
Abstract
Significance:
The architecture of the mitochondrial network and cristae critically impact cell differentiation and identity. Cells undergoing metabolic reprogramming to aerobic glycolysis (Warburg effect), such as immune cells, stem cells, and cancer cells, go through controlled modifications in mitochondrial architecture, which is critical for achieving the resulting cellular phenotype.
Recent Advances:
Recent studies in immunometabolism have shown that the manipulation of mitochondrial network dynamics and cristae shape directly affects T cell phenotype and macrophage polarization through altering energy metabolism. Similar manipulations also alter the specific metabolic phenotypes that accompany somatic reprogramming, stem cell differentiation, and cancer cells. The modulation of oxidative phosphorylation activity, accompanied by changes in metabolite signaling, reactive oxygen species generation, and adenosine triphosphate levels, is the shared underlying mechanism.
Critical Issues:
The plasticity of mitochondrial architecture is particularly vital for metabolic reprogramming. Consequently, failure to adapt the appropriate mitochondrial morphology often compromises the differentiation and identity of the cell. Immune, stem, and tumor cells exhibit striking similarities in their coordination of mitochondrial morphology with metabolic pathways. However, although many general unifying principles can be observed, their validity is not absolute, and the mechanistic links thus need to be further explored.
Future Directions:
Better knowledge of the molecular mechanisms involved and their relationships to both mitochondrial network and cristae morphology will not only further deepen our understanding of energy metabolism but may also contribute to improved therapeutic manipulation of cell viability, differentiation, proliferation, and identity in many different cell types. Antioxid. Redox Signal. 39, 684–707.
Introduction
The spatial arrangement of mitochondria is closely related to their metabolic function. Unlike its frequent depiction as a kidney bean-shaped organelle in many textbooks, the real morphology of mitochondria is a highly dynamic reticular network, constantly undergoing fusion and fission, thereby changing between elongated/branched networks and small spherical structures (Friedman and Nunnari, 2014). Mitochondrial morphological changes are one of the important drivers of cellular energy metabolism.
The two major sources of adenosine triphosphate (ATP) in the cell are substrate-level phosphorylation by glycolysis in the cytoplasm and oxidative phosphorylation (OXPHOS) inside the mitochondria. Fluxes through each of these pathways are dynamically rewired based on cellular demands and contexts. Mitochondrial morphology can contribute to such metabolic reprogramming by directly impacting OXPHOS activity and efficiency. Perhaps the most prominent example of metabolic reprogramming is aerobic glycolysis or the Warburg effect.
First described in cancer cells almost a century ago (Warburg et al., 1927), it was later found that such a switch to favor glycolysis is not unique to cancer cells; similar phenomena are seen in actively proliferating cells, such as pluripotent stem cells (PSCs) and antigen-activated immune cells (Abdel-Haleem et al., 2017). In these cells, the mode of metabolism is critical for proper differentiation and function. Therefore, the disturbance of mitochondrial morphology results in disturbed energy metabolism, manifesting as impaired cell proliferation and cell fate determination.
A more recent addition to this picture, especially thanks to the introduction of novel microscopic techniques such as stimulated emission depletion, is the dynamic changes in cristae shape (Kondadi et al., 2020b). A growing body of evidence ties cristae integrity directly to OXPHOS activity, suggesting it to be another potential contributor to or sensor of metabolic reprogramming.
In this review, we summarize the current findings on how mitochondrial architecture impacts the metabolic switching between aerobic glycolysis and OXPHOS. Importantly, metabolism and mitochondrial architecture exhibit mutual crosstalk; therefore, metabolic changes can also modify mitochondrial structure. Therefore, we first review various signals, including metabolic/nutrient signals that impact mitochondrial architecture.
This is followed by an overview of how changes in mitochondrial architecture, in turn, impact metabolism. We review T cells, macrophages, stem cells, and cancer cells as examples of mitochondrial architecture regulating cell metabolism and phenotype. In all these cell types, mitochondrial architecture has emerged as a critical regulator of metabolic phenotype.
The Machinery of Mitochondrial Dynamics and Cristae Shape Regulation
The molecular machinery that regulates mitochondrial shape has been well reviewed (Cogliati et al., 2016; Giacomello et al., 2020), and only a summary will be mentioned here. A group of membrane-shaping Dynamin family guanosine triphosphatases (GTPase) regulates mitochondrial morphology: the fission-inducing dynamin-related protein 1 (DRP1), the fusion-inducing mitofusin 1, 2 (MFN1, 2), and optic atrophy type 1 protein (OPA1) (shown in Fig. 1A).
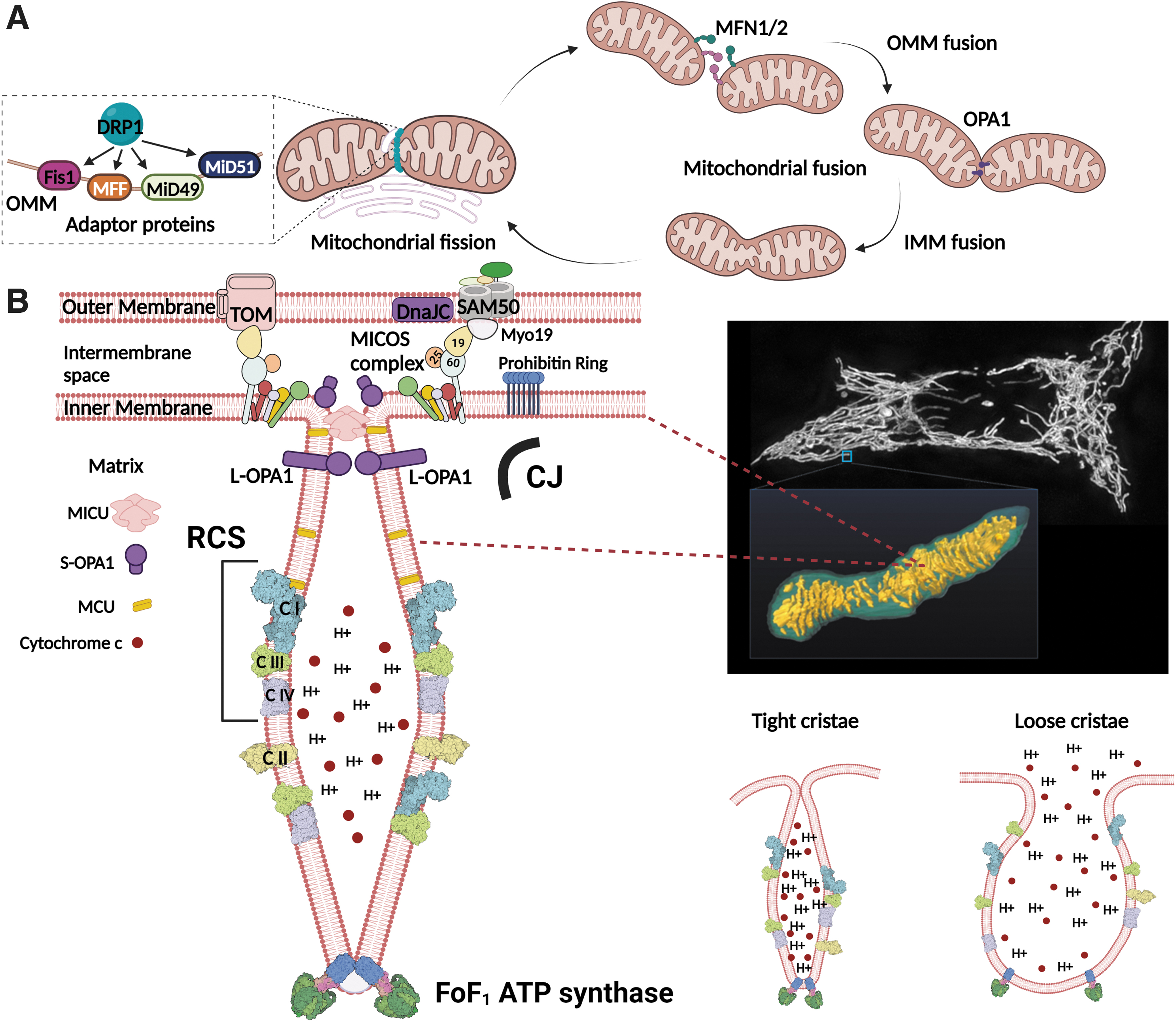
DRP1 is localized to the cytoplasm and remains inactive when it is phosphorylated at Ser637 by protein kinase A, but is activated, moved, and polymerized to rings that squeeze the outer mitochondrial membrane (OMM) tubular surface on Ser637 dephosphorylation by calcineurin (Cereghetti et al., 2008; Chang and Blackstone, 2007; Cribbs and Strack, 2007). Phosphorylation at Ser616 by cyclin-dependent kinase 1/5 or extracellular signal-regulated kinase 1/2 (ERK1/2) also has an activating effect (Jahani-Asl et al., 2015; Kashatus et al., 2015; Prieto et al., 2016; Serasinghe et al., 2015; Taguchi et al., 2007).
DRP1 then binds to adaptor proteins known as receptors, namely mitochondrial fission 1 protein (Fis1), mitochondrial fission factor, MiD49, and MiD51 (James et al., 2003; Otera et al., 2010; Palmer et al., 2011). Subsequently, DRP1 oligomerizes and constricts mitochondrial OMM, powered by GTP hydrolysis (Kalia et al., 2018).
Mitochondrial fusion is a two-step process consisting of OMM fusion mediated by MFN1/2 and inner mitochondrial membrane (IMM) fusion mediated by OPA1. MFN1 and MFN2 share significant sequence and structural similarity, but they seem to play distinct cellular functions (Schrepfer and Scorrano, 2016). MFN1 mediates mitochondrial fusion more efficiently, probably thanks to having an approximately eightfold higher GTPase activity (Ishihara et al., 2004).
OPA1 also depends on MFN1 but not MFN2 for promoting fusion (Cipolat et al., 2004). Meanwhile, MFN2 tethers mitochondria to the endoplasmic reticulum (ER), although the full spectrum of its function remains to be explored (de Brito and Scorrano, 2008).
Numerous OPA1 isoforms have two categories: long-OPA1 (L-OPA1) and short-OPA1 (S-OPA1). L-OPA1 protrudes from the IMM, and it forms oligomers in cristae membranes (CMs) below cristae junctions (CJs). Meanwhile, S-OPA1 is a soluble proteolytic cleavage product of L-OPA1 that is localized to the intracristal space. Cleavage is done most importantly by the zinc metalloendopeptidase OMA1 (Ehses et al., 2009; Head et al., 2009) and ATP-dependent i-AAA protease YME1L (Song et al., 2007).
All eight human isoforms of OPA1 contain the S1 cleavage site for OMA1, whereas only four (isoforms 4, 6, 7, 8) contain an additional S2 cleavage site for YME1L (Delettre et al., 2001; Ishihara et al., 2006). Recently, another cleavage site S3 was identified for YME1L (Wang et al., 2021a). Crucially, differences in the cleavage site affect the functionality of OPA1. Normally, YME1L constitutively cleaves a portion of L-OPA1 into S-OPA1, and its activity is enhanced as the cellular metabolic profile shifts from glycolysis to enhanced OXPHOS (Mishra et al., 2014).
Although L-OPA1 has been reported to be fusion-competent, efficient fusion still requires a moderate quantity of S-OPA1 (Ge et al., 2020). Therefore, the constitutive cleavage of L-OPA1 by YME1L and OMA1 is likely to contribute to a moderate level of S-OPA1 formation that favors mitochondrial fusion. Under stress, OMA1 activity obscures YME1L, completely cleaving OPA1 into S-OPA1 and resulting in mitochondrial fragmentation (Anand et al., 2014; Ohba et al., 2020).
A relatively novel aspect is unraveling mitochondrial ultramorphology, which deals with the cristae shape. The CM is where most respiratory chain machinery is located (Gilkerson et al., 2003). Unsurprisingly, the mitochondrial membrane potential (ΔΨm) is higher in cristae than in the inner boundary membrane (IBM) region of the IMM (Wolf et al., 2019).
Cristae are electrically insulated by mitochondrial contact site and cristae organizing system-sorting and assembly machinery (MICOS-SAM) complexes surrounding the outlet of the crista at its transition to the IBM. As a result, each crista independently maintains ΔΨm (Wolf et al., 2019). Different cristae can merge with or separate from each other over seconds through a movement of CJs and hypothetical concomitant membrane remodeling, redistributing ΔΨm between them (Kondadi et al., 2020a).
Cristae integrity is maintained by IMM proteins, primarily the MICOS complex, and FoF1 ATP synthase dimers/higher oligomers (Quintana-Cabrera et al., 2018) and various OPA1 isoforms or oligomers (shown in Fig. 1B). OPA1 stability directly correlates with the existence of cristae lumen and CJ, as its deficiency results in cristae widening, whereas overexpression tightens cristae (Glytsou et al., 2016; Griparic et al., 2004). A complete loss of OPA1 results in the absence of most of the cristae.
The MICOS complex that forms CJ by joining the SAM complex also maintains cristae shape. In humans, the MICOS complex consists of seven subunits: Mic60, Mic19, Mic25, Mic10, Mic13, Mic26, and Mic27 (Colina-Tenorio et al., 2020). Mic60 and OPA1 physically interact with each other to form CJ, although Mic60 is able to maintain CJ alone without OPA1 (Barrera et al., 2016). Mic19 and Mic60 of the MICOS complex are tethered to the OMM by the OMM protein SAM 50 kDa subunit (SAM50), which is also important for cristae shape (Ding et al., 2015; Tang et al., 2019).
In addition, another OMM protein known as myosin 19 was recently reported to associate with the SAM-MICOS supercomplex and play a key role in maintaining cristae structure and OXPHOS activity (Shi et al., 2022). Mitochondrial proteases can also target MICOS complex proteins. Stress conditions can activate OMA1, which cleaves Mic19 at the N-terminus, thereby disrupting this connection and cristae morphology (Tang et al., 2019). Meanwhile, Mic60 homeostasis could be regulated by YME1L (Li et al., 2015). It is worth noting that MICOS complex proteins can also play individual roles outside the complex. A recent example is Mic10, which was shown to stabilize FoF1 ATP synthase dimeric oligomer assembly and thereby contribute to respiratory growth (Rampelt et al., 2022).
Lastly, FoF1 ATP synthases are the sources of most cellular ATP, but besides their catalytic function, their dimers are organized into higher oligomeric complexes that form rows along the cristae edges (Blum et al., 2019; Davies et al., 2012). This oligomerization could be promoted by the binding of the ATPase inhibitory factor 1 (IF1) (García-Bermúdez and Cuezva, 2016). FoF1 ATP synthases are localized predominantly in the cristae, but recent evidence suggests that there are also FoF1 ATP synthases located in the IBM, and that this subpopulation has different mobility and might preferentially mediate ATP hydrolysis (Salewskij et al., 2020).
It is worth noting that IMM morphology is also influenced by its lipid composition, which is highly unique. IMM contains a high level (approximately half) of non-bilayer cone-shaped phospholipids, namely phosphatidylethanolamine (34%) and cardiolipin (18%), which promotes a curved membrane structure (Horvath and Daum, 2013). In particular, cardiolipin has been computationally and experimentally shown to strongly prefer negative curvatures, as it relaxes curvature frustration (Beltrán-Heredia et al., 2019; Federico Elías-Wolff et al., 2019).
Such a curvature-sensing property of cardiolipin is likely to be important for cristae structure, as both the crista tip and CJ exhibit significant curvature (Ikon and Ryan, 2017). Cardiolipin also interacts with other cristae-forming proteins, namely OPA1 (Ban et al., 2017), Mic27 (Weber et al., 2013), Mic10 oligomers (Rampelt et al., 2018), and ATP synthase (Eble et al., 1990), aiding in their functions.
Signals That Impact Mitochondrial Architecture
Nutrient signals
Dynamic changes in bioenergetic demand can be communicated to mitochondrial morphological proteins through various signaling pathways and post-translational modifications, especially those connected to nutrient availability and stress (Sabouny and Shutt, 2020). For example, protein acetylation and O-linked-N-acetylglucosaminylation (O-GlcNAcylation) are sensitive to nutrient availability and can directly modify mitochondrial morphological proteins. These pathways could be involved in regulating mitochondrial morphology during metabolic reprogramming.
DRP1, MFN1/2, and OPA1 can all be acetylated to promote mitochondrial fission; acetylation induces DRP1 activation and mitochondrial translocation (Hu et al., 2020) as well as reduced MFN1/2 and OPA1 activity (Biel et al., 2015; Lee et al., 2014; Samant et al., 2014). Meanwhile, deacetylation can be mediated by histone deacetylases (HDAC), particularly sirtuins (SIRT), under caloric restriction (Haigis and Guarente, 2006).
Mitochondrial SIRT3 deacetylates matrix-exposed OPA1 (Samant et al., 2014) whereas extramitochondrial SIRT1 deacetylates OMM proteins, including both MFN1 and MFN2 (Biel et al., 2015; Oanh et al., 2017), enhancing their activity. Glucose starvation also triggers MFN1 deacetylation and activation by HDAC6 (Lee et al., 2014). HDAC6 has also been reported to interact with MFN2, enhancing its stability against hypoxic stress-induced degradation (Kim et al., 2015).
These deacetylases are often downregulated in many types of cancer, suggesting their potential involvement in cancer metabolic reprogramming (Chalkiadaki and Guarente, 2015). Further, the acetylation state can also impact mitochondrial morphology through upstream signaling. Cytoplasmic SIRT2 downregulation aids in the glycolytic switch during somatic reprogramming by increasing the acetylation of mitogen-activated protein kinase (MAPK) kinase-1 (MEK1), which activates ERK to phosphorylate and activate DRP1 (Cha et al., 2021). In addition, SIRT2 downregulation also leads to Akt hyperacetylation and inactivation, which further contributes to DRP1 activation (Cha et al., 2021).
Nutrient excess can also be sensed by the hexosamine biosynthetic pathway, which regulates O-GlcNAcylation. This is because the synthesis of the substrate for protein O-GlcNAcylation, uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), depends on uridine triphosphate, glucose, glutamine, and acetyl-coenzyme A (CoA) derived from various nutrients (nucleotides, glucose, amino acids, and fatty acids) (Akella et al., 2019).
Both DRP1 and OPA1 have been reported to undergo O-GlcNAcylation, which promotes mitochondrial fission (Gawlowski et al., 2012; Makino et al., 2011; Park et al., 2021). O-GlcNAcylation is generally increased in many cancer cells (Ferrer et al., 2016), and it is also required for pluripotency acquisition (Jang et al., 2012) and T cell activation (Lund et al., 2016). Since all these cell types exhibit the Warburg effect, O-GlcNAcylation could participate in mitochondrial remodeling that promotes the glycolytic switch.
Another important nutrient-sensing pathway involves the mammalian target of rapamycin (mTOR). mTOR complex 1 (mTORC1) is stimulated by increased intracellular amino acids, glucose, and oxygen availability, the latter two via the inhibition of adenosine monophosphate-activated protein kinase (AMPK). Insulin, which signals systemic nutrient availability, also activates mTORC1 via Akt signaling (Howell and Manning, 2011).
mTORC1 increases mitochondrial fission through the increased translation of mitochondrial fission process 1 (MTFP1). MTFP1, in turn, increases the activating Ser616 phosphorylation of DRP1 while reducing inhibitory Ser637 phosphorylation, leading to increased DRP1 recruitment to mitochondria (Fig. 2A) (Morita et al., 2017).
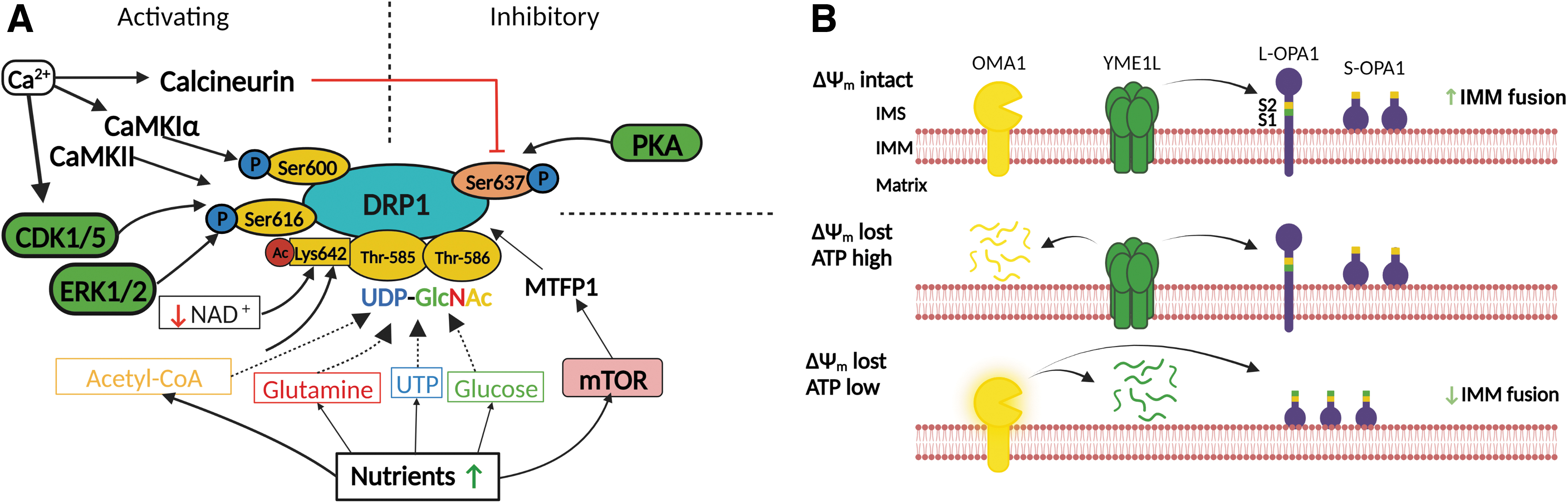
Ca2+ signaling
Ca2+ can induce both OXPHOS increase and morphological changes in mitochondria. Mitochondrial Ca2+ activates several mitochondrial dehydrogenases, namely glycerol-3-phosphate dehydrogenase, matrix complexes of pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase, leading to increased nicotinamide adenine dinucleotide hydrogen (NADH), increased electron transport chain (ETC) activity, and increased ATP production (Jouaville et al., 1999).
However, excessive calcium accumulation can open the mitochondrial permeability transition pore and lead to cell death (Baumgartner et al., 2009; Ichas and Mazat, 1998). Meanwhile, cytoplasmic Ca2+ can favor mitochondrial fission through various mechanisms; calcineurin dephosphorylates DRP1 at Ser537 (Cereghetti et al., 2008), calmodulin-dependent protein kinase Iα (CaMKIα) phosphorylates DRP1 at Ser600 (Han et al., 2008), and CaMKII phosphorylates DRP1 at Ser616 (Bo et al., 2018), all of which activate fission. Notably, Ca2+ homeostasis can, in turn, be impacted by changes in mitochondrial dynamics. Mitochondrial fission decreases mitochondrial Ca2+ uptake rates and capacity, whereas mitochondrial fusion increases them (Kowaltowski et al., 2019).
Cristae shape can also be impacted by Ca2+ via mitochondrial calcium uptake 1 (MICU1). It is hypothesized that CJ is synergistically closed by both OPA1 and MICU1, preventing mitochondrial calcium uniporters (MCU) located in the CM from accessing Ca2+ (Gottschalk et al., 2019). When CJ is disrupted by the knockdown of OPA1 or MICU1, Ca2+ can now freely pass through MCUs to reach the mitochondrial dehydrogenases, resulting in increased OXPHOS (Fülöp et al., 2011; Gottschalk et al., 2019). A similar CJ disruption and OXPHOS enhancement can take place on an increase in cytosolic Ca2+ level, which disassembles MICU1 at the CJ (Gottschalk et al., 2022). The MCU is then disseminated into the IBM, allowing further Ca2+ uptake into the matrix (Gottschalk et al., 2019).
Redox signaling and oxidative stress
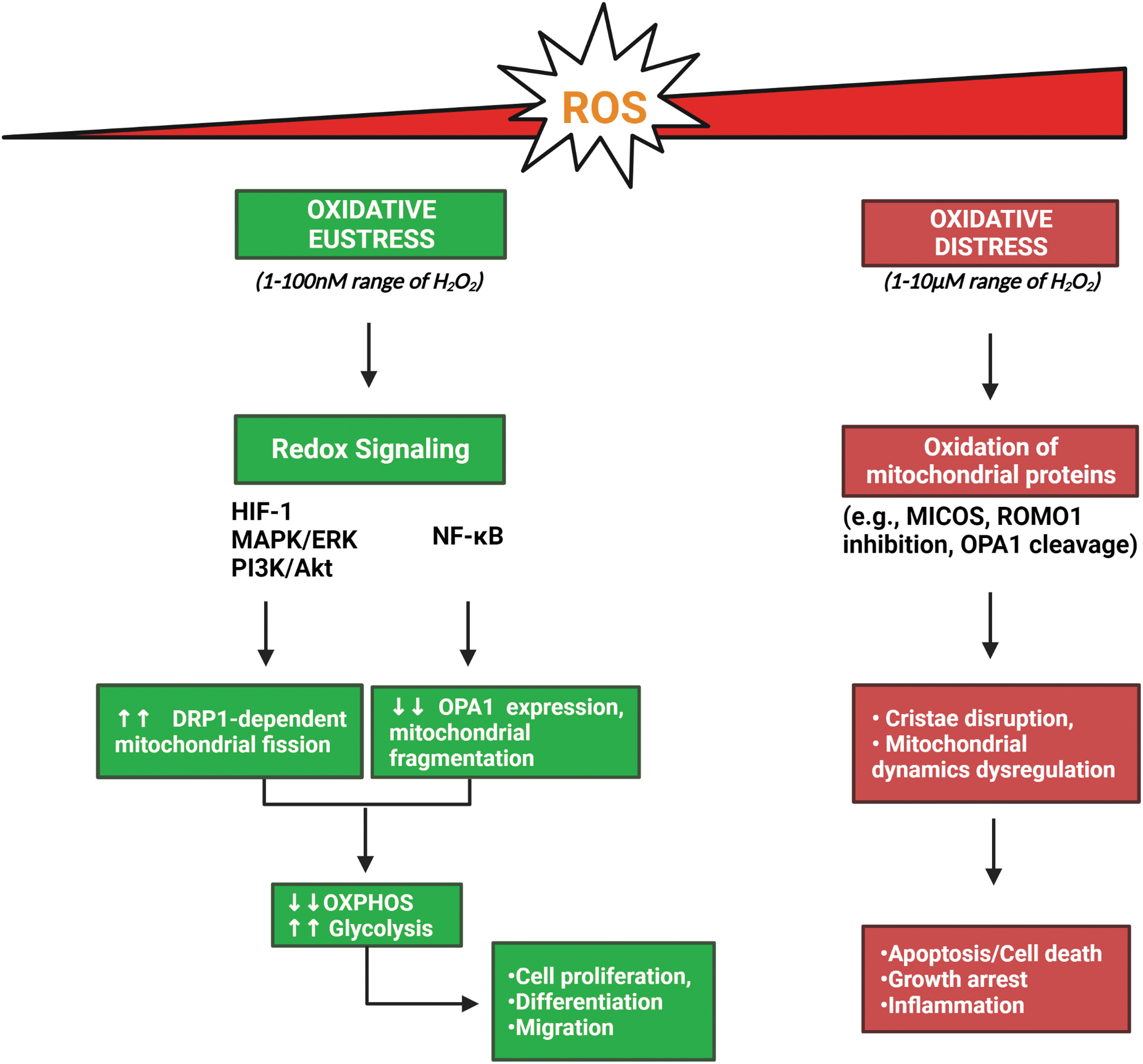
Mitochondria are the major cellular source of reactive oxygen species (ROS). Early research primarily associated ROS with cellular damage due to the unwanted oxidation of biomolecules, resulting in the concept of oxidative stress. However, later findings suggested that not all ROS generations result in unwanted deleterious consequences. Therefore, oxidative stress is now recognized in two patterns, known as “oxidative eustress” and “oxidative distress” (Sies and Jones, 2020).
Under oxidative eustress, low levels of physiological ROS generation activate redox signaling that promotes cell proliferation, differentiation, and migration. In particular, hydrogen peroxide (H2O2) and superoxide anion radicals (O2 ·−) are thought to impact cell signaling through the redox modification of protein thiol groups (Ježek et al., 2020). An estimated 1–100 nM of H2O2 is thought to be relevant for redox signaling, although this range can be impacted by, for example, cellular antioxidant levels (Sies, 2017).
At higher levels of H2O2, cellular stress responses and adaptive changes occur, such as the enhanced expression of antioxidants. Even higher levels of ROS (into the 1–10 μM range) overwhelm cellular defense mechanisms against ROS, thereby inducing inflammation, growth arrest, and cell death; this process corresponds to oxidative distress (Schieber and Chandel, 2014; Sies and Jones, 2020).
How mitochondrial architecture is impacted by ROS is not completely understood. Many mitochondrial morphological proteins are sensitive to oxidation by ROS. For example, Mic60 and Mic19 are both sensitive to cell redox states; oxidized Mic19 connects the two MICOS subcomplexes and support cristae shape, and oxidized Mic60 leads to delayed mitophagy and defective respiration (Li et al., 2021; Sakowska et al., 2015). Several studies indicate that oxidative stress impairs OPA1 function, by OPA1 cleavage (Garcia et al., 2018) and by the inactivation of reactive oxygen species modulator 1, which is indispensable for proper OPA1 function (Norton et al., 2014; Richter et al., 2019). Whether these changes are relevant in physiological contexts require further studies.
Besides the direct oxidation of mitochondrial morphological proteins, various redox signaling pathways can also regulate mitochondrial dynamics (Ježek et al., 2018a; Willems et al., 2015). For example, the hypoxia-inducible factor-1 (HIF-1), MAPK/ERK, phosphoinositide 3′-kinase (PI3K)/Akt, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways are activated by moderate levels of ROS (Schieber and Chandel, 2014; Sies and Jones, 2020), and they can all influence mitochondrial dynamics, mostly by increasing mitochondrial fission.
Specifically, the HIF-1 (Marsboom et al., 2012; Wan et al., 2014), MAPK/ERK (Kashatus et al., 2015; Serasinghe et al., 2015), and PI3K/Akt (Kim et al., 2016; Tondera et al., 2004) pathways increase DRP1-dependent mitochondrial fission, whereas the NF-κB pathway reduces OPA1 expression and increases mitochondrial fragmentation (Fig. 3) (Laforge et al., 2016). Crucially, some of them also contribute to metabolic reprogramming; HIF-1, PI3K/Akt, and NF-κB signaling pathways can all directly modulate glycolytic and OXPHOS activities and promote the Warburg phenotype (Elstrom et al., 2004; Mauro et al., 2011; Papandreou et al., 2006; Semenza et al., 1994).
Mitochondrial membrane potential
Yet another signal that impacts mitochondrial morphology is ΔΨm, which is maintained by the continuous activity of ETC and its consumption by ATP synthesis due to H+ backflow via the c-ring of ATP-synthase. Typically, the maximum ΔΨm is established at nearly zero ATP synthesis, since ΔΨm is only consumed by H+ leak. With increasing ATP synthesis, H+ backflow via the ATP synthase decreases ΔΨm from the maximum by several millivolts. In severe states, for example, in fragments containing mutant mitochondrial deoxyribonucleic acid (mtDNA), ΔΨm declines even further below the values established at ATP synthesis. Note that mitophagy cleans these fragments.
Significantly low ΔΨm activates OMA1, which cleaves its target proteins in the intermembrane space (IMS), most importantly OPA1 (Anand et al., 2014; Head et al., 2009) and Mic19 (Tang et al., 2019). OPA1 and Mic19 cleavage loosens the CJ and widens the cristae, thereby releasing cytochrome c from the cristae lumen, potentiating the cell toward apoptosis. To prevent low ΔΨm-induced apoptosis, some glycolytic cells even reverse mitochondrial FoF1-ATP synthase, effectively using it as an ATPase that hydrolyzes glycolytic ATP shunted into the matrix, for example, by the adenine nucleotide translocator (ANT), to maintain ΔΨm.
This is seen in activated macrophages (Garedew et al., 2010), human PSCs (Zhang et al., 2011), and cancer cells (Depaoli et al., 2018). It is worth noting that cancer cells could alter IMM ATP/ADP exchange from the ANT to the ATP-Mg/phosphate carrier, which decreases the cytosolic ATP/ADP ratio to favor aerobic glycolysis (Lemasters, 2021). In addition, IF1 may also inhibit ATP hydrolysis to prevent ATP depletion during OXPHOS inactivation (Chen et al., 2014).
It is worth noting that a low ΔΨm does not necessarily lead to OMA1-induced OPA1 cleavage and mitochondrial remodeling, and cellular differentiation and cellular ATP level can also influence OPA1 processing. For example, in H9c2 cardiomyoblasts, OMA1-mediated L-OPA1 cleavage due to low ΔΨm was absent in undifferentiated, glycolytic cardiomyoblasts but was robust in differentiated cells that rely on OXPHOS (Garcia et al., 2021).
Interestingly, when the mitochondrial membrane is depolarized under cellular stress, YME1L and OMA1 can degrade each other depending on the ATP availability. In depolarized mitochondria with a low level of cellular ATP, as seen in cells under acute oxidative stress, OMA1 is autocatalytically activated and degrades YME1L (Rainbolt et al., 2015). Meanwhile, if mitochondrial depolarization is accompanied by a high ATP level, YME1L degrades OMA1 (shown in Fig. 2B) (Rainbolt et al., 2016).
Extracellular acidosis
Extracellular acidosis, a frequent product of anaerobic glycolysis, can regulate both mitochondrial morphology and OXPHOS. Acidosis (pH = 6.5) was reported to trigger mitochondrial fusion and cristae tightening irrespective of oxygen availability (Khacho et al., 2014). This is mediated by DRP1 inhibition and activation of the stress-induced mitochondrial hyperfusion pathway, which utilizes OPA1, MFN1, and Stomatin-like protein 2 to promote mitochondrial fusion (Khacho et al., 2014; Tondera et al., 2009). OPA1 processing and the disruption of OPA1 oligomeric complexes are both prevented under acidosis (Khacho et al., 2014). As a result, respiratory chain supercomplexes (RCS) integrity improves and OXPHOS is enhanced.
How Mitochondrial Architecture Impacts Metabolism
OXPHOS activity
The dynamic fusion and fission of mitochondria alter the respiratory activity to meet the ever-changing bioenergetic demands of the cell. Fusion enables mtDNA in the matrix as well as IMM respiratory proteins to mix between mitochondria. This mixing contributes to the integrity of the respiratory chain and mtDNA, enabling efficient OXPHOS. Thus, it is often seen in cells with increased ATP demand, such as cells under caloric restriction or mild stress (Liesa and Shirihai, 2013; Wai and Langer, 2016).
Meanwhile, fissioned mitochondria are seen in cells that do not require high respiratory activity (Westermann, 2012). Enhancing mitochondrial fission promotes a glycolytic shift, potentially driven by altered redox signaling (Guido et al., 2012). Further, fragmented mitochondria also enable the elimination of damaged mitochondria through mitophagy, thereby maintaining the quality of the bioenergetic machinery (Twig et al., 2008).
The quality check is based on ΔΨm and fragments with low ΔΨm, that is, those with highly oxidized proteins that are unable to provide an optimum function and those with mutant mtDNA and hence mutated subunits of the complex I membrane part, or one of complex III or two subunits of the membrane FO moiety of ATP-synthase. In contrast, surprisingly, Yu et al. (2019) found that when mitochondrial fusion was enhanced in pancreatic ductal adenocarcinoma cells through DRP1 inhibition or MFN2 overexpression, OXPHOS decreased due to enhanced mitophagy.
This result may be unique to pancreatic ductal adenocarcinoma cells, but it also indicates that the correlation between fusion and enhanced OXPHOS may be context-dependent, and not a universal finding.
Cristae shape also undergoes dynamic morphological alterations to adjust the OXPHOS rate (Cogliati et al., 2016; Kondadi et al., 2020b; Patten et al., 2014). The efficiency of OXPHOS is maximum in the folded, narrow cristae, where cytochrome c remains inside the cristae and rows of ATP synthase dimers line the edge of the cristae. This cristae conformation allows for the optimal arrangement of RCS, which is an assembly of multiple respiratory complexes consisting of complex I, III, and IV that enables more efficient electron transfer (Cogliati et al., 2013).
The special arrangement of cristae and ATP synthase dimers has also been suggested to promote local pH gradients that can enhance proton-motive force (Strauss et al., 2008). However, others concluded that the pH difference is too low to drive increased ATP synthesis, and instead the kinetic coupling through physical proximity between complexes is more important for ATP synthesis (Toth et al., 2020). Cristae shape also responds to nutrient availability and respiratory activity.
Cells that received high glucose treatment (11–20 mM) or hypoxia (5% oxygen, 72 h) exhibit widened cristae with corresponding changes in Mic60 distribution (Dlasková et al., 2018; Plecitá-Hlavatá et al., 2016). When respiration is increased with dimethyl-2-oxoglutarate, these hypoxia-adapted cristae instantly narrow (Dlasková et al., 2019). These findings suggest that cristae tightness is closely coordinated with the cellular OXPHOS activity.
With increased mitochondrial fission and a shift to aerobic glycolysis, mitochondrial ATP synthesis might be attenuated, and this could impact the ATP level of the entire cell. However, there is little evidence to suggest that cellular ATP depletion takes place by morphological manipulation. For example, the impairment of mitochondrial fusion by MFN2 knockdown did not deplete cellular ATP despite attenuating OXPHOS (Yao et al., 2019).
This could be thanks to the fast rate of glycolytic ATP synthesis, despite being inefficient per glucose molecule (Liberti and Locasale, 2016; Vaupel et al., 2021). In fact, ATP levels might even set an upper limit to glycolytic flux and tricarboxylic acid (TCA) cycle flux through allosteric inhibition, restricting biosynthesis (Nelson and Cox, 2021). In this context, attenuating mitochondrial ATP synthesis by mitochondrial fission and even consuming a part of glycolytic ATP to maintain ΔΨm could benefit proliferating cells by increasing the fluxes of biosynthetic pathways.
Redox signaling
Changes in mitochondrial dynamics, specifically mitochondrial fission, may produce ROS to initiate redox signaling. For example, in hepatocellular carcinoma (HCC) cells, mitochondrial fission induced by the forced expression of DRP1 or knockdown of MFN1 resulted in increased ROS production and subsequent Akt activation as well as increased NF-κB transcriptional activity (Huang et al., 2016). Although not mentioned in this study, the activation of Akt and NF-κB likely contributes to the Warburg phenotype of HCC (Elstrom et al., 2004; Mauro et al., 2011). Notably, mitochondrial dynamics might not always be accompanied by changes in ROS generation. For instance, the transient overexpression of either DRP1 or MFN2 in human lung adenocarcinoma cells did not alter ROS generation (Wu et al., 2011).
Cristae integrity and dynamics could also influence ROS generation via modulating RCS stability (Lopez-Fabuel et al., 2016; Maranzana et al., 2013). A recent study showed that OPA1 can reduce mitochondrial ROS accumulation in an ATP synthase dimer-dependent fashion, likely through improving of RCS efficiency (Quintana-Cabrera et al., 2021). In addition, Mic60 was recently suggested to be involved in ROS generation through its interaction with ferrochelatase, the terminal enzyme for heme synthesis (Dietz et al., 2021). However, whether these cristae-regulating proteins can activate redox signaling remains to be studied.
Aspartate metabolism
Aspartate is particularly important in supporting the biosynthetic demands of proliferating cells such as cancer cells (Garcia-Bermudez et al., 2018), hematopoietic stem cells (HSCs) (Qi et al., 2021), and proliferating lymphocytes (Newsholme and Calder, 1997). This is due to aspartate being the precursor for purine/pyrimidine and protein synthesis. Somewhat surprisingly, sufficient aspartate synthesis is dependent on a robust OXPHOS promoted by mitochondrial fusion.
Impairing mitochondrial fusion by MFN2 knockdown attenuated OXPHOS as well as aspartate synthesis, reducing proliferation in non-transformed mouse fibroblasts (Yao et al., 2019). Further, OPA1 disruption leads to the depletion of aspartate and its related metabolites (glutamate and α-ketoglutarate) (Bocca et al., 2018; Chao de la Barca et al., 2019). These findings may be explained by mitochondrial fusion enabling efficient ETC activity and regeneration of NAD+.
The protonated NADH directly inhibits rate-limiting enzymes of the TCA cycle, bringing it to a halt. An active TCA cycle is required to replenish oxaloacetate, the precursor for aspartate biosynthesis. Indeed, it has been reported that ETC inhibition can compromise aspartate synthesis (Birsoy et al., 2015; Sullivan et al., 2015). The loss of fusion proteins does not fully impair ETC activity, but proliferating cells with high aspartate demand seem to require proportionately robust ETC activity achieved by mitochondrial fusion to support biosynthesis.
Mitochondrial Architecture in Cells That Undergo Metabolic Reprogramming
Mitochondrial shape and dynamics in T cells
Recent advances in immunometabolism have shown that there are distinct metabolic profiles between dormant naïve/memory cells and activated effector cells. In quiescent cells such as naive or memory T (TM) cells, regulatory T (Treg) cells, or alternatively activated (M2) macrophages, fatty acid oxidation (FAO) and OXPHOS predominate. Meanwhile activated immune cells such as effector T (TE) cells and classically activated (M1) macrophages adopt aerobic glycolysis (Buck et al., 2016; O'Neill et al., 2016).
This metabolic difference might be explained evolutionarily by the optimization of resource allocation known as the life history theory (Wang et al., 2019). During cellular growth, proliferation, and defense, signaling pathways such as pro-inflammatory cytokine pathways favor anabolic reactions to generate the required biomass. Meanwhile, during dormancy triggered by various anti-inflammatory signals, catabolic reactions such as OXPHOS and FAO become dominant to maintain efficient ATP production.
Mitochondrial morphology regulation is likely another part of this picture, providing a mechanistic connection between the signaling pathways and the alteration in metabolism. Specifically, mitochondrial fragmentation and cristae loosening support the anabolic shift to aerobic glycolysis by suppressing OXPHOS activity (Ježek et al., 2018b; Plecitá-Hlavatá et al., 2016; Plecitá-Hlavatá et al., 2015). Mitochondrial fusion, or perhaps more importantly cristae tightening, counters this through supporting catabolic OXPHOS and FAO (Buck et al., 2016; Geltink et al., 2017).
Mitochondrial morphology and subsequent metabolic reprogramming have emerged as an essential driver indispensable for proper T cell maturation and function (shown in Fig. 4A). For example, OPA1 is required for T cell maturation during the OXPHOS-dependent cluster of differentiation (CD)4 CD8 double-negative (DN)3 stage but not during the aerobic glycolysis-dependent DN4 stage (Corrado et al., 2021). OPA1 deletion impairs cellular respiration and makes DN3 cells more prone to apoptosis (Corrado et al., 2021).
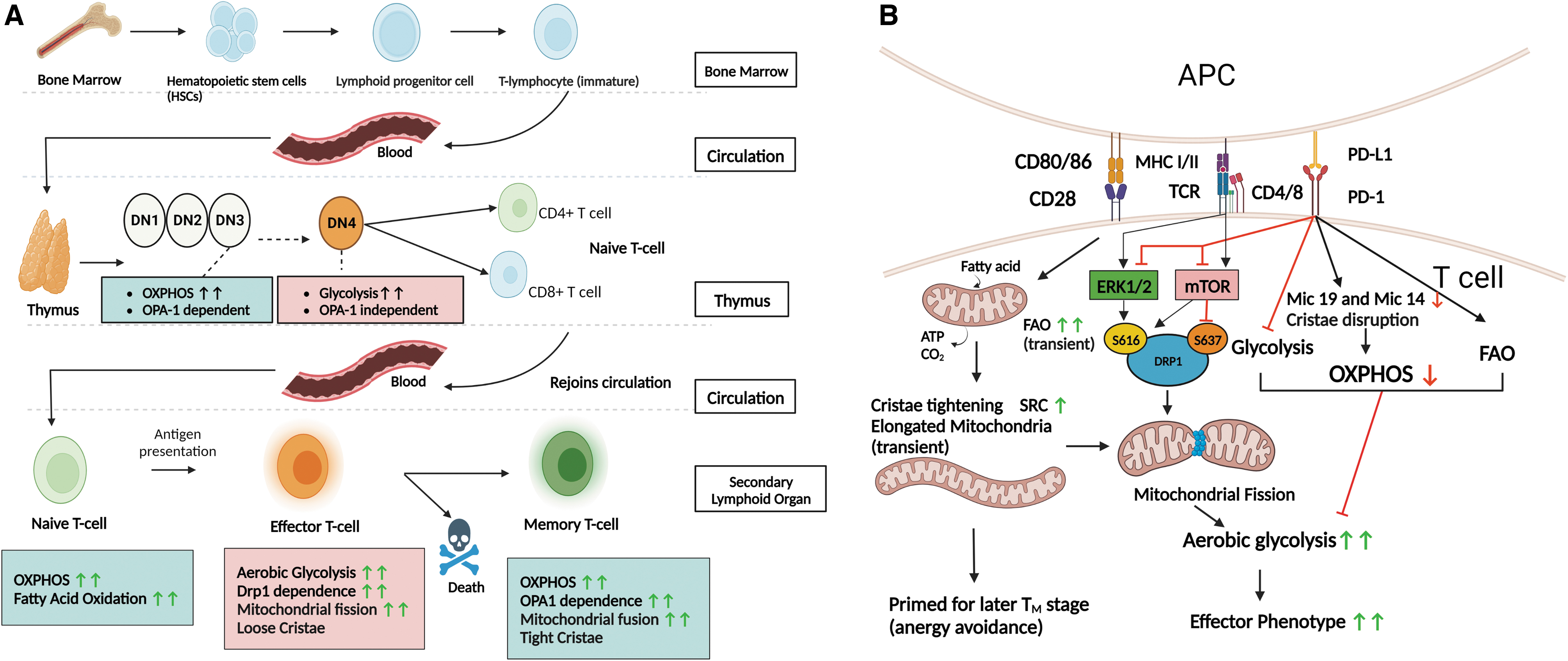
Surviving T cells that join the circulation system constitutively exhibited an effector memory T cell phenotype without antigenic stimulation, and were unable to switch into long-term TM cells, suggesting a lack of the metabolic flexibility required for normal T cell differentiation (Corrado et al., 2021).
Importantly, the antigen activation of naive T cells depends on a glycolytic switch coordinated by mitochondrial fragmentation. On T cell activation, DRP1 is activated to mediate mitochondrial fission and redistribution to the immune synapse (Baixauli et al., 2011). DRP1-deficient mature T cells not only fail to fragment their mitochondria, but also exhibit impaired metabolic reprogramming to aerobic glycolysis via altered AMPK-mTOR-c-Myc signaling (Simula et al., 2018).
As a result, DRP1 knockout T cells fail to acquire the TE cell phenotype, and instead exhibit a memory-like phenotype on activation in vitro (Simula et al., 2018). This metabolic alteration occurred independent of mitochondrial mass, ΔΨm, glucose uptake, and ATP levels, indicating that mitochondrial fission is sufficient to induce the change.
Once the causative antigen is eliminated, TE cells mostly die out, leaving a small population of quiescent TM cells that rely primarily on OXPHOS. OPA1 was found to be essential for more oxidative TM cells, which exhibit more fused mitochondria, but not in glycolytic TE cells with fissioned mitochondria (Buck et al., 2016), which is consistent with the fact that OPA1-induced fusion and cristae tightening favors FAO and OXPHOS. Interestingly, the loss of MFN1/2 does not cause a similar impairment in TM cells, indicating that non-fusion-related functions of OPA1, especially cristae maintenance, could be more important for TM identity (Rambold and Pearce, 2018).
T cell identity is also critically regulated by the surface receptors co-stimulator CD28 and immune checkpoint programmed cell death-1 (PD-1). CD28 is required to form non-anergic TM cells, whereas PD-1 is an inhibitor of activated T cells that directly antagonizes CD28. Interestingly, both alter T cell identity by changing mitochondrial morphology and altering metabolism (shown in Fig. 4B). CD28 increases both glycolysis and OXPHOS through increased glucose uptake and mitochondrial fusion (Beckermann et al., 2020; Frauwirth et al., 2002).
Further, with CD28 stimulation, mitochondrial FAO is transiently activated before the first cell division, preventing cristae loosening. This gives T cell mitochondria the subsequent capacity to support the OXPHOS-predominant profile in TM cells (Geltink et al., 2017). After this mitochondrial priming, mitochondria are fragmented and cristae loosen, enabling a higher glycolytic activity that supports the TE state.
As TM cells differentiate from the surviving TE cells, mitochondrial fusion takes place, but only CD28-primed cells exhibit tight cristae and a non-anergic phenotype (Geltink et al., 2017). The authors of the study also point out that tight cristae are not necessarily required for FAO, but it seems to be required for spare respiratory capacity (SRC). SRC is the reserve respiratory capacity available in case of increased demand or stress, which seems to be indispensable for later TM cell identity (Geltink et al., 2017).
Indeed, CD8+ TM cells but not CD8+ TE cells exhibit significant SRC (Windt et al., 2012). The importance of cristae structure in T cells is further implied by recent findings that showed cardiolipin and fatty acid-binding protein 5, both of which regulate cristae shape, to be indispensable for T cell activity (Corrado et al., 2020; Field et al., 2020).
Meanwhile, PD-1 attenuates both glycolysis and OXPHOS, and at the same time augments FAO, resulting in relative OXPHOS dominance in activated T cells (Patsoukis et al., 2015). Unable to shift to aerobic glycolysis, these T cells fail to trigger a robust inflammatory reaction. Interestingly, PD-1 primarily targets OXPHOS by reducing the expression of MICOS complex proteins Mic19 and Mic14, thereby reducing cristae number and length (Ogando et al., 2019).
Mic19 silencing mimics the action of PD-1, with the reduced mitochondrial polarization and interferon-γ release usually associated with T cell activation (Ogando et al., 2019). This result suggests that mitochondrial cristae integrity is indispensable for the normal activated state in CD8+ T cells, implying that some OXPHOS is required in TE cells. Further, a recent report showed that PD-1 may also downregulate DRP1 activity via attenuating the ERK1/2 and mTOR pathways (Simula et al., 2022).
Mitochondrial shape and dynamics in macrophages
A macrophage is another example of metabolic reprogramming on encounter with an antigen (Koo and Garg, 2019). In an unprimed M0 state, macrophages proliferate slowly and depend primarily on OXPHOS. On encountering pathogen-associated molecular patterns such as lipopolysaccharide (LPS) or T helper type 1 (TH1) cytokines, macrophages are polarized to a pro-inflammatory classically activated M1 state.
Metabolically, this is associated with a shift to aerobic glycolysis and TCA cycle breakage at two points to favor succinate and itaconate accumulation (O'Neill, 2015). Meanwhile, when macrophages are stimulated by T helper type 2 (TH2) cytokines, they polarize to an anti-inflammatory alternatively activated M2 state. This is associated with OXPHOS-predominant metabolism. It is important to note that the picture presented here is somewhat simplified, and there are a broad spectrum of macrophage polarizations along the M1/M2 dimension (Koo and Garg, 2019).
The morphology of mitochondria is closely correlated with macrophage polarization (Fig. 5). Glycolytic M1 macrophages were found to possess fragmented mitochondria with underdeveloped cristae, whereas OXPHOS-predominant M2 macrophages possessed more fused mitochondria packed with dense, well-developed cristae (Li et al., 2020). Unsurprisingly, pro-fission DRP1 has been reported to play a central role in pro-inflammatory macrophage response.
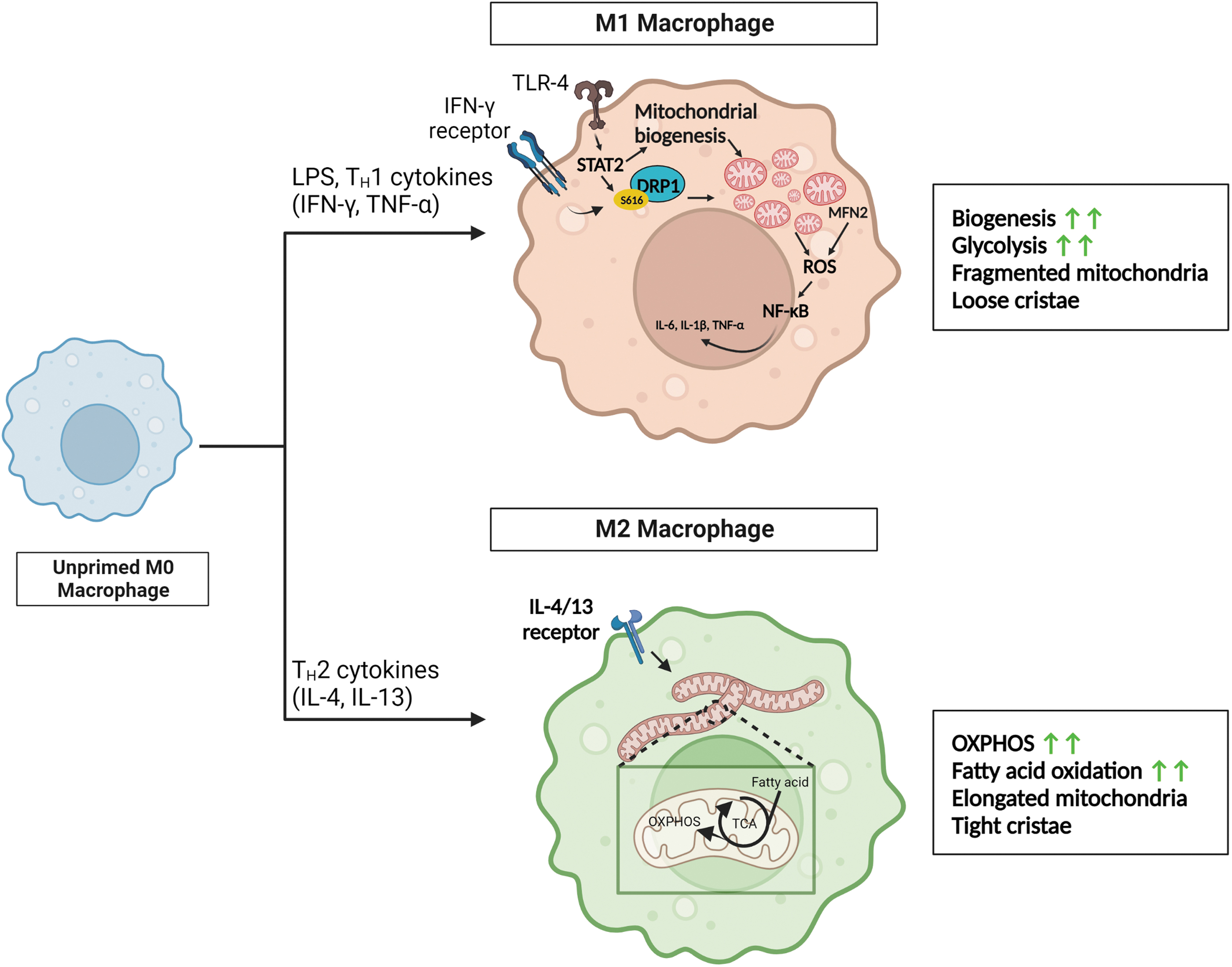
When macrophages bind to LPS, the upregulation of signal transducers and activators of transcription 2 promotes DRP1 phosphorylation and activation, leading to enhanced mitochondrial fission as well as increased mitochondrial biogenesis (Yu et al., 2020). When DRP1 is constantly active, macrophages exhibit elevated pro-inflammatory activity even in the absence of LPS, whereas the loss or inhibition of DRP1 attenuates M1 polarization (Umezu et al., 2020; Yu et al., 2020).
It is worth noting that DRP1-dependent mitochondrial fission is also critical for other aspects of the pro-inflammatory response of macrophages such as apoptotic cell phagocytosis or efferocytosis (Wang et al., 2017b) and positive post-transcriptional regulation of the pro-inflammatory cytokine tumor necrosis factor-α (Gao et al., 2021), although these effects are likely not mediated via glycolytic switching.
The LPS-activated M1 macrophages increase their mitochondrial mass but utilize mitochondria for ROS production rather than ATP synthesis (Mills et al., 2016; Yu et al., 2020). The ROS production is thanks to succinate oxidation and glycolytic ATP production enabling ΔΨm elevation, and ROS is important for pro-inflammatory NF-κB signaling (Mills et al., 2016; Yu et al., 2020).
In this line, surprisingly, pro-fusion MFN2 was found to be essential for LPS-induced ROS production and pro-inflammatory signaling, as well as other pro-inflammatory actions of macrophages such as cytokine release, phagocytosis, and antigen processing (Tur et al., 2020). However, ROS production was not altered by MFN1 loss, indicating the non-fusion-related functions of MFN2 such as ER-mitochondrial contact to be more important in pro-inflammatory ROS production (Tur et al., 2020).
OPA1 deletion in macrophages was recently studied in a myeloid-conditional knockout mouse model (Sánchez-Rodríguez et al., 2023). Along with fragmented mitochondria with sparse and loose cristae, the authors found defective RCS assembly, a significant reduction in OXPHOS, enhanced glycolysis, and the accumulation of TCA cycle intermediates including extracellular succinate and α-ketoglutarate. Surprisingly, this resulted in impaired NF-κB signaling and compromised M1 polarization.
OPA1 loss in macrophage also increased the pro-inflammatory cytokine interleukin-1β, likely due to the NLR family pyrin domain containing 3 inflammasome activation facilitated by mitochondrial dysfunction and ROS generation (Sánchez-Rodríguez et al., 2023). In summary, this study shows that the disruption of mitochondrial morphology can impact metabolic reprogramming that is critical for macrophage polarization.
Meanwhile, mitochondrial morphology that favors OXPHOS seems to counteract macrophage inflammatory response. Attenuating M1 polarization in the THP-1 macrophage cell line by inhibiting dipeptidyl peptidase 8 or 9 was associated with Mic19 and Mic60 upregulation and therefore tighter cristae, pro-fusion annexin A6 induction, and prohibitin-2 (PHB2) upregulation, which prevents OPA1 processing (Suski et al., 2020). These changes collectively lead to increased fusion and cristae tightening, augmenting OXPHOS efficiency while reducing the glycolytic profile, presumably thereby reducing M1 activation.
Mitochondrial shape and dynamics in PSCs
Manipulations of mitochondrial dynamics are also critical for metabolic changes that define stem cell identity and fate (shown in Fig. 6). PSCs, namely embryonic stem cells (ESCs) and induced PSCs (iPSCs), are a group of cells that can differentiate into any cell type from all three germ layers, and therefore have great therapeutic potential. These PSCs exhibit a distinct metabolism that heavily relies on glycolysis, whereas somatic cells depend on OXPHOS (Folmes et al., 2011).
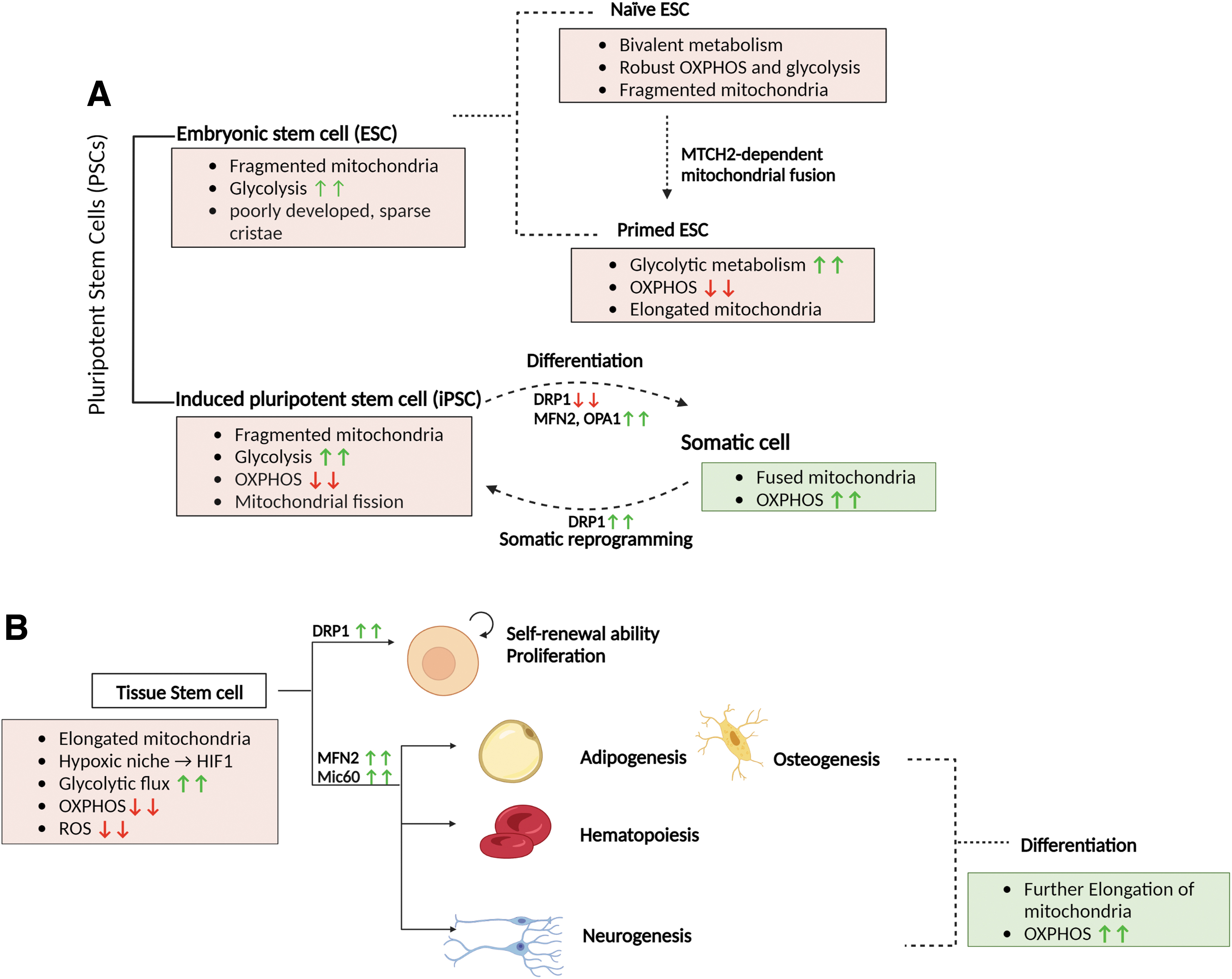
In fact, on the creation of iPSCs from somatic cells, a considerable metabolic switching from OXPHOS to glycolysis takes place (Folmes et al., 2011). Consistent with this switch, PSCs exhibit underdeveloped, fragmented mitochondria with poorly developed, sparse cristae located primarily in the perinuclear area (Chung et al., 2007; Folmes et al., 2011).
Mitochondrial fragmentation is a coordinated event in PSCs that is essential for glycolytic switch and pluripotency. On somatic reprogramming, DRP1 is activated via ERK1/2 activation and Akt inactivation, partially thanks to SIRT2 downregulation (Cha et al., 2021; Prieto et al., 2016). When DRP1 activity is enhanced by the reduction expression 1-cyclin B1/B2 axis, reprogramming efficiency also significantly improves (Son et al., 2013). On the other hand, DRP1 loss compromises the glycolytic switch and impairs the stemness of PSCs (Prieto et al., 2016; Son et al., 2013). Mitochondrial fission in pluripotency induction is also mediated by de novo fatty acid synthesis (Wang et al., 2017a).
The expression of the lipogenic enzyme acetyl-CoA carboxylase 1 (ACC1) is increased in PSCs. ACC1 stabilizes the pro-fission Fis1 by reducing its acetylation and proteasome-mediated degradation (Wang et al., 2017a). Enhancing fatty acid synthesis through ACC1 favored iPSC reprogramming in human fibroblasts through the action of Fis1, further supporting the importance of mitochondrial fission in the establishment of PSCs (Wang et al., 2017a).
Meanwhile, fused, tubular mitochondria and enhanced OXPHOS are associated with the loss of pluripotency, suggesting that the perturbation of mitochondrial morphology is sufficient to prevent normal pluripotency induction (Varum et al., 2011). Notably, other mechanisms besides mitochondrial fragmentation can also contribute to the glycolytic switch. For example, uncoupling protein 2 suppresses mitochondrial glucose oxidation in PSCs, enabling higher glycolytic flux via lactate formation (Zhang et al., 2011).
Pluripotency is now recognized in two distinct states, called naive and primed. The naive state corresponds to ESCs originating from epiblasts before implantation, whereas the primed state corresponds to ESCs derived from epiblasts after implantation, with further maturation and some differentiation bias (Nichols and Smith, 2009). Reflecting the metabolic profiles of embryonic cells of origin, naive pluripotent cells exhibit a more bivalent metabolism with both robust OXPHOS and glycolysis, whereas primed cells exhibit a more glycolytic metabolism (Takashima et al., 2014; Zhou et al., 2012).
This metabolic transition seen as cells proceed from the naive to the primed pluripotent state can be considered to be preparation for implantation to the hypoxic uterine wall. The ability to fuse mitochondrial seems to be critical for this transition, as the loss of fusion-mediating mitochondrial carrier homolog 2 (MTCH2) led to mitochondrial fragmentation in naive state mouse ESCs, delaying its transition to the primed state (Bahat et al., 2018).
Augmented MFN2 expression or a dominant-negative form of DRP1 was sufficient to overcome MTCH2 deficiency and induced transition from the naive state to the primed state (Bahat et al., 2018). MTCH2 loss also resulted in a reduction in mtDNA copy number as well as mitochondrial respiration. The authors propose that this initial respiratory suppression is compensated in HSCs and skeletal muscles by other mechanisms, as MTCH deletion led to increased respiration in these cells (Buzaglo-Azriel et al., 2016; Maryanovich et al., 2015).
Interestingly, despite relatively suppressed OXPHOS activity, primed ESCs paradoxically exhibit more fused and tubular mitochondria with better-developed cristae than naive ESCs, suggesting a potential non-energetic function of mitochondrial fusion and cristae shape in primed cells (a summary is shown in Fig. 6A) (Ware et al., 2014; Zhou et al., 2012).
When PSCs differentiate, mitochondrial OXPHOS is re-awakened (Cho et al., 2006; Facucho-Oliveira et al., 2007). Evidence suggests that mitochondrial fusion promotes this process. Indeed, pro-fission DRP1 inhibition promotes iPSC differentiation (Hoque et al., 2018), whereas pro-fusion MFN2 and OPA1 loss impairs it, although non-fusion-related actions of MFN2 and OPA1 might also participate (Fang et al., 2016; Kasahara et al., 2013).
Interestingly, DRP1 is at least required for PSC differentiation into neural lineage cells (Wang et al., 2014). Excessive fission, meanwhile, impairs iPSC self-renewal and neuronal differentiation (Zhong et al., 2019). Mechanistically, mitochondrial fission increases cytosolic Ca2+ level and Ca2+/CaMKII activity, leading to the ubiquitination and degradation of β-catenin, which is critical for normal stem cell differentiation (Zhong et al., 2019). In addition to morphological changes, the UPC2 expression level must be attenuated to aid in the OXPHOS transition required for PSC differentiation (Zhang et al., 2011).
Mitochondrial shape and dynamics in tissue stem cells
The iPSCs and ESCs constantly divide, whereas most tissue stem cells (TSCs) reside in hypoxic niches and are quiescent. They also exhibit a high glycolytic flux, mainly thanks to the action of HIF-1 (Ishihara et al., 2009; Wakabayashi et al., 2009). Attenuated or dormant OXPHOS in quiescent TSCs may help to reduce ROS production and prevent cellular aging. Meanwhile, TSC proliferation and differentiation is stimulated by increased oxygen availability and growth factor kinase signaling, which increase OXPHOS and ROS production.
A moderate amount of ROS is required to activate redox-sensitive signaling pathways that promote cell proliferation and differentiation (Holmström and Finkel, 2014; Schieber and Chandel, 2014; Shyh-Chang and Ng, 2017). When OXPHOS was completely impaired through the loss of cardiolipin synthesizing enzyme PTPMT1, the differentiation ability of HSCs was lost, suggesting that OXPHOS is indispensable for this metabolic re-awakening (Yu et al., 2013).
The TSCs such as mesenchymal stem cells (MSCs), HSCs, and neural stem cells (NSCs) all exhibit relatively fused and elongated mitochondria, despite being predominantly glycolytic (Chen et al., 2008; Khacho et al., 2016; Simsek et al., 2010). With increased differentiation, all these stem cells become progressively dependent on OXPHOS. Accordingly, MSCs and HSCs exhibit increased MFN2 expression and further mitochondrial fusion on differentiation (Forni et al., 2016; Luchsinger et al., 2016).
Interestingly, MFN1/2 upregulation and mitochondrial fusion are seen in the early stages of adipogenesis and osteogenesis from MSCs, whereas DRP1, Fis1/2 upregulation, and fission are seen in chondrogenesis, indicating that the differential regulation of mitochondrial dynamics influences the fate of MSCs (Forni et al., 2016; Li et al., 2017). Further, when Mic60 is upregulated by resveratrol treatment that pharmacologically mimics starvation, bone marrow-derived MSCs exhibited improved differentiation and reduced senescence, indicating that starvation-induced cristae remodeling could directly influence MSC differentiation (Lv et al., 2018).
In HSCs, MFN2 action is attributed to Ca2+ buffering and reduced calcineurin activity leading to reduced DRP1 activation (Luchsinger et al., 2016). Ca2+ signaling promoting mitochondrial activity is also implicated in HSC division (Umemoto et al., 2018). A recent study showed that with each division, HSCs irreversibly remodel their mitochondrial network due to DRP1 loss (Hinge et al., 2020). Thus, after many divisions, dysfunctional mitochondria accumulate in one of the daughters cells through asymmetric division (Hinge et al., 2020).
In essence, mitochondrial dynamics maintain a form of HSC divisional memory, making HSCs less potent with more divisions. When DRP1 is genetically or pharmacologically inactivated and therefore fission-inhibited, the quiescence of HSC was still preserved, but the ability for self-renewal was lost (Hinge et al., 2020).
Further, the glycolysis-OXPHOS transition also takes place as NSCs differentiate, and MFN2 is also indispensable for iPSC-derived cortical neuron development (a summary is shown in Fig. 6B) (Fang et al., 2016; O'Brien et al., 2015).
Mitochondrial elongation and maturation are also critical in germline stem cell (GSC) differentiation, although these were reported in non-mammalian model organisms (Charmpilas and Tavernarakis, 2019). Interestingly, in GSCs, FoF1-ATP synthase was found to be indispensable for differentiation, although not through its action on OXPHOS, but instead through the maintenance of cristae structure (Teixeira et al., 2015). However, how cristae are organized in TSCs is still poorly understood, and it remains to be tested whether manipulations of cristae shape are sufficient to influence TSC multipotent state and differentiation.
Although TSC differentiation is associated with mitochondrial fusion, the exit from quiescence and activation of proliferation is associated with mitochondrial fission, as was recently shown in adult muscle stem cells (MuSCs). On activation, systemic hepatocyte growth factor-mTOR signaling fragments the mitochondria in MuSCs (Baker et al., 2022).
When mitochondrial fragmentation is promoted by OPA1 loss, MuSCs were activated prematurely via increased mitochondrial ROS and subsequent glutathione-redox signaling, which promoted nuclear gene expression that favored cell cycle progression and myogenesis (Baker et al., 2022).
Recent evidence suggests that manipulations of mitochondrial morphological proteins also influence epigenetics via DNA/histone methylation and acetylation. OPA1 haploinsufficiency impaired DNA methylation and reduced the expression of a necessary transcription factor for neural differentiation (Caglayan et al., 2020), whereas DRP1 inhibition impaired histone acetylation at the promoters of genes critical for fibroblast transdifferentiation (Wang et al., 2020).
Although the mechanism connecting mitochondrial morphological changes and epigenetics is largely elusive, altered ROS levels may be involved (Caglayan et al., 2020; Wang et al., 2020). Since methylation and acetylation are sensitive to cellular metabolic changes, altered levels of metabolites may also contribute (Su et al., 2016).
Mitochondrial fission/fusion in cancer cells
Mitochondrial morphology and dynamics regulate many cellular processes critical for cancer, such as cell survival against apoptotic signals, aerobic glycolysis, genome instability through ROS generation, metastasis, and even angiogenesis (Herkenne et al., 2020). Undifferentiated cancer cells utilize more glucose, as measured by 18F-fluorodeoxyglucose positron emission tomography, suggesting the possibility that the Warburg phenotype can be considered to be the persistence of stem cell metabolism (Riester et al., 2018). Consistent with the glycolytic profile, mitochondria fission tends to be favored in cancer cells, resulting in an overall fragmented morphology (Dai and Jiang, 2019).
Not surprisingly, in many human cancers, pro-fusion MFN1/2 or OPA1 are downregulated, whereas pro-fission DRP1 is upregulated compared with non-cancerous cells, for example, in lung cancer and breast cancer (Rehman et al., 2012; Zhao et al., 2012).
Mitochondrial fission is driven by various oncogenic signaling mechanisms such as MAPK/ERK and PI3K-Akt signaling (Trotta and Chipuk, 2017). For example, activating oncogenic Ras signaling leads to ERK1/2-induced DRP1 phosphorylation at Ser616 and activation (Kashatus et al., 2015; Serasinghe et al., 2015). In fact, when DRP1 is lost, Ras-induced tumor growth is inhibited (Kashatus et al., 2015; Nagdas et al., 2019; Serasinghe et al., 2015). Meanwhile, oncometabolites may also promote mitochondrial fission; for example, succinate can activate DRP1 via GPR91 (Ko et al., 2017; Lu et al., 2018).
Manipulations of GTPases to favor fusion, that is, MFN2 overexpression or DRP1 inhibition, generally leads to the inhibition of cancer cell proliferation (Cheng et al., 2013; Rehman et al., 2012). Interestingly, the arthritis drug leflunomide has been reported to induce mitochondrial fusion in an MFN1/2-dependent manner (Miret-Casals et al., 2018). Leflunomide achieves this by inhibiting dihydroorotate dehydrogenase (DHODH), an enzyme involved in de novo pyrimidine synthesis. The leflunomide treatment of pancreatic cancer forced mitochondrial fusion and subsequent mitophagy, resulting in overall bioenergetic compromise and cancer cell death (Yu et al., 2019).
Beyond supporting proliferation, increased DRP1-induced mitochondrial fission also favors various other features of malignancy, namely cancer cell migration and metastasis, cancer cell stemness, and genome instability (Cao et al., 2022; Peiris-Pagès et al., 2018; Zhao et al., 2012). DRP1-dependent mitochondrial fission supports lamellipodia formation by promoting the recruitment of mitochondria to lamellipodial regions (Zhao et al., 2012).
The inhibition of DRP1 also impairs cancer stem cell signaling, thereby suppressing the stem-like phenotype (Peiris-Pagès et al., 2018). Further, recent evidence showed that mitochondrial fission also promotes oncogenic DNA damage by recruiting Bcl-2-associated X protein (BAX) to OMM. This potentiates mitochondria fragmentation, limited OMM permeabilization, and non-lethal caspase and caspase-activated DNase activation, culminating in DNA damage (Cao et al., 2022). It is worth noting that both mitochondrial fission (Cai et al., 2016; Tomková et al., 2019) and fusion (Decker et al., 2020; Wang et al., 2021b) have been implicated in cancer chemoresistance, depending on the specific chemotherapeutic agents and the tissue of origin.
Meanwhile, mitochondrial fusion and increased OXPHOS activity can still support cancer cell proliferation, not as the predominant ATP source, but instead as an aspartate source (Yao et al., 2019). Simultaneously, OXPHOS is also required for DHODH activity in cancer cells (Bajzikova et al., 2019). Together, aspartate and DHODH can support de novo pyrimidine synthesis, which can be limiting for proliferation (vander Heiden and DeBerardinis, 2017).
Cristae shape in cancer
Cristae shape regulation is important in cancer cells for the regulation of respiratory efficiency, ROS production, and perhaps most importantly, avoiding apoptosis (Cogliati et al., 2013; Frezza et al., 2006). Cristae shape in cancer cells seems to be variable depending on the cancer type. In ovarian cancer, mitochondria possessed narrow cristae with a reduced CJ diameter, with elevated OPA1 and PHB2 levels (Signorile et al., 2019).
Meanwhile, in gliomas, mitochondria were found to be swollen, and cristae were disorganized (Arismendi-Morillo, 2011). In the latter, the aberrant cristae shape could be attributed to the fact that gliomas are highly glycolytic, and mitochondrial OXPHOS is likely severely impaired (Oudard et al., 1997).
Apart from its pro-fusion activity, OPA1 is also particularly important for the maintenance of cristae integrity and sequestering cytochrome c (Frezza et al., 2006; Scorrano et al., 2002). A reduced OPA1 level is associated with increased apoptosis (Chen et al., 2009; Olichon et al., 2006). When cells go through apoptosis, BH3-only BCL-2 family proteins such as BID and BIM induce OPA1 disassembly and the widening of CJ, leading to the release of cytochrome c into the IMS (Frezza et al., 2006; Yamaguchi et al., 2008).
This is followed by OMM perforation by BAX/BAK. Cytochrome c is subsequently released into the cytoplasm to be assembled into apoptosomes, setting off further downstream cascades that execute apoptosis. Crucially, BAX/BAK aggregation triggers OMA1 activation, which cleaves OPA1, further promoting cristae disruption (Jiang et al., 2014). OPA1 is also connected to the tumor suppressor protein p53. P53 activates BAX/BAK assembly on the OMM while binding to PHB to release OMA1 (Fusaro et al., 2003; Kong et al., 2014).
OMA1 also participates in the regulation of IMM and OMM contact by physically associating with Mic60 (Viana et al., 2021). Activated OMA1 can cleave Mic19, thereby disrupting the Sam50-Mic19-Mic60 axis that mediates IMM and OMM contact (Tang et al., 2019). This disruption also contributes to pro-apoptotic cristae remodeling.
Such pro-apoptotic OPA1 processing by OMA1 is inhibited by IF1, potentially by IF1 protecting intracellular ATP levels (Faccenda et al., 2017). In addition, IF1 can also induce a metabolic shift to aerobic glycolysis through the direct inhibition of FoF1-ATP synthase (Sánchez-Cenizo et al., 2010). In cancer cells, both actions could be highly advantageous; indeed, IF1 is highly overexpressed in a wide variety of cancer cell lines, including colon, lung, breast, and ovarian cancer cells (Sánchez-Aragó et al., 2013; Sánchez-Cenizo et al., 2010).
However, IF1 overexpression does not always promote cancer phenotype. In a cohort of colorectal carcinoma cells, IF1 overexpression was associated with less aggressive behavior with lower in vivo metastatic potential (González-Llorente et al., 2019). Therefore, whether IF1 functions as an oncogene or a tumor suppressor may be context-dependent.
Cristae shape might serve a particularly important role in cancer cell survival in hypoxia. Hypoxia induces chemotherapy resistance, and at least part of the mechanism seems to involve HIF-1-dependent MFN1-mediated mitochondrial fusion and enlargement (Chiche et al., 2010). Although this atypical enlargement resembles mitochondrial swelling, they nonetheless preserve their ΔΨm and were found to resist apoptosis (Chiche et al., 2010).
The IMM is remodeled in these hypoxic cells as well, altering cristae shape and obliterating the IMS, thereby potentially relocalizing cytochrome c and preventing their leakage into the cytoplasm to initiate apoptosis (Chiche et al., 2010).
Further, apart from apoptosis resistance, hypoxia-induced cristae remodeling may also coordinate metabolic reprogramming in cancer. Hypoxic stress in a glycolytic HCC cell line of HepG2 cells led to cristae widening and blunted edges due to an ∼20% downregulation of Mic60 and reduced fraction of FoF1-ATP synthase dimerization (Plecitá-Hlavatá et al., 2016). This hypoxia-induced crista widening likely helps in the attenuation of OXPHOS, enhancing glycolysis. An interesting recent study showed that HIF-1 activation influences mitochondrial dynamics and activity via the hairy/enhancer-of-split related with YRPW motif protein 1 (HEY1)-phosphatase and tensin homolog-induced kinase 1 (PINK1) axis (Kung-Chun Chiu et al., 2019).
HEY1 is transcriptionally activated by HIF-1 and it, in turn, transcriptionally represses PINK1, a critical regulator of mitochondrial biogenesis and OXPHOS activity (Kung-Chun Chiu et al., 2019). This leads to reduced hypoxia-induced oxidative stress and favors HCC growth (Kung-Chun Chiu et al., 2019). This HEY1 elevation and PINK1 attenuation are also associated with reduced, malformed mitochondrial cristae compared with normoxia, as the normal cristae shape depends on PINK1 expression (Kung-Chun Chiu et al., 2019).
Another recent finding was that hypoxia activates OMA1, leading to OPA1 cleavage and the suppression of OXPHOS (Wu et al., 2021). Simultaneously, OMA1 increases mitochondrial ROS to stabilize HIF-1α, thereby enhancing glycolysis (Wu et al., 2021). The loss of OMA1 suppressed colorectal cancer development, suggesting its essential role in carcinogenesis (Wu et al., 2021). In summary, the cristae shape coordinates both cell survival and energy metabolism in hypoxia, contributing to cancer growth.
Conclusion
Mitochondria ensure a plethora of functions in cells. Apart from producing ATP, they serve as redox signaling hubs, Ca2+ signaling modulatory and buffering domains, sites of heme biosynthesis, regulators of apoptosis, sources of epigenetic signals, and many more. The complexity of their function is reflected in their elaborate morphology and frequent membrane interactions with other organelles.
Recent developments in fluorescence and electron microscopy techniques have not only allowed a fine description of mitochondrial morphology in different cell types but also addressed its dynamic changes in response to various nutrient and stress signals. Thus, a growing number of studies document that mitochondrial structure and metabolic activity are closely linked and mutually regulated.
The plasticity of mitochondrial architecture is particularly vital for metabolic reprogramming. Immune, stem, and tumor cells, which reprogram their metabolism as they change cell identity, exhibit striking similarities in the coordination of mitochondrial morphology with metabolic pathways. Many signals that induce a shift to glycolytic metabolism, such as nutrient sensing pathways and redox signaling pathways, also lead to increased mitochondrial fission.
Generally, fused mitochondria with tight cristae maximize the OXPHOS efficiency of the cell, which supports non-activated, differentiated cell phenotypes (such as non-activated immune cells or differentiated somatic cells). On antigen activation or somatic reprogramming, DRP1-induced mitochondrial fission (as well as cristae loosening in some contexts) attenuates OXPHOS, favoring enhanced glycolytic flux.
When mitochondrial morphological proteins, particularly OPA1, MFN1/2, DRP1, and MICOS complex proteins, are disrupted, cells fail to adopt the appropriate mitochondrial shapes required for metabolic reprogramming. However, although many general unifying principles can be observed, their validity is not absolute, and the mechanistic links thus need to be further explored.
For example, mitochondrial fusion or fission does not always correlate with the predominance of OXPHOS or glycolysis, respectively, such as in the transition from naive to primary PSCs and glycolytic TSCs (Bahat et al., 2018; Chen et al., 2008; Khacho et al., 2016; Simsek et al., 2010; Ware et al., 2014; Zhou et al., 2012).
Cristae architecture and mainly its dynamics are new additions to the picture, and their role in metabolic programming is largely yet to be studied. Gaining new insights in this area will closely depend on the further development of groundbreaking super-resolution microscopy techniques, the resolution of which will have to be below 20 nm to enable the live-cell imaging of cristae.
In summary, the field of mitochondrial morphology and its relationship to metabolic reprogramming holds promising potential, and a deeper understanding of the molecular mechanisms will not only further deepen our understanding of energy metabolism, but may also contribute to an improved therapeutic manipulation of cell viability, differentiation, proliferation, and identity in many different cell types.
Footnotes
Acknowledgments
The authors gratefully acknowledge the Imaging Methods Core Facility at BIOCEV (Faculty of Sciences, Charles University) supported by the National Infrastructure for Biological and Medical Imaging (Czech-BioImaging–LM2015062). All images were created with
Authors' Contributions
Conception: A.D. Data collection and analysis/interpretation: I.K. Writing the original draft: I.K., A.D., B.B., and P.J. Preparation of figures and tables: I.K., B.B. Critical revision of the article: A.D., P.J. All authors have read and agreed to the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The project was funded by a grant from the Grant Agency of the Czech Republic (No. GA22-02203S to Andrea Dlasková and No. 21-01205S to Petr Ježek).