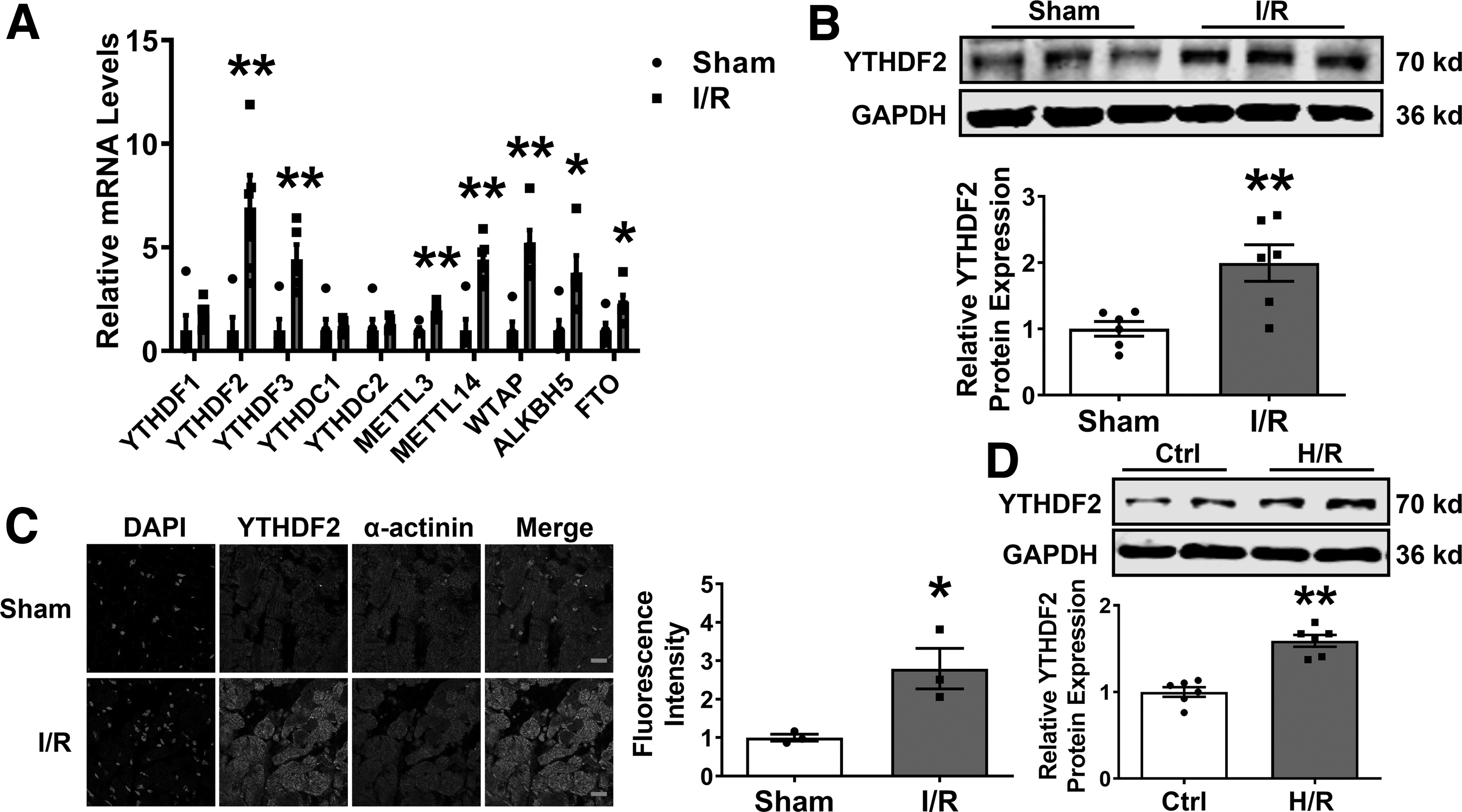
Immunofluorescence staining revealed that YTHDF2 expression increased after I/R injury (Fig. 1C). Consistent with the in vivo results, mRNA and protein levels of YTHDF2 were elevated in neonatal mouse ventricular cardiomyocytes (NMVCs) subjected to hypoxia-reoxygenation (H/R) treatment (Supplementary Fig. S1 and Fig. 1D). Therefore, we assumed that the m6A reader YTHDF2 is generally upregulated and may be linked to the pathological process of I/R.
Inhibition of YTHDF2 attenuates I/R-induced cardiac injury in mice
To determine whether YTHDF2 is involved in I/R, we conducted loss-of-function studies using shYTHDF2-V to silence YTHDF2 via tail vein injection (Fig. 2A). Successful delivery and knockdown of shYTHDF2-V were verified by GFP staining, Western blotting, and polymerase chain reaction (PCR) (Supplementary Fig. S2A–D) in mouse hearts. YTHDF2 upregulation in I/R mice was significantly reversed by sh-YTHDF2 (Fig. 2B).
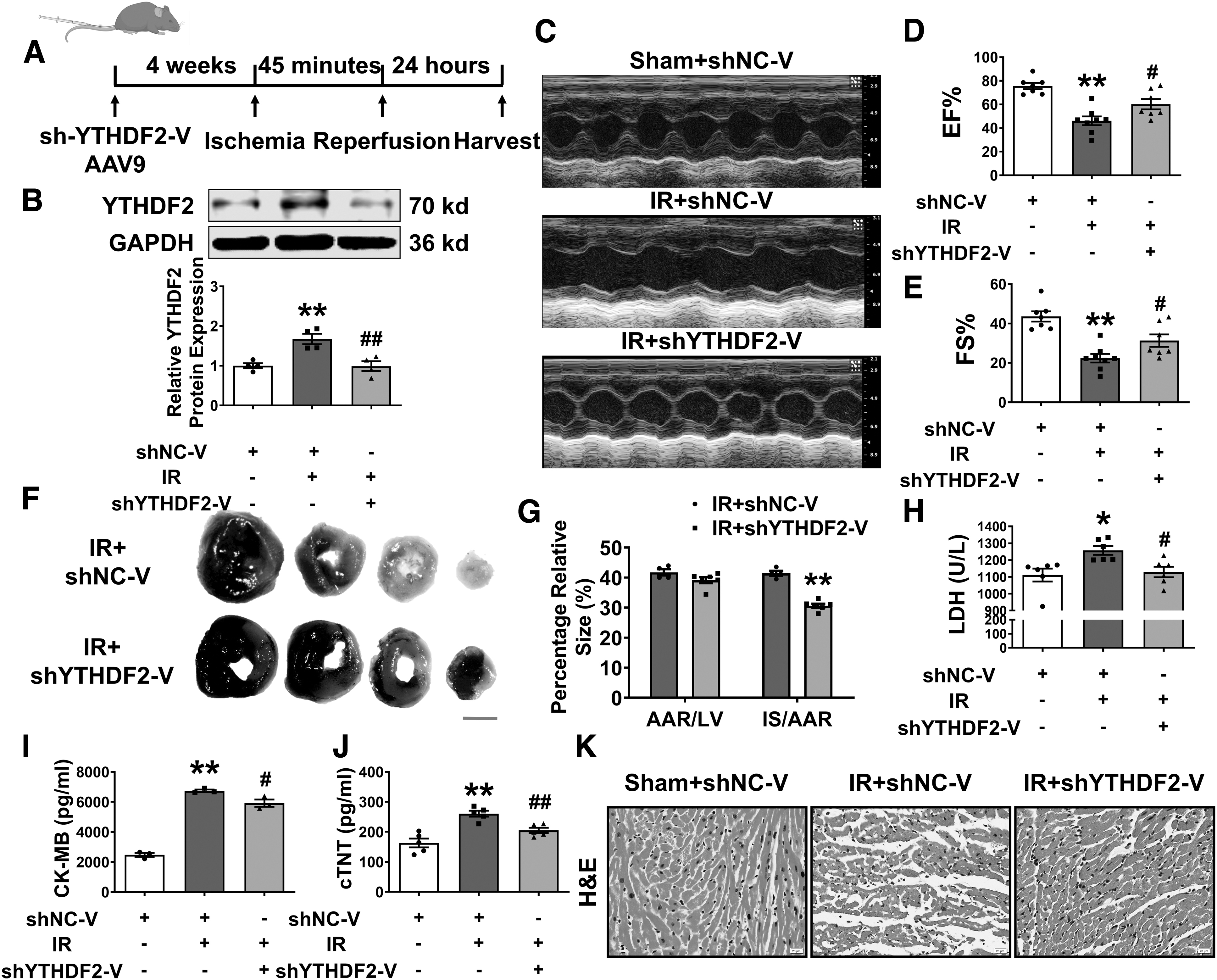
To assess the biological function of YTHDF2 in mice with cardiac injury, we measured cardiac function and found that YTHDF2 deficiency had no effect on normal mice (Supplementary Fig. S2E, F) but rescued the impaired ejection fraction (EF), fractional shortening (FS), and left ventricular internal dimension at systole (LVID; s) in the hearts of I/R mice (Fig. 2C–E and Supplementary Fig. S2G). Evans blue/2,3,5-triphenyl tetrazolium chloride (TTC) staining showed that the infarct size was reduced by shYTHDF2 in I/R-injured mice (Fig. 2F, G).
Lactate dehydrogenase (LDH), creatine kinase-MB (CK-MB), and cardiac troponin-T (cTNT) are biomarkers of cardiac injury (Zhang et al., 2021b); consistent with expectations, the upregulated levels of LDH, CK-MB (Xiao et al., 2022; Yang et al., 2023; Zhang et al., 2021b), and cTNT post-I/R were significantly reversed by silencing YTHDF2 (Fig. 2H–J).
Hematoxylin and eosin (H&E) staining revealed that the downregulation of YTHDF2 alleviated I/R-induced disorder in cardiomyocyte arrangement (Fig. 2K). Together, these data indicated that decreased YTHDF2 levels have a cardioprotective effect against I/R injury.
Forced expression of YTHDF2 aggravates cardiac I/R injury
To demonstrate whether YTHDF2 is detrimental to the myocardium, we performed gain-of-function studies by overexpressing YTHDF2 with YTHDF2-V for our subsequent experiments (Fig. 3A). GFP fluorescence intensity was significantly enhanced (Supplementary Fig. S3A, B), with higher protein and mRNA levels of YTHDF2 than those in the NC-V group in mouse hearts (Supplementary Fig. S3C, D). YTHDF2 upregulation in I/R mice was significantly enhanced by YTHDF2-V treatment (Fig. 3B).
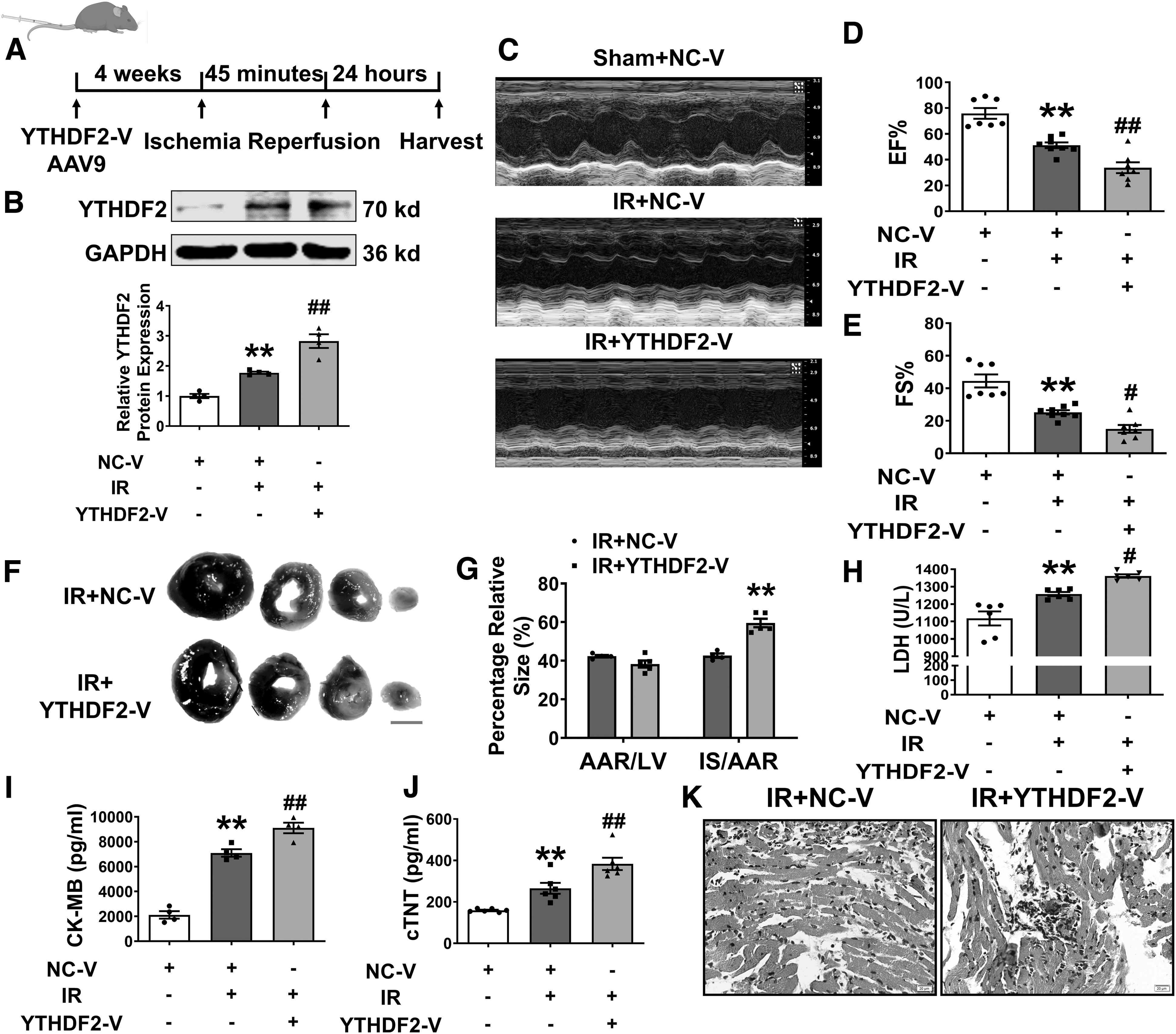
We investigated whether YTHDF2 caused hazardous effects similar to those of I/R injury in normal mice. Echocardiography confirmed that YTHDF2 did not affect EF (%) or FS (%) compared with those in the NC-V group (Supplementary Fig. S3E, F). YTHDF2 impaired cardiac function (Fig. 3C–E and Supplementary Fig. S3G) and increased the infarct size (Fig. 3F, G) and levels of LDH, CK-MB, and cTNT (Fig. 3H–J) post-I/R injury. Consistently, the pernicious histological effects of I/R on the myocardial tissue were aggravated by YTHDF2 (Fig. 3K). These results showed that the forced expression of YTHDF2 facilitates cardiac I/R injury.
YTHDF2 mediates ferroptosis and regulates cardiac injury induced by I/R
To explore the molecular mechanism by which YTHDF2 regulates cardiac I/R injury, we performed RNA-Seq using I/R+shNC-V and I/R+shYTHDF2-V myocardia. According to the RNA-Seq results, the expression levels of 439 genes, including 213 upregulated and 226 downregulated genes, were significantly altered compared with those in the I/R+shNC-V group (Supplementary Fig. S4).
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed a strong correlation between ferroptosis and shYTHDF2 post-I/R (Fig. 4A). The morphological characteristics of ferroptosis are mainly manifested in the mitochondria, including an increase in membrane density and a decrease or even disappearance of the mitochondrial cristae (Dixon et al., 2012; Yagoda et al., 2007; Yang and Stockwell, 2008).
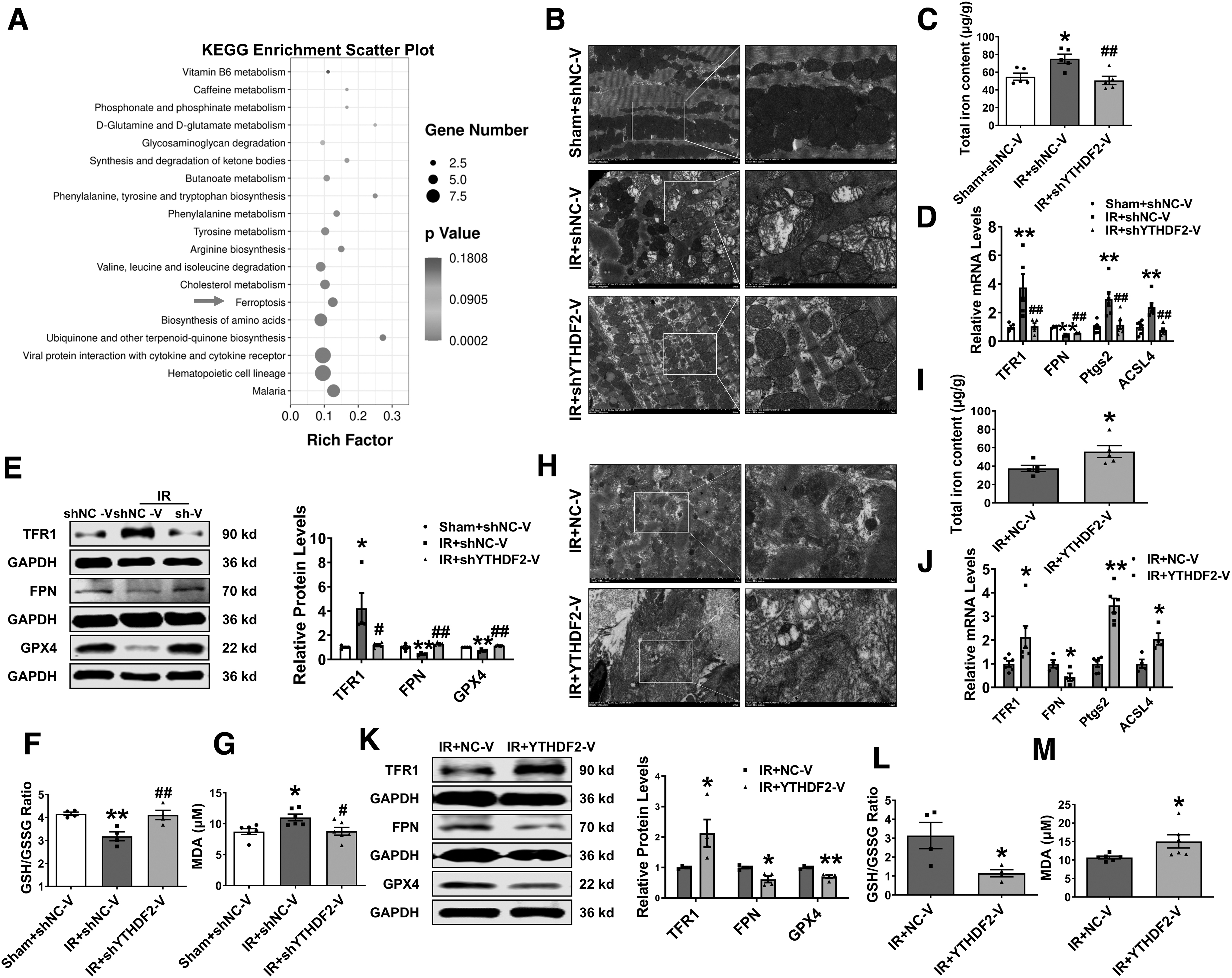
Therefore, we used electron microscopy to assess the mitochondrial ultrastructural architecture. Promiscuous mitochondria and reduced cristae density in I/R mice were abolished by shYTHDF2 (Fig. 4B). YTHDF2 deficiency reversed the increase in iron content in I/R heart tissue (Fig. 4C). mRNA and protein levels of transferrin receptor 1 (TFR1), which imports iron from the extracellular space into cells (Yang and Stockwell, 2008), and iron exporter ferroportin (FPN), which transfers iron out of the cell (Donovan et al., 2005), were reversed by shYTHDF2 in I/R mice (Fig. 4D, E).
We then measured the expression levels of acyl-CoA synthetase long-chain family member (ACSL4) and prostaglandin-endoperoxide synthase 2 (Ptgs2), two biomarkers of ferroptosis (Yang et al., 2014), and found that the deletion of YTHDF2 downregulated ACSL4 and Ptgs2 mRNA levels in I/R hearts (Fig. 4D). Consistently, the protein expression of GPX4, an endogenous lipid peroxidation inhibitor, was inhibited by cardiac I/R injury and upregulated by shYTHDF2 (Fig. 4E).
Decreased GSH levels result in ferroptosis, and we found that the ratio of GSH/oxidized glutathione (GSSG) was reduced in I/R mice and returned to normal levels by silencing YTHDF2 (Fig. 4F). Aberrant levels of the lipid peroxide malondialdehyde (MDA) in I/R mice were reversed by shYTHDF2 (Fig. 4G).
YTHDF2 overexpression enhanced mitochondrial disarrangement and iron accumulation (Fig. 4H, I). Further, changes in ferroptosis-related genes at the mRNA and protein levels were altered by YTHDF2 in I/R mice (Fig. 4J, K). Abnormal levels of GSH/GSSG and MDA were intensified by YTHDF2 (Fig. 4L, M). Collectively, these data revealed that YTHDF2 participates in regulating ferroptosis and aggravating cardiac I/R injury.
Deficiency of YTHDF2 alleviates cardiomyocyte injury after H/R treatment through ferroptosis
These results raise the question of whether inhibiting YTHDF2 in NMVCs exposed to H/R produces the same results in vivo. To clarify this, we performed a loss-of-function experiment using siRNAs. mRNA and protein levels of YTHDF2 were lower after transfection with si-YTHDF2 than in the negative control (NC) group (Supplementary Fig. S5A and Fig. 5A).
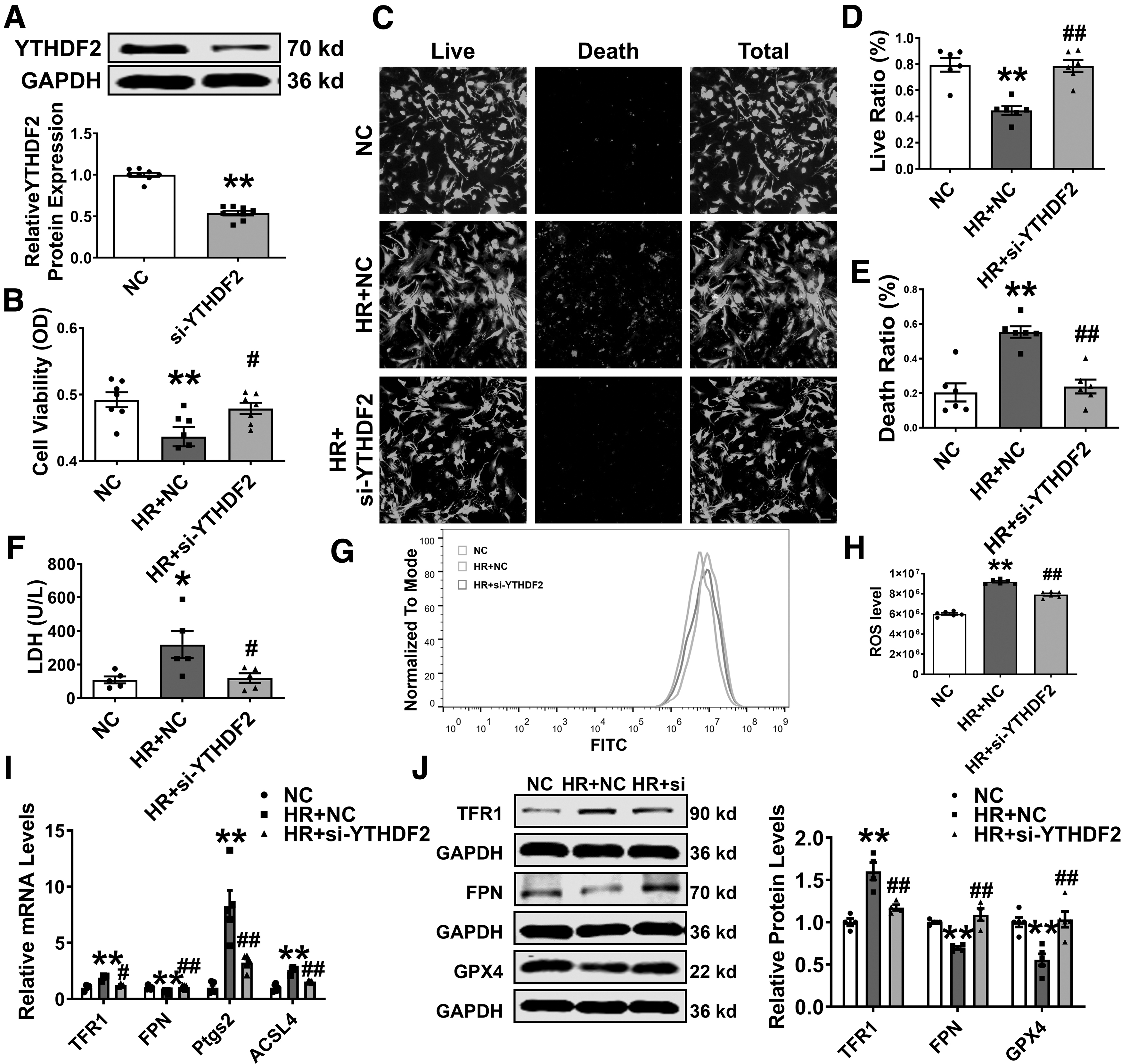
Moreover, the detrimental effects, including decreased cell viability accompanied by high levels of LDH after H/R damage, were countered by si-YTHDF2 (Fig. 5B–F). Further experiments revealed that YTHDF2 knockdown attenuated the H/R-induced production of ROS, a characteristic of ferroptosis (Fig. 5G, H, and Supplementary Fig. S5B, C).
Consistent with the in vivo results, the effects of H/R on TFR1, FPN, Ptgs2, and ACSL4 mRNA levels were significantly altered on YTHDF2 silencing (Fig. 5I). Moreover, it abolished the abnormal protein expression levels of TFR1, FPN, and GPX4 in H/R-treated cardiomyocytes (Fig. 5J).
Silencing YTHDF2 represses erastin-induced cardiomyocyte ferroptosis
To investigate whether the downregulation of YTHDF2 protects against cardiac I/R injury in response to ferroptosis, erastin, a ferroptosis activator that targets the cystine/glutamate antiporter (system Xc −), was used in further experiments (Dixon et al., 2012). Similar to what was observed in H/R-induced cardiomyocytes, the inhibition of YTHDF2 had a protective effect against erastin-induced injury, including reduced cell viability, increased LDH levels, and ROS accumulation (Fig. 6A–G and Supplementary Fig. S5D, E).
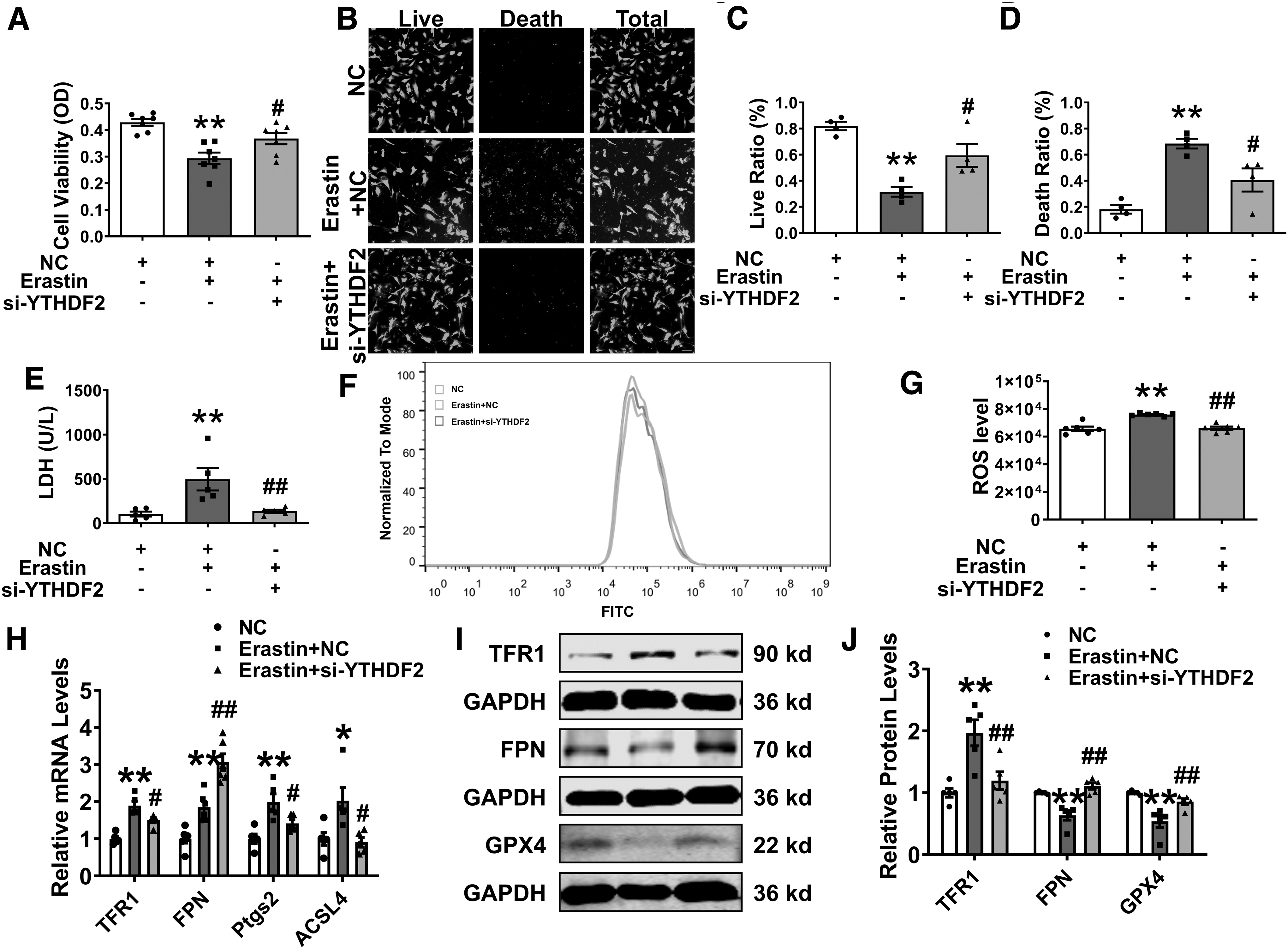
The inhibition of YTHDF2 reversed the abnormal mRNA levels of TFR1, FPN, Ptgs2, and ACSL4 after treatment with erastin (Fig. 6H). Western blotting showed that the abnormal protein expression levels of TFR1, FPN, and GPX4 were reversed by YTHDF2 inhibition (Fig. 6I, J). These results indicated that YTHDF2 knockdown mitigates erastin-induced ferroptosis in cardiomyocytes.
YTHDF2 facilitates the degradation of SLC7A11 mRNA
To uncover the downstream mechanism of the regulation of cardiac ferroptosis by YTHDF2, we used a heatmap to analyze the differentially expressed genes in the I/R+shNC-V and I/R+shYTHDF2-V myocardium (Fig. 7A). The differential expression of SLC7A11 protein and mRNA in I/R mice and H/R cardiomyocytes was reversed by the overexpression or inhibition of YTHDF2 (Fig. 7B–D and Supplementary Fig. S6A–C).
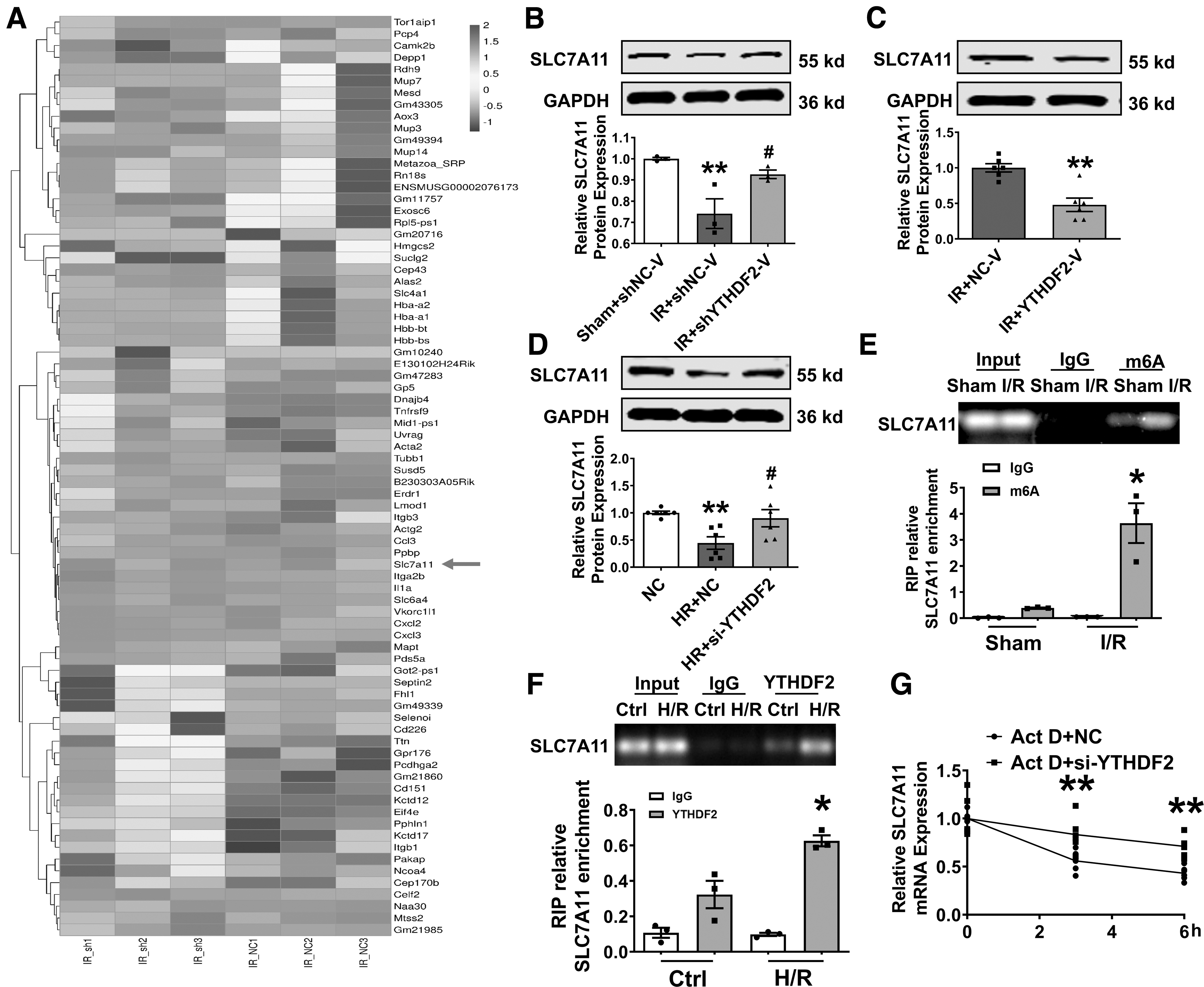
Next, we verified that there were many m6A modification sites on SLC7A11 mRNA using SRAMP, and RNA immunoprecipitation results confirmed that the binding of m6A to SLC7A11 was enhanced by I/R injury (Supplementary Fig. S7A and Fig. 7E). YTHDF2 accelerates the degradation of target mRNAs by recognizing m6A modification sites (Wang et al., 2014).
The RNA immunoprecipitation data confirmed that SLC7A11 combined with YTHDF2 in mouse hearts and was more strongly identified by YTHDF2 in H/R-induced cardiomyocytes (Supplementary Fig. S7B and Fig. 7F). To examine whether YTHDF2 influences SLC7A11 mRNA stability, actinomycin D (Act D) was used to inhibit RNA transcription. PCR showed that silencing YTHDF2 significantly repressed the degradation of SLC7A11 mRNA in cardiomyocytes treated with Act D (Fig. 7G).
YTHDF2 targets SLC7A11 to regulate ferroptosis in I/R mice and H/R cardiomyocytes
We hypothesized that the beneficial effect of sh-YTHDF2 in I/R mice is mediated by SLC7A11. To clarify this, we silenced SLC7A11 using shSLC7A11-V in YTHDF2 knockdown mice and found that silencing SLC7A11 did not affect EF (%) and FS (%) (Supplementary Fig. S8A, B). Western blotting and PCR (Supplementary Fig. S8C, D) confirmed the successful knockdown of SLC7A11 in mouse hearts.
The beneficial effect of YTHDF2 inhibition on cardiac function (Fig. 8A–C) was reversed by silencing SLC7A11. Suppression of SLC7A11 also abrogated the decrease in infarct size and LDH level (Fig. 8D–F). siRNAs were designed to suppress the expression of SLC7A11 (Supplementary Fig. S8E, F) in vitro. Co-transfection with si-YTHDF2 and SLC7A11-specific siRNA (si-SLC7A11) intensified H/R-induced cardiomyocyte injury, which promoted cell death and increased the release of LDH and ROS compared with si-YTHDF2 (Fig. 8G–L and Supplementary Fig. S8G).
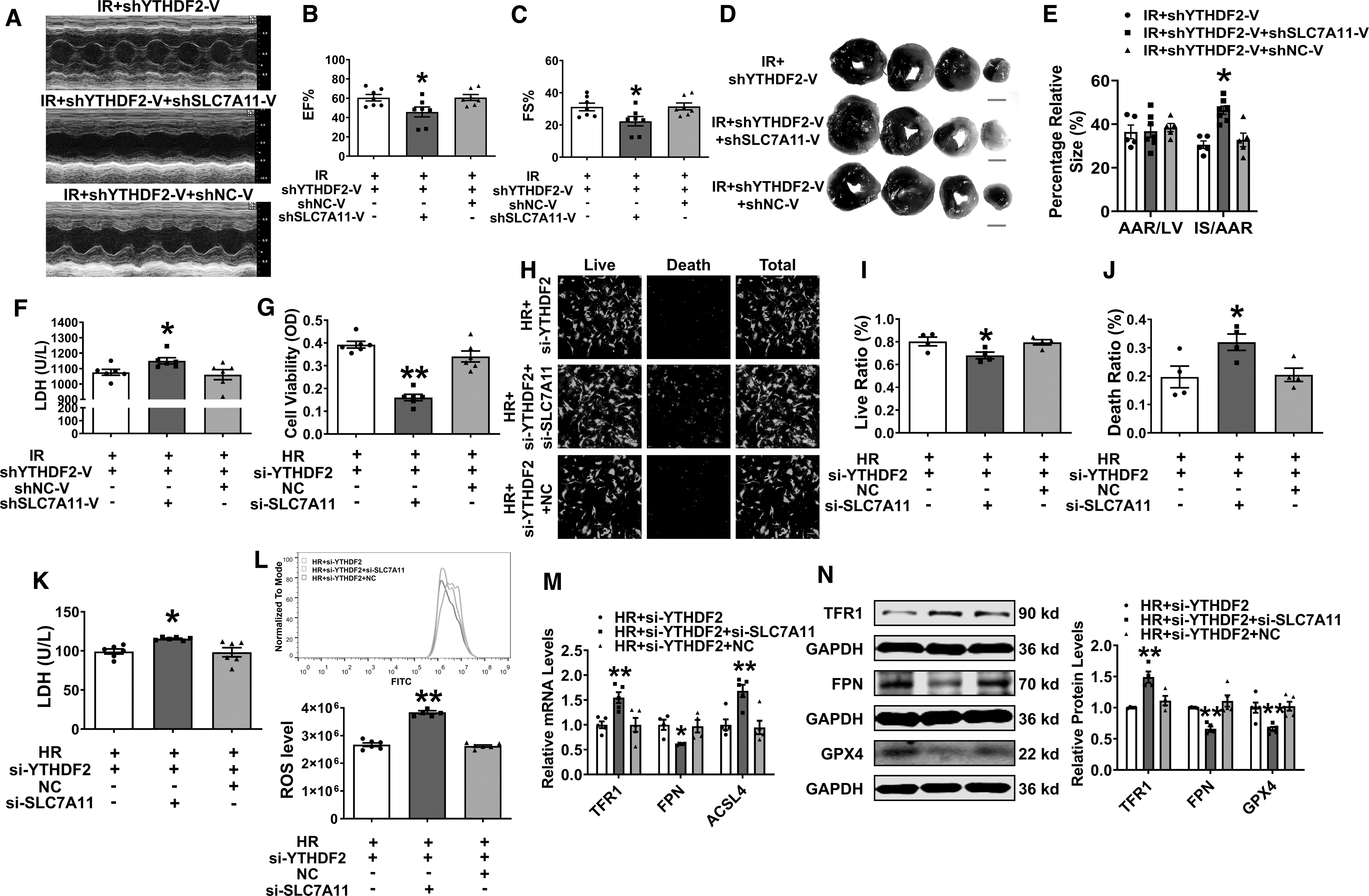
Further, the disadvantageous expressions of ferroptosis-related genes at mRNA and protein levels were aggravated after co-transfection with SLC7A11 and YTHDF2 siRNAs (Fig. 8M, N). Taken together, the deficiency of YTHDF2 attenuates cardiac ferroptosis by stabilizing SLC7A11 m6A modification.
Discussion
The aim of this study was to elucidate the regulatory role and potential mechanisms of YTHDF2 in cardiac ferroptosis and I/R injury. Initially, we found that protein and mRNA levels of YTHDF2 increased and exerted detrimental effects on I/R-treated hearts and H/R-induced cardiomyocytes. Forced expression of YTHDF2 resulted in infarct size enlargement and cardiac dysfunction, whereas knockdown of YTHDF2 reduced these effects after cardiac I/R injury.
Extensive research has demonstrated that the cardioprotective effects of silencing YTHDF2 partly occur by recognizing the m6A modification sites of SLC7A11 and inhibiting its mRNA degradation and subsequent ferroptosis. Briefly, these findings prompted us to delineate this process as follows: I/R or H/R → YTHDF2 ↑ → SLC7A11 stability ↓ → ROS ↑ and iron overload → cardiac ferroptosis → aggravated cardiac injury, suggesting therapeutic targets for treating ischemic cardiac diseases.
The targeting of mRNA using m6A modification relies on reading protein, which comprises the YTH domain family (YTHDF1–3, YTHDC1–2) and IGF2 mRNA-binding protein family (Frye et al., 2018). Multiple studies have shown that the abnormal expression of m6A modification-reading proteins participates in some physiological and pathological processes, such as those involved in biological individual growth and development, sperm development and maturation, DNA damage repair, and various types of disease (Deng et al., 2018; Frye et al., 2018).
Recent studies have demonstrated that m6A methylation plays a critical role in cardiovascular diseases. For example, METTL3 regulates cardiac homeostasis and hypertrophy and FTO mitigates cardiac function during remodeling and repair (Dorn et al., 2019; Mathiyalagan et al., 2019). However, the effect of YTHDF2 on myocardial I/R injury remains unclear. In the present study, we explored the role and regulatory mechanisms of YTHDF2 in heart damage.
We found that the mRNA and protein expression of YTHDF2 increased during cardiac I/R injury, which steadily worsened cardiac function. In addition, the results demonstrated that the inhibition of YTHDF2 ameliorated impaired EF (%) and FS (%) and increased infarct size, LDH release, and CK-MB and cTNT levels post-I/R, whereas YTHDF2 aggravated cardiac damage.
To investigate the underlying mechanism, we used RNA-Seq and KEGG analysis. An additional consequence of this observation is the involvement of ferroptosis in YTHDF2-mediated myocardial injury. There is evidence to suggest that ferroptosis plays an essential role in tumors (Kim et al., 2016), neurodegenerative diseases (Ishii et al., 2019), renal failure (Adedoyin et al., 2018), and cardiovascular diseases (Fang et al., 2019).
In one study, in which the differentially expressed genes between treatment and control groups in a doxorubicin-induced mouse cardiomyopathy model were analyzed, Hmox1 was upregulated, which mediated the release of iron ions in heme to induce ferroptosis and even heart failure owing to iron overload (Fang et al., 2019). In addition, rapamycin (mTOR) inhibits ferroptosis in mouse cardiomyocytes during I/R injury; this may be because mTOR inhibits ROS production, although the underlying mechanism remains to be elucidated (Baba et al., 2018).
Another study revealed that fer-1 (an iron death inhibitor) reduces cardiomyocyte death in heart transplantation and myocardial I/R models, while improving left ventricular systolic function and ventricular remodeling in I/R models (Li et al., 2019). Consistent with these studies, we demonstrated that ferroptosis was activated by cardiac I/R injury, as evidenced by changes in morphological characteristics and levels of ferroptosis markers.
We found that YTHDF2 knockdown impeded promiscuous mitochondria and reduced cristae density, reversing ferroptosis in I/R mice and H/R cardiomyocytes, whereas YTHDF2 overexpression facilitated these harmful effects.
Iron homeostasis regulates multiple iron transport-related proteins. The Andrews Laboratory of Duke University in the United States reported for the first time that the cardiac-specific knockout of TFR1 induces a lethal phenotype, which was attributed to mitochondrial dysfunction caused by severe iron deficiency in the myocardium (Xu et al., 2015).
FPN1, the only cellular iron efflux protein found in mammals, has been specifically knocked out in mouse hearts, which induces cardiac iron metabolism imbalances and cardiac insufficiency (Lakhal-Littleton et al., 2015). Disorders in cardiac FPN1 can increase iron efflux, which accounts for fatal iron deficiency (Gao et al., 2015).
Iron homeostasis plays a vital role in cardiovascular disease. Iron overload triggers ferroptosis by increasing ROS accumulation, which leads to cardiovascular diseases. Our data showed that silencing YTHDF2 reversed iron overload in I/R mice, as evidenced by increased iron content and TFR1 expression and decreased FPN expression. Consistently, YTHDF2 deficiency mitigated erastin-induced ferroptosis in cardiomyocytes. The overexpression of YTHDF2 facilitates these harmful effects. Overall, our gain- and loss-of-function experiments demonstrated that YTHDF2 regulates cardiomyocyte injury via ferroptosis.
Many studies have indicated that YTHDF2 induces the degradation of target mRNA and that differentiation-related mRNAs with the m6A modification in neurons undergo degradation, impairing the self-renewal of neural stem/progenitor cells and development of neurons, resulting in neurodevelopmental defects (Li et al., 2018; Wang et al., 2014).
In hepatocellular carcinoma, YTHDF2 reduces the stability of EGFR mRNA by binding to the m6A site of the 3′UTR (untranslated region) of EGFR and inhibits the ERK/MAPK signaling pathway, thereby exerting a tumor suppressor effect (Zhong et al., 2019). YTHDF2 also reduces the level of UBXN1 by recognizing the m6A modification of UBXN1 mRNA mediated by METTL3, which activates NF-κB, facilitating target gene transcription and the development of glioma (Chai et al., 2021).
Moreover, YTHDF2 recognizes the METTL3-mediated m6A modification of PTEN mRNA and promotes the degradation of PTEN, which leads to excessive PASMC proliferation via the activation of the PI3K/Akt signaling pathway (Qin et al., 2021). Consistent with this, YTHDF2 is upregulated in cardiac hypertrophy. However, YTHDF2 alleviates this injury by promoting the degradation of Myh7 mRNA.
The pathological process of myocardial hypertrophy involves enlarged cardiomyocytes and the proliferation of interstitial cells and connective tissue, which reduces myocardial contractility, obstructs the blood supply, and increases oxygen consumption and heart failure (HF) (Xu et al., 2021).
Myocardial I/R-induced cell death is primarily caused by peroxidation, inflammation, and intracellular calcium overload (Talukder et al., 2009). Therefore, we speculated that YTHDF2 recognizes various m6A modification sites and plays different roles in HF and I/R injury to facilitate mRNA degradation.
Volcano plot analysis showed that SLC7A11 was upregulated by shYTHDF2-V in I/R mice. Ferroptosis, a newly discovered form of iron-dependent cell death, is caused by the inhibition of the cystine/glutamate antiporter (system Xc −) (Dixon et al., 2012). Deferoxamine reduces ferroptosis by inhibiting glutamine metabolism in an isolated mouse heart I/R model (Gao et al., 2015).
SLC7A11 inhibits iron overload-induced ferroptosis by influencing cysteine uptake, decreasing ROS production, and conferring protection against ferroptosis during iron overload (Wang et al., 2017). YTHDC2 destabilizes SLC7A11 mRNA through its m6A reading YTH domain, restraining cystine uptake and downstream antioxidant programs and inhibiting pulmonary adenocarcinoma effects (Ma et al., 2021).
We found that YTHDF2 inhibition significantly increased the stability of SLC7A11 mRNA by recognizing multiple m6A binding sites and relieving cardiomyocyte damage. Moreover, the inhibition of SLC7A11 counterbalanced the beneficial effects of YTHDF2 knockdown on I/R and H/R injury.
In conclusion, our initial observations revealed a cardinal regulatory relationship between YTHDF2 and ferroptosis during cardiac I/R injury. YTHDF2 can inhibit SLC7A11, affecting the transport of glutamate and cystine and reducing the synthesis of GPX4.
However, this can cause iron overload. The synergistic effects of these two factors trigger ferroptosis and aggravate myocardial ischemia–reperfusion injury. Consequently, YTHDF2 targeting SLC7A11 to regulate ferroptosis has novel therapeutic value for myocardial I/R injury and provides potential applications for clinical treatment.
This study has the following shortcomings: (1) The reason for the upregulation of YTHDF2 is currently unclear, and we speculate that there may be transcription factors or noncoding RNAs that regulate the expression of YTHDF2 in cardiac I/R mice. (2) In addition to SLC7A11, YTHDF2 may affect the stability of other RNA that regulate myocardial I/R injury. However, these questions require further investigation.
Materials and Methods
Mouse model of cardiac I/R injury
This study was approved by the Institutional Animal Care and Use Committee of Harbin Medical University. All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Eight-week-old male C57BL/6 mice weighing 20–22 g were intraperitoneally injected with a solution containing avertin to achieve anesthesia (0.2 g/kg; Sigma–Aldrich Corporation).
After intubation of the trachea, the left descending coronary artery (LAD) was ligated with 7-0 nylon for 45 min, and blood perfusion was gently restored for 24 h to establish a mouse model of cardiac I/R injury. The sutures were passed through the LAD of other mice without ligation, as in the sham group. Echocardiography was performed to evaluate the heart function, followed by dissection to obtain tissue samples encompassing the infarcted regions for other experiments.
Gene delivery in mice
C57BL/6 mice were injected with the virus solution adeno-associated virus 9 (AAV9) carrying YTHDF2 and SLC7A11 special short hairpin RNA (shRNA) or YTHDF2 (1 × 1011 virus particles; shYTHDF2-V, shSLC7A11-V, shNC-V, YTHDF2-V, or NC-V) via tail vein injection, and a cTNT promoter conjugated with the GFP gene was constructed (Hanbio Biotechnology). After 4 weeks, I/R surgery was performed.
The sequences of shYTHDF2-V were: sense 5′-CUAGAGAACAACGAGAAUATT-3′ and antisense 5′-UAUUCUCGUUGUUCUCUAGTT-3′. The sequences of shSLC7A11-V were: sense 5′-UGACAAACGUGGCCUAUUUTT-3′ and antisense 5′-AAAUAGGCCACGUUUGUCATT-3′. The expression of YTHDF2 and SLC7A11 was determined using PCR and Western blotting.
Echocardiographic analysis
Mice were anesthetized with 2% avertin 24 h after reperfusion, followed by M-mode echocardiography using a Vevo2100 echocardiographic system (Visualsonics, Toronto, Canada) at a probe frequency of 10 MHz to assess left ventricle (LV) function. The LV of the mice was vertically shot with a noninvasive ultrasound beam, followed by measurement of the LV internal dimensions at EF, FS and LVID for an average of at least three consecutive cardiac cycles.
Evans blue and TTC staining
After 24 h of reperfusion, the LAD ligature was removed and 2% Evans blue dye was injected into the mouse abdominal aorta. Whole hearts obtained from euthanized mice were frozen in a −80°C refrigerator. After hardening, the hearts were cut into four slices with a thickness of 1 mm and stained with 2% TTC dyeing buffer (Solarbio, Beijing, China) at 37°C for 20 min.
Finally, the sections were photographed and ImageJ (NIH) was used to calculate the infarcted area of the hearts.
Electron microscopy
A patch of the heart obtained from C57BL/6 mice was fixed in 2.5% glutaraldehyde and washed with Tris-buffered saline. After fixation using 1% osmium tetroxide at 4°C, the tissues were stained with 1% uranyl acetate. Ethanol was used for dehydration, and epoxy resin was used to embed the samples. Finally, the samples were stained with uranyl acetate and lead citrate, and photographs were captured using a JEM-1200 electron microscope (FEI, Hillsboro, OR).
Immunofluorescence
The heart sections were permeabilized with 0.5% Triton X-100 at room temperature for 30–40 min in the dark and subsequently blocked with 10% normal goat serum at 37°C for 1 h. Afterward, the primary antibody YTHDF2 or α-actinin was added to each slide, which were incubated overnight at 4°C, and then incubated with the corresponding secondary antibody for 1 h in the dark.
After rinsing three times with phosphate buffered saline (PBS), the nuclei were stained with DAPI (Beyotime Institute of Biotechnology, Shanghai, China) for 5–8 min, and immunofluorescence was analyzed using a laser scanning confocal microscope.
H&E staining
The heart sections were dewaxed and dehydrated with xylene and alcohol. Sections were stained with hematoxylin and eosin (Solarbio) according to the manufacturer's instructions.
Measurement of labile iron level
Iron concentrations were measured using an iron assay kit (No. ab83366; Abcam, Cambridge, MA) according to the manufacturer's instructions. Iron buffer or iron reducer was added to the heart samples to detect iron or total iron, which were incubated with the iron probe for 60 min. Finally, the absorbance was measured using a colorimetric microplate reader at OD 593 nm to determine the iron concentration.
Assay kits
LDH, GSH, GSSG, and MDA levels were measured using LDH, GSH, GSSG, and MDA detection assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). CK-MB and cTNT levels were measured using CK-MB and cTNT enzyme-linked immunosorbent assay (ELISA) kits (Elabscience, Wuhan, China), as previously described (Zhang et al., 2021b).
Primary cardiomyocyte culture
NMVCs from 1- to 3-day-old mice were digested in a solution containing trypsin (Solarbio) and Dulbecco's modified Eagle's medium (Biological Industries, Haemek, Israel) for 12 h at 4°C with rotation. The tissues were then digested with collagenase Type II (Thermo Fisher Scientific, Waltham, MA) until the hearts disappeared, and the lysates were collected and centrifuged at 1500 rpm for 10 min. The precipitated cells were resuspended in the culture medium. Dissociated cardiomyocytes were seeded onto plates for follow-up experiments.
Cell transfection and treatment
YTHDF2-specific small interfering RNA (si-YTHDF2), si-SLC7A11, and negative control siRNA (si-NC) were purchased from RiboBio (Guangzhou, China). Transfection with siRNAs at a final concentration of 50 nM was performed using the X-treme GENE Transfection Reagent (Roche, Basel, Switzerland). The sequences of siRNAs for mouse si-YTHDF2 were: sense 5′-CUAGAGAACAACGAGAAUATT-3′ and antisense 5′-UAUUCUCGUUGUUCUCUAGTT-3′.
The sequences of the siRNAs for mouse si-SLC7A11 were: sense 5′-UGACAAACGUGGCCUAUUUTT-3′ and antisense 5′-AAAUAGGCCACGUUUGUCA-3′. Forty-eight hours after transfection, cardiomyocytes were cultured under hypoxic conditions (5% CO2 and 95% N2) for 12 h, followed by reoxygenation for 24 h to establish the H/R model.
Cell viability by cell counting kit-8 assay
Cardiomyocytes were incubated with cell counting kit-8 (CCK-8) solution (Beijing Labgic Technology Co., Ltd., Beijing, China) for 1.5 h in the dark in a 37°C incubator. Absorbance was measured at 450 nm using a colorimetric microplate reader to evaluate cell viability.
Live/dead and ROS staining
The viability and ROS content of the cardiomyocytes were assessed using a live/dead assay kit (No. L3224; Life Technologies) and a ROS assay kit, respectively, according to the manufacturer's instructions. The live and dead cells were stained green and red, respectively.
The NMVCs were incubated with 10 μM DCFH-DA (No. S0033S-1, Beyotime Institute of Biotechnology) for ROS staining at 37°C for 30–60 min. Finally, the fluorescence intensity of ROS was measured using a laser scanning confocal microscope or flow cytometer, as previously described (Anselmino et al., 2020; Valerio et al., 2022; Zhu et al., 2021).
Western blotting
A bicinchoninic acid protein kit (Beyotime Institute of Biotechnology) was used to measure the protein concentration. Proteins were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes.
The membranes were blocked in confining liquid (A mixed with B), followed by incubation overnight at 4°C with primary antibodies against YTHDF2 (No. 24744-1-AP, 1:500; Proteintech, Wuhan, China), GPX4 (No. 67763-1-Ig, 1:500; Proteintech), FPN (No. 26601-1-AP, 1:1000; Proteintech), TFR1 (No. 13-6800, 1:1000; Invitrogen, Carlsbad, CA), SLC7A11 (No. 175186, 1:1000; Abcam, Cambridge, United Kingdom), and GAPDH (No. TA-08, 1:1000; ZsBio, Beijing, China). Next, horseradish peroxidase-labeled anti-rabbit/mouse IgG (1:10,000; LI-COR Bioscience, Lincoln, NE) was incubated for 1 h at room temperature in the dark.
The membranes were washed three times with Tris-buffered saline and Tween-20 for 8 min each at the beginning and end of secondary antibody incubation. The membranes were scanned and analyzed using an Odyssey infrared imaging system (LI-COR Bioscience), and protein bands were assessed and quantified. GAPDH was used as an internal control.
RNA isolation and quantitative real-time RT-PCR
Total RNA was extracted from the cardiac tissues and cardiomyocytes using TRIzol reagent (Invitrogen). The RNA concentration was measured using a NanoDrop ND-8000 (Thermo Fisher Scientific). The total RNA mass was controlled at 500 ng, and DNA was synthesized using a reverse transcription kit (Toyobo). Real-time PCR was performed using SYBR Green Master Mix (Toyobo) to quantify YTHDF2, SLC7A11, FPN, TFR1, Ptgs2, and ACSL4 levels on an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA).
Expression was normalized to that of GAPDH or 18S as an internal control. The 2−△△Ct method was used to determine the relative RNA levels. The primers used in this study were: GAPDH-F 5′-AAGAAGGTGGTGAAGCAGGC-3′, GAPDH-R 5′-TCCACCACCCTGTTGCTGTA-3′, 18S-F 5′-CCTGGATACCGCAGCTAGGA-3′, 18S-R 5′-GCGGCGCAATACGAATGCCCC-3′, SLC7A11-F 5′-TCAAAAGCTTGGCCATCTGC-3′, SLC7A11-R 5′-CTGTGAGCTTGCCTCACTGTA-3′, ACSL4-F 5′-CCACACTTATGGCCGCTGTT-3′, ACSL4-R 5′-GGGCGTCATAGCCTTTCTTG-3′, Ptgs2-F 5′-CTGCGCCTTTTCAAGGATGG-3′, Ptgs2-R 5′-GGGGATACACCTCTCCACCA-3′, FPN-F 5′-GTGGAGTACTTCTTGCTCTGG-3′, FPN-R 5′-CTGCTTCAGTTCTGACTCCTC-3′, YTHDF2-F 5′-TAGCCAACTGCGACACATTC-3′, YTHDF2-R 5′-CACGACCTTGACGTTCCTTT-3′, TFR1-F 5′-GGAGCATGCCGAGAAACTGA-3′, and TFR1-R 5′-CTCTCCCAGTCATCACGGTC-3′.
Act D treatment
The NMVCs were plated in 6-well plates at a density of 1 × 106 cells/well. After transfection for 48 h, the cells were treated with Act D (5 g/mL; MedChemExpress) and dimethyl sulfoxide (DMSO; BioFroxx) to inhibit transcription, followed by collection at different time points.
RNA-binding protein immunoprecipitation assay
N6-methyladenine RNA-binding protein immunoprecipitation (Me-RIP) and YTHDF2-RNA-binding protein immunoprecipitation (RIP) were performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer's protocol.
In summary, fresh hearts were homogenized in RIP lysis buffer, incubated overnight with 50 μL of protein-A/G agarose beads (Roche, USA) and antibodies against m6A (No. 202003, 5 μg; Synaptic Systems, Goettingen, Germany) or YTHDF2 (No. 24744-1-AP, 5 μg; Proteintech), transiently stained, and washed six times with RIP washing buffer. Finally, the immunoprecipitated RNA was purified and subjected to PCR.
RNA-Seq
Hearts were delivered to a company that constructed the mRNA library and sequenced RNA using LC Bio (Hangzhou, China). KEGG enrichment was used to assess the expression changes in the quantified genes under different conditions to screen for differentially expressed genes. KEGG enrichment analysis, heatmaps, and volcano plots were constructed as described by LC Bio.
Statistical analysis
All data are presented as the means ± standard error of measurement of at least three independent experiments and were statistically analyzed using GraphPad Prism 7.0. Student's t-test was used to analyze two groups, and one-way analysis of variance with Dunnett's correction was applied for multiple-group comparisons. A p value of <0.05 indicated a significant difference.
An electronic laboratory notebook was not used.
Data Availability
The datasets used and/or analyzed to support the findings of this study are available in this article or in the Supplementary Figures S1–S8. All the raw data supporting the results of this study are available from the corresponding author on request.
Footnotes
Authors' Contributions
B.Y., N.W., and Y.B. supervised and designed the study. P.P., W.S., H.W., J.J., K.L., Y.J., H.D., L.Z., W.J., Y.Y., and Y.X. performed all experiments. Y.B., P.P., W.S., and H.W. wrote the article. X.K., Z.Z., F.Z., J.S., and C.W. collected and analyzed the data. All the authors approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (Nos. 82104168 and U21A20339), China Postdoctoral Science Foundation (No. 2021M693832), and Heilongjiang Province Postdoctoral Science Foundation (No. LBH-Z20174).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.