
Abstract
Significance:
Excess oxidative stress and neuroinflammation are risk factors in the onset and progression of Alzheimer's disease (AD) and its association with amyloid-β plaque accumulation. Oxidative stress impairs acetylcholine (ACH) and N-methyl-
Recent Advances:
Some clinical trials have shown a benefit of monotherapy with vitamin D (VD), whose deficiency is linked to AD or with
Future Directions:
Clinical trials are needed to determine whether safe low-cost dietary supplements, using combined VD+LC, have the potential to alleviate elevated AChE, oxidative stress, and inflammation levels, thereby halting the onset of AD.
Goal of Review:
The goal of this review is to highlight the pathological hallmarks and current Food and Drug Administration-approved treatments for AD, and discuss the potential therapeutic effect that cosupplementation with VD+LC could manifest by increasing GSH levels in patients. Antioxid. Redox Signal. 40, 663–678.
Introduction
Alzheimer's disease (AD), a progressive brain disorder, characterized by acquired and progressive cognitive decline, disability, and increased mortality, is one of the most common causes of dementia. AD is a major public health issue worldwide, seen more frequently in people over the age of 65 and in African Americans (AA) (Narayanan et al., 2020; O'Brien and Wong, 2011; Tang et al., 2001). As the size of the aging population increases, the number of people living with dementia is expected to more than triple to >120 million by the year 2050.
While the exact cause of AD onset is still unclear, the prevailing hypothesis is that the increase in amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain leads to synaptic dysfunction, and thus the onset of AD (Dias et al., 2014; Nakamura et al., 2018; Selkoe and Hardy, 2016). Given the enormous potential social, economic, and health care impact of AD, novel approaches are needed in the development of effective therapeutics and prevention of AD.
Oxidative stress-induced inflammation is linked to many chronic diseases, including AD (Galasko and Montine, 2010; Gella and Durany, 2009). Glutathione (GSH), the most abundant antioxidant in the human body, plays a pivotal role in protecting against oxidative stress. Decreased GSH levels have been shown to correlate with cognitive decline (Mandal et al., 2015; Mandal et al., 2012), and preclinical studies have reported that GSH can protect against neuronal damage by different mechanisms, including activation of nerve growth factor, glial cell-derived neurotrophic factor, nitric oxide synthase, and choline acetyl transferase (Haddad et al., 2021; Iskusnykh et al., 2022; Nakamura et al., 2021; Zhang et al., 2021b).
Systematic reviews conclude that a low serum concentration of vitamin D (VD) is linked to an increased risk of dementia and AD (Annweiler et al., 2013a; Annweiler et al., 2013b; Balion et al., 2012). However, clinical trials involving VD supplementation have demonstrated limited success in protection against dementia and AD onset (Bischoff-Ferrari et al., 2020; Gall and Szekely, 2021; Lewis and Laing, 2015; Liu et al., 2022). Animal studies have shown that combined ingestion of VD and
The goal of this review is to highlight the pathological hallmarks and current treatment options for AD, and then to discuss the potential therapeutic effect that cosupplementation with VD+LC can exert by increasing GSH levels in these patients, resulting in decreased oxidative stress and inflammation.
Sporadic Versus Familial Forms
AD is divided into two forms, familial and sporadic, with most cases being the latter (Piaceri et al., 2013). The sporadic form occurs mostly in people over the age of 65 and is largely influenced by the apolipoprotein E (APOE) gene (Piaceri et al., 2013; Raulin et al., 2022). The APOE gene has several functions in the brain, including cholesterol transport and efflux of Aβ. The gene exists in three forms, APOE2, APOE3, and APOE4, with APOE3 being the most common. Each of these isoforms has a different amino acid sequence, which influences how it interacts with proteins and peptides. The differences among the isoforms have been explained in detail by Butterfield and Mattson (2020).
Inheritance of the APOE4 allele greatly enhances the risk of late onset AD since it results in increased deposition of Aβ in the brain. On the contrary, the APOE2 allele serves a protective role in the development of AD (Butterfield and Mattson, 2020; Raulin et al., 2022;). The familial form of AD exhibits an autosomal dominant inheritance pattern and is associated with early disease onset, with people commonly becoming symptomatic ∼30–60 years of age (Kriebs, 2023). To date, mutations in three genes, amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), have been associated with the familial form of AD (Rujeedawa et al., 2021).
Pathophysiology of AD
The pathophysiology of AD is complex and still not completely understood. Despite this, it is widely accepted that certain hallmark characteristics play critical roles in the development of AD. Oxidative stress and inflammation have received much attention for their contribution to AD progression. Accumulation of two proteins, Aβ and Tau, is a classic hallmark in brains of AD patients. While Aβ accumulates extracellularly, forming plaques (Bloom, 2014), hyperphosphorylated Tau accumulates intracellularly, forming twisted NFT.
The accumulation of these two proteins results in direct damage and destruction of the synapses that mediate memory and cognition, thus making them drivers of the well-known symptoms of AD, including memory loss, behavioral problems, and cognitive impairment (Bloom, 2014). Decreased levels of acetylcholine (ACH), a neurotransmitter important in brain function, are also seen in AD patients. In addition, upregulation of the N-methyl-
APP and Aβ
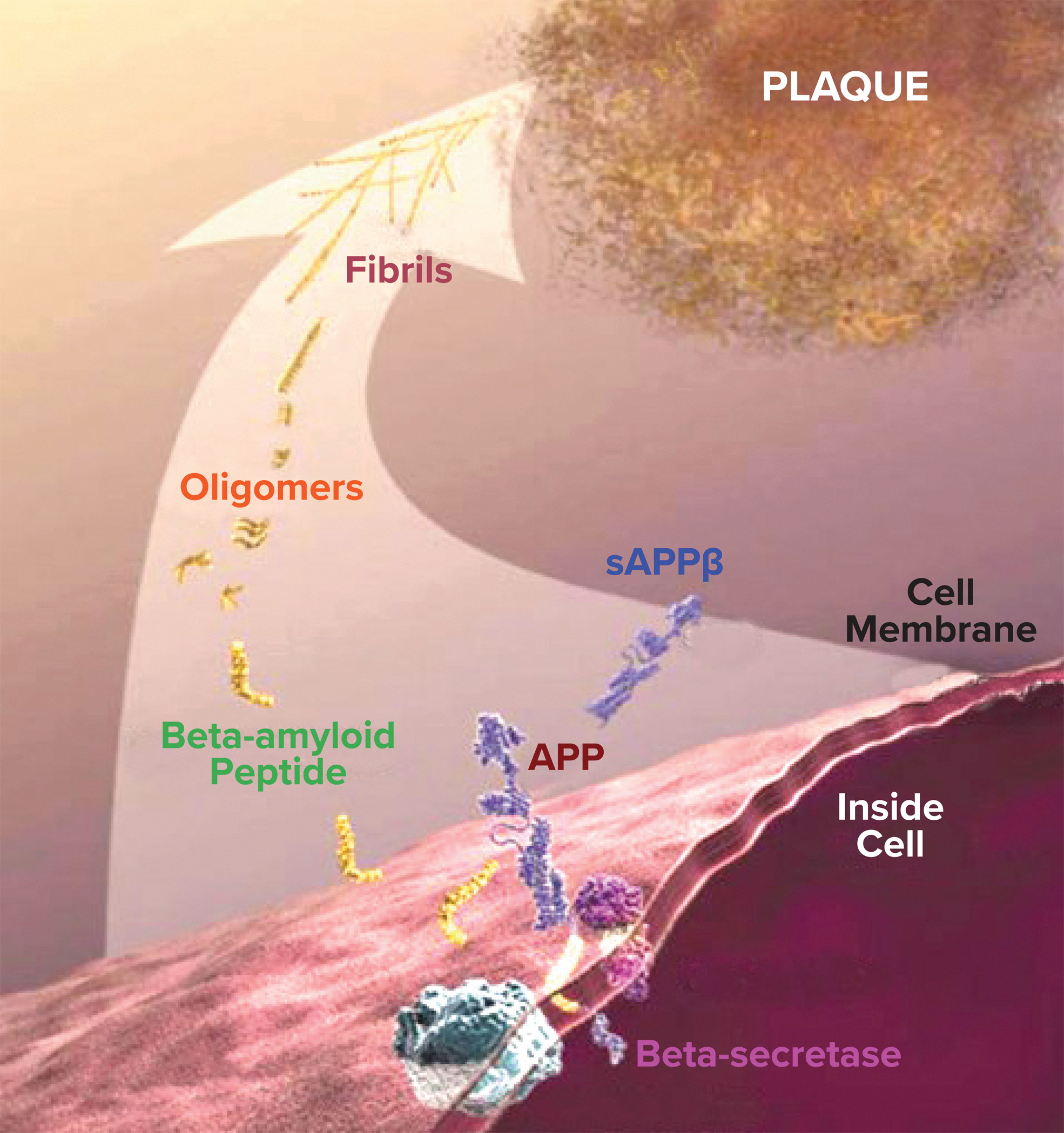
APP is a transmembrane protein produced in large quantities by neurons in the brain. APP is broken down by two enzymes, β-secretase, also known as β-site APP cleaving enzyme 1 (BACE1), and γ-secretase, a multiprotein complex composed of PSEN1 and PSEN2 and three other subunits. Through complex mechanisms, these secretases catalyze the generation of Aβ peptides, with Aβ peptides 1–42 and 1–40 being the most common. Another APP metabolism pathway is catalyzed by metalloprotease 10 (ADAM10), an α-secretase. ADAM10 cleaves APP to generate neuroprotective soluble APPα, which precludes the generation of Aβ.
Aβ exists in different forms, including aggregated, fibrillary, and oligomeric (Caughey and Lansbury, 2003; Dahlgren et al., 2002). The oligomeric form, considered the most toxic form, is strongly associated with oxidative stress (Dahlgren et al., 2002; Drake et al., 2003; Fawzi et al., 2007; Walsh et al., 2001). Low levels of Aβ are required for physiological function (Pearson and Peers, 2006). However, when present in normal concentrations, Aβ does not induce oxidative stress, but when the amount of Aβ increases significantly, as it does in AD, it evolves into a pro-oxidant that enhances oxidative stress, leading to disease progression (Rani et al., 2017). In the brains of AD patients, abnormal levels of Aβ clump together to form plaques that disrupt normal cell function. The pathophysiology of Aβ plaque formation is shown in Figure 1.
Oxidative stress
Oxidative stress was first described by Sies (1985) and is defined as an imbalance between the production of reactive oxygen species (ROS) and antioxidant defense. Classic examples of ROS include superoxide and hydrogen peroxide (Galasko and Montine, 2010). Abnormally high levels of ROS can be detrimental by damaging biomolecules. In addition, increased levels of ROS can trigger chronic inflammation by upregulating inflammatory pathways (Lushchak and Storey, 2021; Poljsak et al., 2013; Shields et al., 2021).
Oxidative stress-induced brain damage is associated with aging and is usually involved in the development of AD pathology. Enhanced oxidative stress has been observed in the brains (Alawode et al., 2021; Hansson, 2021) and peripheral tissues and cells (e.g., blood cells) of AD patients (Burroughs Pena et al., 2019; Gordon et al., 2017). Many studies have shown both overproduction of oxidant compounds and impairment of antioxidant systems, as well as increased oxidative damage, in several brain regions of patients with both mild cognitive impairment (MCI) and AD (Luca et al., 2015; Sliwinska et al., 2016; Torres et al., 2011).
The fact that high levels of peripheral markers of oxidative–inflammatory stress were detected in individuals with MCI lead us to believe that oxidative stress and inflammation are early events in the pathogenesis of AD, and that they precede Aβ deposits and the onset of AD symptoms, which has been shown in experimental models of AD (Ferretti et al., 2012; Wright et al., 2013). Oxidative stress has also been reported to stimulate β-secretase and γ-secretase, leading to increased levels of Aβ deposits (Tamagno et al., 2008).
Inflammation
Inflammation, a natural reaction that occurs in the body in response to injurious stimuli, is the beginning of the process used by the body to restore tissue integrity and homeostasis. It is now widely accepted that chronic inflammation contributes to the pathogenesis of neurodegenerative disorders, including AD (Di Benedetto et al., 2022; Minghetti, 2005). The initial inflammatory reaction serves a protective role by enhancing tissue repair and clearance of toxic stimuli. However, as the diseased state progresses, chronic inflammation causes harmful effects, leading to neurodegeneration (Di Benedetto et al., 2022).
In the brain, two major immune cells regulate the immune response, microglia and astrocytes (Kwon and Koh, 2020). Studies have shown that microglia and astrocytes become activated around senile plaques, present in AD brains, and in response to oxidative stress, leading to a neuroinflammatory response that eventually induces neurodegeneration (Bouvier et al., 2016; Di Benedetto et al., 2022; Perez-Nievas and Serrano-Pozo, 2018).
Microglia express pattern recognition receptors on their surfaces. These receptors can become activated by binding different species of Aβ, including the toxic oligomeric form. Once Aβ stimulates the microglia, proinflammatory cytokines, such as interleukin (IL)-1β, are released from the microglia and induce their inflammatory properties, leading to neurotoxicity and neurodegeneration (Di Benedetto et al., 2022; Hickman et al., 2018). Overactivated microglia have also shown to increase concentrations of Aβ and Tau, which in turn further activate microglia (Guan et al., 2022).
Astrocytes are cells resident in the central nervous system (CNS) that participate in a wide array of cellular processes, including inflammation (Vasile et al., 2017). In the presence of Aβ, astrocytes are activated and release proinflammatory cytokines, leading to neuroinflammation. In addition, since astrocytes react to proinflammatory cytokines by increasing Aβ production, the presence of inflammation can lead to increased levels of Aβ in the brain (Gonzalez-Reyes et al., 2017). Therefore, finding ways to decrease oxidative stress and inflammation may be potentially therapeutic for AD patients.
Tau protein
Elevated concentrations of Tau, a protein found predominantly in neurons that plays an important role in the stabilization of microtubules, are also seen in AD patients (Qu et al., 2021). Tau hyperphosphorylation leads to the dissociation of Tau from microtubules and the formation of clumps of phosphorylated Tau (pTau) known as NFT, which cause neurofibrillary degeneration (Zhang et al., 2021a). Aβ can affect Tau aggregation and the formation of Tau oligomers, an intermediate form of Tau that forms before NFT and induces toxicity by potentiating neuronal damage (Shafiei et al., 2017).
The presence of ROS has been shown to increase the capacity of Aβ aggregation and oligomerization, thus facilitating neuronal damage (Cheignon et al., 2018). The interaction between Aβ and Tau has also been shown to facilitate neuronal loss and synaptic damage, which is associated with cognitive decline and changes in behavior in AD patients (von Bernhardi and Eugenin, 2012). Two clinical studies carried out in 2017 and 2018, involving healthy older individuals, showed that those with altered levels of Aβ and Tau had greater cognitive decline (Clark et al., 2018; Schindler et al., 2017).
A recent study comparing the amount of plasma pTau in cognitively impaired and cognitively normal patients reported a continuously escalating trend for those who were more cognitively impaired (Xiao et al., 2021). Postmortem studies have also revealed that Tau pathology likely has a higher association with the magnitude of cognitive impairment than Aβ does (Murray et al., 2015; Nelson et al., 2012; Vogels et al., 2020). In addition, compounds that decrease the phosphorylation status of Tau have been recognized as potential therapeutic agents for AD patients (Shin et al., 2021).
Neurofilament light protein
Neurofilaments are intermediate filaments found in neurons that play a role in the conduction process along the neuron's axon. Neurofilament light (NFL) protein is a protein that comprises neurofilaments (Yamasaki et al., 1991); elevated levels of NFL are often seen in neurodegenerative diseases such as AD (Alagaratnam et al., 2021; Khalil et al., 2018; Narayanan et al., 2021). Since NFL is only expressed in the cytoplasm of neurons and reaches abnormal levels when axons are damaged, such as those observed in neurodegenerative diseases, it has recently gained significant attention as a potential biomarker for detecting neurological disorders (Khalil et al., 2018).
Pathological processes that result in axonal damage cause the release of neurofilament proteins into the cerebral spinal fluid (CSF) and peripheral blood. Therefore, increased levels of neurofilament proteins, such as NFL, can serve as indicators for axonal damage and progression of neurological disease (Khalil et al., 2018). Studies that examined the plasma concentration of NFL in two different cohorts, one from King's College in London (n = 805) and other from the Swedish BioFINDER study (n = 1464), concluded that plasma NFL was helpful in detecting cognitive impairment in these individuals (Ashton et al., 2021).
Higher levels of NFL have also been associated with an increased likelihood of dementia and poorer global cognition and attention/processing speed (Gonzales et al., 2021). In essence, elevated levels of NFL in CSF and plasma seem to be appropriate indicators of neurodegeneration and progression of neurodegenerative disease due to axonal damage with the subsequent release of NFL into the CSF and blood.
ACH levels
ACH, a neurotransmitter released by cholinergic neurons, is involved in cognition, learning, memory, and the suppression of inflammation (Chen et al., 2022). It is synthesized by choline acetyltransferase and stored in presynaptic vesicles at the terminal end of neurons. Propagation of an action potential along the entire length of the axon leads to the fusion of the vesicles with the presynaptic membrane and the subsequent release of ACH from these presynaptic vesicles. Once in the synapse, ACH binds to postsynaptic ACH receptors, causing depolarization (Sam and Bordoni, 2022). ACH esterase (AChE), an enzyme that breaks down ACH to choline and acetate in the synapse, is the target of several drugs currently used to treat AD (Blotnick-Rubin and Anglister, 2018; Sam and Bordoni, 2022).
It is well known that a defect in cholinergic transmission contributes to the memory impairment seen in AD patients (Chen et al., 2022; Marucci et al., 2021). Studies have shown the possibility that Aβ and oxidative stress contribute to defective cholinergic transmission. Melo et al. (2003) found that Aβ-induced oxidative stress increased AChE levels, leading to decreased ACH signaling. Oxidative stress has also been shown to decrease recycling of choline from the synapse (Wong et al., 2020) and the number of ACH receptors (Arora and Hess, 1985), both of which lead to ACH deficiency.
NMDA receptor
The NMDA receptor, a glutamate receptor and ion channel found in neurons, is the primary excitatory neurotransmitter receptor in the human brain. Activation of the receptor depends on the binding of glycine and glutamate and AMPA receptor-mediated depolarization of the postsynaptic membrane, which relieves the voltage-dependent channel blocked by Mg2+ (Bledsoe et al., 2017). Activation and opening of the receptor channel thus allow the flow of K+, Na+, and Ca2+ ions, and the influx of Ca2+ that triggers intracellular signaling pathways (Danysz and Parsons, 2003; Johnson and Kotermanski, 2006). The NMDA receptor is thought to be very important for controlling synaptic plasticity, and mediating learning and memory functions (Li and Tsien, 2009).
NMDA receptor dysfunction is believed to be involved in a wide variety of CNS conditions, including many that are neurodegenerative (Zhou and Sheng, 2013). It has been observed in humans that a more severe degree of NMDA receptor dysfunction is present in the AD brain than in those of age-matched controls (Ulas and Cotman, 1997). Multiple studies from different laboratories have also shown an association between NMDA receptor dysfunction and increasing age (Gonzales et al., 1991; Magnusson, 1998; Newcomer et al., 2000; Saransaari and Oja, 1995).
As stated before, oxidative stress increases with age; in theory, as oxidative stress increases, NMDA receptor dysfunction is likely to increase (Fig. 2). This relationship between oxidative stress and NMDA receptor dysfunction has been reported in the literature (Betzen et al., 2009; Hasam-Henderson et al., 2018; Kamat et al., 2016; Ma et al., 2020; Reyes et al., 2012).
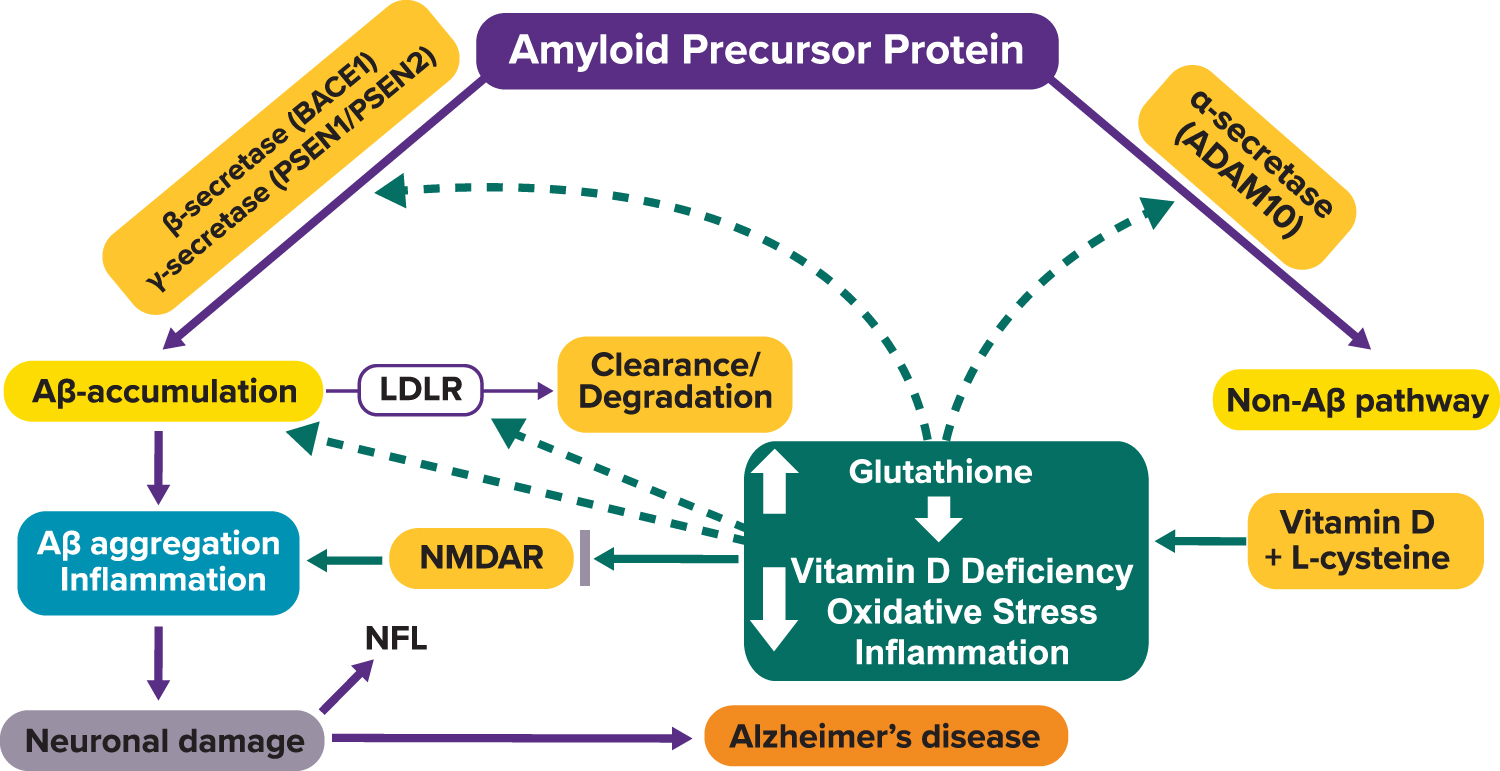
In individuals at risk of developing AD, amyloidopathy and oxidative stress can interact to increase the burden of NMDA receptor dysfunction. The first neurodegenerative stage seems to entail the deposition of low concentrations of amyloid in the brain and the interaction of amyloid with certain NMDA receptors in a manner that increases the sensitivity of these receptors to glutamate. Excessive stimulation of NMDA receptors by glutamate results in abnormally high amounts of intracellular calcium. This intense increase in intracellular calcium causes neurotoxicity, leading to neuronal cell death (Wang and Reddy, 2017). As these neurons degenerate, amyloid plaques may form and incorporate portions of the degenerating neurons and other neural and glial processes into the immediate environment (Newcomer et al., 2000).
The Connection Between GSH, LC, and VD Levels and AD
GSH, the most abundant and main antioxidant in the blood, defends against oxidative stress (Aw et al., 1991; Badaloo et al., 2002; Bizzozero et al., 2007; Curtis et al., 2012; Dalle-Donne et al., 2003; De Mattia et al., 1998; Grimsrud et al., 2008; Nkhoma et al., 2009). LC is a precursor of GSH, and cosupplementation with LC+VD has been shown to decrease oxidative stress by increasing GSH status (Jain et al., 2021; Jenkins et al., 2021). Numerous studies have shown that VD deficiency, which is associated with increased inflammation, is common in patients with AD (Di Somma et al., 2017; Littlejohns et al., 2014; Slinin et al., 2010). This section will focus on how each of these factors contributes to the pathological process of AD.
GSH and LC levels
GSH is formed from LC, glycine, and glutamate by the enzymatic actions of glutamate-cysteine ligase and GSH synthetase (Franklin et al., 2009; Richie et al., 2015). LC is the rate-limiting factor in GSH synthesis (Vina et al., 1989; Yin et al., 2016). Lower GSH levels can occur because LC is not available in the food consumed or as a result of consumption of an energy-rich diet, which increases ROS and oxidative stress, impaired reduction of oxidized glutathione disulfide (GSSG) to GSH, and/or increased utilization of GSH relative to its biosynthesis.
A proper ratio of GSH (reduced form) to GSSG (oxidized/disulfide form) is vital for healthy cells. When this ratio is reduced (higher GSSG and lower GSH), it is associated with negative outcomes, such as increased oxidative stress and many diseased states, including AD (Haddad et al., 2021; Zhou et al., 2014). Brains of patients with AD and MCI have consistently been shown to express decreased concentrations of antioxidant enzymes, making them more susceptible to the toxic effects of Aβ.
One study that investigated blood GSH levels of 30 AD patients, 35 vascular dementia patients, and 40 control patients found significantly lower GSH levels in both the AD and vascular dementia groups compared with the control group (Krishnan and Rani, 2014). Another study reported elevated levels of IL-6, TNF-α, and reduced levels of GSH reductase, the enzyme responsible for producing reduced GSH, and total antioxidant capacity in the blood of AD patients (n = 21) compared with that of controls (n = 10) (Gubandru et al., 2013). Puertas et al. (2012) reported similar results, observing lower GSH levels in AD patients compared with those in the control group. A linear correlation between increased GSSG levels and decreased cognitive status in AD patients has also been reported (Lloret et al., 2009).
Supplementation with GSH and LC has been shown to successfully improve GSH status in blood and tissues, including the brain, while lowering inflammation and oxidative stress in human and animal studies (Aw et al., 1991; Badaloo et al., 2002; Clemente Plaza et al., 2018; De Mattia et al., 1998; Jain et al., 2009; McPherson and Hardy, 2011; Richie et al., 2015; Sinha-Hikim et al., 2013; Vina et al., 1989; Yin et al., 2016). LC supplementation decreases levels of oxidative stress and Aβ toxicity in the brain (Pocernich et al., 2011; Yan and Greene, 1998). N-acetyl cysteine (NAC) decreased Aβ levels in mice that overexpressed the APP gene responsible for Aβ formation (Tucker et al., 2006).
GSH supplementation in an APP/PS1 mouse model of AD completely reversed the development of impaired spatial memory and long-term cognitive/cued-recall, Aβ deposition, and oxidative stress indicators (Ramassamy et al., 2000). A controlled trial in which NAC was administered to late-stage AD patients showed that the NAC-treated group had improved memory (Adair et al., 2001). N-acetyl-
Effect of Cysteine Supplementation on Biomarkers of Dementia and Alzheimer's Disease
Aβ, amyloid-β; AD, Alzheimer's disease; GSH, glutathione; MMSE, Mini-Mental State Examination; NAC, N-acetyl cysteine.
VD levels
VD is a fat-soluble vitamin that humans can obtain from diet or exposure to sunlight. Upon entering the body, VD is hydroxylated twice, first in the liver and then in the kidney, to produce calcitriol, the active form of VD (Warner et al., 2021). VD supplementation increases GSH, and reduces oxidative stress by upregulating GSH reductase and GSH peroxidase (Ansari et al., 2020; Jain and Micinski, 2013; Uttara et al., 2009). VD deficiency has been identified as a potential risk factor for neurocognitive impairment, dementia, and brain health (Gezen-Ak et al., 2014; Spiro and Buttriss, 2014).
A study done in 2020, in which 350 subjects aged ≥65 were enrolled into 3 groups, healthy subjects (n = 103), MCI subjects (n = 109), and AD subjects (n = 138), showed that serum VD levels were significantly decreased in the MCI and AD groups compared with the healthy subject group. The authors also observed that subjects who progressed from MCI to AD had lower VD levels than those who continued to experience only MCI (Mavraki et al., 2020). A meta-analysis performed in 2017 reported that individuals with low VD levels, <25 nM, were at increased risk of dementia (Sommer et al., 2017).
A large prospective population-based study of 405 subjects concluded that severe VD deficiency, especially in the elderly, was independently associated with a future risk of MCI and dementia (Moon et al., 2015). Another study done in mice reported that the VD receptor plays an important role in reducing Aβ plaque buildup, and that the VD receptor may be a potential target in the prevention and treatment of AD (Durk et al., 2014). VD has been shown to increase the clearance of Aβ by stimulating macrophages (Masoumi et al., 2009; Mizwicki et al., 2012), an important observation since aggregation and decreased clearance of Aβ from the brain play a prominent role in AD (Durk et al., 2014).
In addition, VD deficiency increases pTau (Sommer et al., 2017), and promotes synaptic dystrophy and neuronal loss in the brains of APP/PS1 mice. We have also shown that improved GSH status upregulates the genes of VD metabolism and the VD receptor (Jain et al., 2018).
As stated, increased inflammation is common in AD patients and is thought to contribute to its development. Studies have shown increased inflammation in VD-deficient individuals, and that VD exerts anti-inflammatory properties, suggesting an association between these two variables (Calton et al., 2015; Mellenthin et al., 2014; Zhou and Hypponen, 2023). Zhou and Hypponen (2023) recently investigated the causality and direction of the association between VD status and inflammation, measured by C-reactive protein (CRP) levels, and concluded that it was likely that the increase in CRP levels was due to insufficient VD levels.
In a cross-sectional study consisting of 5870 adults, lower VD levels were associated with increased CRP and white blood cell levels (de Oliveira et al., 2017). Another study consisting of 67 individuals found that VD supplementation after 6 months showed an anti-inflammatory effect by decreasing CRP levels and stabilizing IL-10, an anti-inflammatory cytokine (Krajewska et al., 2022).
As shown by these data, and the fact that as many as 70%–90% of AD patients are VD deficient (Di Somma et al., 2017), it can be hypothesized that VD has the potential to be a beneficial therapeutic agent that helps alleviate the symptoms of AD. Table 2 summarizes various studies in the literature that have shown VD supplementation to have a positive effect on enhancing memory and decreasing AD biomarkers in both humans and animals.
Effect of Vitamin D Supplementation on Biomarkers of Dementia and Alzheimer's Disease
GSH, glutathione; IL, interleukin; NGF, nerve growth factor; P-gp, P-glycoprotein; TNF-α, tumor necrosis factor-α; VD, vitamin D.
Treatment for AD
Due to the detrimental impact AD has on society, much research has been focused on finding therapeutic agents that can relieve the effects of AD. The Food and Drug Administration (FDA) has approved six different drugs to help treat AD. However, a cure for AD is still needed. While antioxidant therapy has shown some promising effects in preclinical studies, it has had poor success in clinical studies. This section will discuss the current treatment options available for AD. In addition, we will justify why VD+LC supplementation could be beneficial in the treatment of AD and which population we hypothesize would benefit the most from this treatment.
Current FDA-approved medications
Currently, six drugs, donepezil, rivastigmine, galantamine, memantine, aducanumab, and lecanemab, have been approved by the FDA for the management of AD (Table 3) (Birks and Harvey, 2018; Dunn et al., 2021; Grossberg, 2003; Lo and Grossberg, 2011; Matsunaga et al., 2018; Patel and Gupta, 2022). Donepezil, rivastigmine, and galantamine all bind reversibly to AChE, thus inhibiting its function.
Current Food and Drug Administration-Approved Medications for Treatment of Alzheimer's
NMDA, N-methyl-
Consequently, ACH accumulates in the synaptic cleft, leading to increased stimulation of post synaptic receptors (Colovic et al., 2013). Memantine differs from donepezil, rivastigmine, and galantamine, in that it exerts its therapeutic effects by slowing the progression of the neurotoxicity thought to be associated with AD rather than increasing ACH levels in the synapse (Rogawski and Wenk, 2003). Memantine acts as a noncompetitive NMDA receptor antagonist, which decreases the excessive excitation of NMDA receptors by glutamate, thus leading to decreased neuronal cell death (Wang and Reddy, 2017).
Aducanumab and lecanemab are the two drugs most recently approved by the FDA for treatment of AD subjects (Cummings and Salloway, 2022). While both drugs are theoretically monoclonal antibodies against Aβ, these drugs function differently due to their different binding targets. Aducanumab, approved by the FDA in 2022, exerts its effects by crossing the blood–brain barrier and selectively binding to Aβ plaques, resulting in reduced plaque concentration (Sevigny et al., 2016). Lecanemab, approved by the FDA in 2023 binds with high affinity to Aβ protofibrils instead of Aβ plaques (Verger et al., 2023).
Justification for GSH optimization to lower the risk of AD by using combined VD+LC supplementation despite lack of antioxidant success in clinical trials
Accumulating evidence suggests that increased levels of oxidative stress contribute greatly to AD; as such, a sound rationale exists for the therapeutic value of increased levels of antioxidants due to their ability to decrease oxidative stress. We hypothesize that combined supplementation with VD+LC will increase endogenous GSH levels, resulting in decreased oxidative stress, and therefore downregulation of many of the characteristic pathological hallmarks of AD responsible for its harmful effects (Fig. 3). We also believe that VD supplementation will be beneficial in protecting against AD pathogenesis in patients who are VD deficient, as VD deficiency is linked to increased inflammation, neuronal damage, and Aβ levels. In addition, optimizing GSH status has the potential to downregulate AChE, resulting in increased ACH levels (Fig. 3).
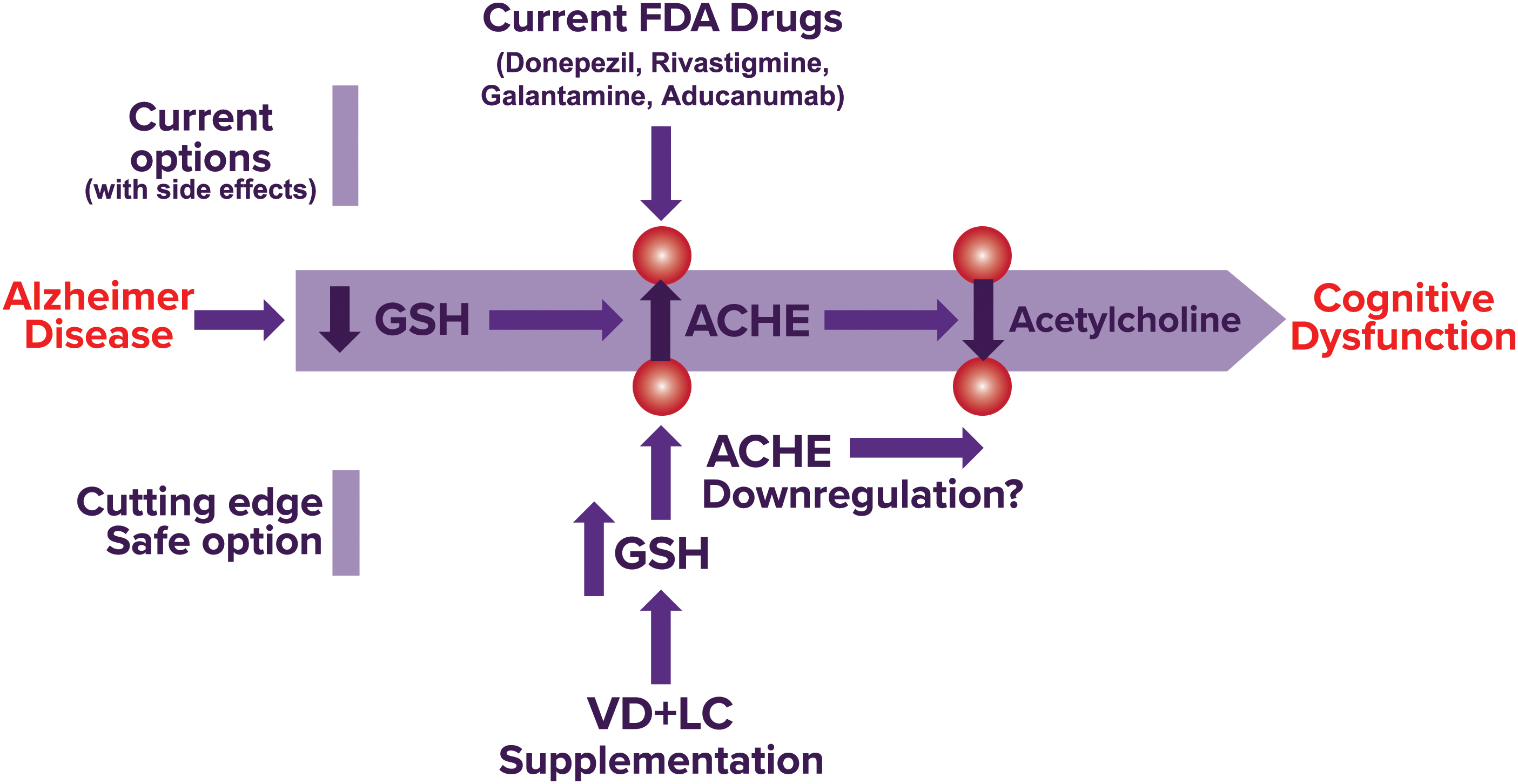
While antioxidant treatment has shown to be beneficial in preclinical studies, the success rate in clinical studies is poor (Kamat et al., 2008). Several reasons for this lack of transitional success have been suggested (Brewer, 2010; Kamat et al., 2008; Persson et al., 2014). Despite the high failure rate of antioxidant therapy in clinical studies, it is important to continue to search for ways to maximize the effects of antioxidant therapy in clinical studies due to the potential benefit increased antioxidant levels may have in AD and other chronic diseases. One hypothesis for the lack of clinical success with antioxidant therapy is that treatment is begun too late in the course of the disease (Brewer, 2010). Once MCI is present, it may be possible that neuronal damage and NFT occur at a rate faster than they can be replaced (Brewer, 2010).
In addition, since oxidative stress naturally increases with age, and it has been suggested that oxidative stress is one of the earliest signs of AD (Persson et al., 2014), initiating treatment before symptomatic onset or even early in the course of the disease may be much more beneficial. We believe that for patients at risk of developing AD, either those known to have inherited the familial form or those who are above age 65, beginning therapy before becoming symptomatic could potentially result in a protective effect by preventing oxidative stress levels from becoming high enough to cause damage.
Possible adverse effects with VD+LC supplementation
As with discussing any therapeutic drug, possible adverse effects are something to be mentioned. While VD toxicity is still a rare occurrence, the number of reported cases has believed to be primarily because of high-dose VD supplementation (Asif and Farooq, 2023). The clinical manifestations of VD toxicity are primarily governed by the consequences of hypercalcemia, which can include fatigue, weakness, anorexia, pancreatitis, and bone pain (Asif and Farooq, 2023). Hypercalcemia can also disturb renal function, leading to polyuria, polydipsia, and nephrolithiasis (Schulster and Goldfarb, 2020). Adverse effects that have been reported with cysteine supplementation include vomiting, diarrhea, nausea, heartburn, and allergic reactions, all of which are rare (Ershad et al., 2023). However, these side effects are rare and in those taking high doses of VD or LC.
Which population would potentially benefit most from this novel treatment?
Both the sporadic and familial forms of AD are associated with increased oxidative stress and Aβ accumulation (Meraz-Ríos et al., 2014; Ramamoorthy et al., 2012). Since the main therapeutic characteristic of our suggested treatment is decreased oxidative stress, which leads to lower levels of Aβ accumulation and downregulation of other pathological hallmarks, we believe that our novel treatment plan, if found beneficial, would be useful in the treatment of both the sporadic and familial forms of AD.
The majority of AD patients (70%–90%) are VD deficient (Keeney and Butterfield, 2015), and AA experience higher rates of VD deficiency and AD compared with other groups (Ames et al., 2021; Barnes and Bennett, 2014; Harris, 2006). AA also tend to have higher rates of GSH deficiency compared with other groups, which means that there may be a link between low levels of GSH and the development of AD (Green et al., 2002; Jain and Parsanathan, 2020; Mayeda et al., 2016; Tang et al., 2001).
A high incidence of glucose-6-phosphate dehydrogenase deficiency in AA (∼14% in AA vs. 1% in non-Hispanic Whites) can amplify GSH impairment due to the inability of cells to produce enough nicotinamide adenine dinucleotide phosphate to regenerate GSH from GSSG (Nkhoma et al., 2009; Tang et al., 2015). Since our novel treatment plan increases levels of GSH and VD, both of which are routinely decreased in AA, we hypothesize that this treatment would be more beneficial for AA. However, it is important to note that we believe that this treatment has the potential to benefit all patients at risk of developing AD regardless of their race or ethnicity.
AD is more prevalent in women than in men (Podcasy and Epperson, 2016). While the reason for this relationship remains uncertain, various hypotheses have been proposed. Since age is the strongest risk factor for AD, it has been suggested that the longer life expectancy in women compared with men is a major reason for this observation (Mielke et al., 2014). Studies have shown that inflammation may also contribute to this, since women have naturally higher concentrations of microglia than men, leading to the increased inflammation and neuronal damage that propagates AD pathogenesis (Hall et al., 2013; Hanamsagar and Bilbo, 2016; Peterson et al., 2015; Podcasy and Epperson, 2016). As a result, our novel treatment plan may be more beneficial in women since they may have naturally increased levels of inflammatory dysregulation.
Conclusion
Elevated levels of oxidative stress have been shown to be a key hallmark in the pathogenesis of AD. GSH is a major physiological antioxidant that protects against oxidative stress, and significant depletion in brain GSH levels correlates with the decline in cognitive function in MCI and AD subjects. Oxidative stress levels are also reduced in subjects supplemented with VD. Future clinical trials are needed to determine whether cosupplementation using a GSH precursor coupled with VD has the potential to lower the risk of dementia and AD when used as a prophylactic treatment or when given early in the course of the disease for patients at risk of developing AD. The development of a safe, low-cost dietary supplement would provide a much needed treatment for dementia.
Footnotes
Acknowledgments
The authors are supported by grants from NIH/NCCIH (5R33AT010637 and 3R33 AT010637-02S1) and the Malcolm Feist Endowed Chair in Diabetes. They thank Ms. Georgia Morgan for excellent editing.
Authors' Contributions
S.K.J. and C.M.S. wrote the article. S.K.J., C.M.S., and S.N.L. edited the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors are supported by grants from NIH/NCCIH (5R33AT010637) and the Malcolm Feist Endowed Chair in Diabetes.