
Abstract
Significance:
The presence of a large number of thioredoxin superfamily members suggests a complex mechanism of redox-based regulation in mammalian cells. However, whether these members are functionally redundant or play separate and distinct roles in each cellular compartment remains to be elucidated.
Recent Advances:
In the mammalian endoplasmic reticulum (ER), ∼20 thioredoxin-like proteins have been identified. Most ER oxidoreductases are soluble proteins located in the luminal compartment, whereas a small family of five thioredoxin-related transmembrane proteins (TMX) also reside in the ER membrane and play crucial roles with specialized functions.
Critical Issues:
In addition to the predicted function of ER protein quality control, several independent studies have suggested the diverse roles of TMX family proteins in the regulation of cellular processes, including calcium homeostasis, bioenergetics, and thiol-disulfide exchange in the extracellular space. Moreover, recent studies have provided evidence of their involvement in the pathogenesis of various diseases.
Future Directions:
Extensive research is required to unravel the physiological roles of TMX family proteins. Given that membrane-associated proteins are prime targets for drug discovery in a variety of human diseases, expanding our knowledge on the mechanistic details of TMX action on the cell membrane will provide the molecular basis for developing novel diagnostic and therapeutic approaches as a potent molecular target in a clinical setting. Antioxid. Redox Signal. 36, 984–1000.
Introduction
Thiol-disulfide oxidoreductases catalyze reduction/oxidation (redox) reactions, leading to the cleavage, formation, or isomerization of disulfide bonds between cysteine residues in substrate proteins. The formation or rearrangement of disulfide bridges is critical for the proper folding and assembly of proteins (7, 9, 41). Further, reversible thiol-disulfide exchange involves a large conformational change, and alteration of the protein redox state can positively or negatively regulate its function, thereby influencing cellular behavior (20, 37, 40, 45).
In mammalian cells, each cellular compartment contains a variety of thiol-disulfide oxidoreductases belonging to the thioredoxin superfamily (6, 31). Although the family members share a characteristic functional thioredoxin-related domain, they differ greatly in size and number of Cys-X-X-Cys (CXXC) active site motifs. The diversity among thioredoxin family members suggests a complex mechanism of redox-based regulation in mammalian cells. However, whether these members share the substrate proteins or exhibit distinctly specific roles remains elusive. Protein disulfide isomerase (PDI) belongs to the thioredoxin superfamily, and more than 20 PDI family proteins have been identified in mammalian cells (23, 64). Most PDIs are soluble proteins located in the luminal compartment of the endoplasmic reticulum (ER), whereas some are integral membrane proteins classified as members of the thioredoxin-related transmembrane protein (TMX) subfamily (27). In this review, we focus on the TMX protein family consisting of five membrane-anchored oxidoreductases (TMX1–5) and discuss the current understanding of their physiological functions, experimental methodology to investigate enzyme–substrate interactions, and implications in human diseases.
TMX1 (TXNDC1, PDIA11)
TMX1 is the founding member of the TMX family. Human TMX1 was identified during a gene-trap screening for novel genes regulated by transforming growth factor-β signaling (1). This gene is located on chromosome 14 and encodes a single-pass type I integral membrane protein with 280 amino acid residues (50) (Fig. 1A). TMX1 has a cleavable N-terminal signal peptide, required for entry into the secretory pathway, yielding a mature protein with a molecular mass of ∼30 kDa. TMX1 predominantly resides in the ER, though it lacks a classical di-lysine (KKXX)-type ER localization signal. An arginine-based motif (RQR) located near the C-terminus of TMX1 is postulated to be responsible for retrieving the protein to the ER, as shown for TMX4 (75). Topological studies have demonstrated that the catalytic thioredoxin-like domain of TMX1 is oriented toward the luminal side of the ER (54). This domain contains a unique CPAC active site motif that shuttles between the oxidized (disulfide) and reduced (dithiol) states in response to external stimuli. The short cytosolic C-terminal tail of TMX1 possesses two phosphorylation sites (Ser247 and Ser253) that have been verified by using global phosphopeptide sequencing (66). However, the biological significance of TMX1 phosphorylation remains unclear.
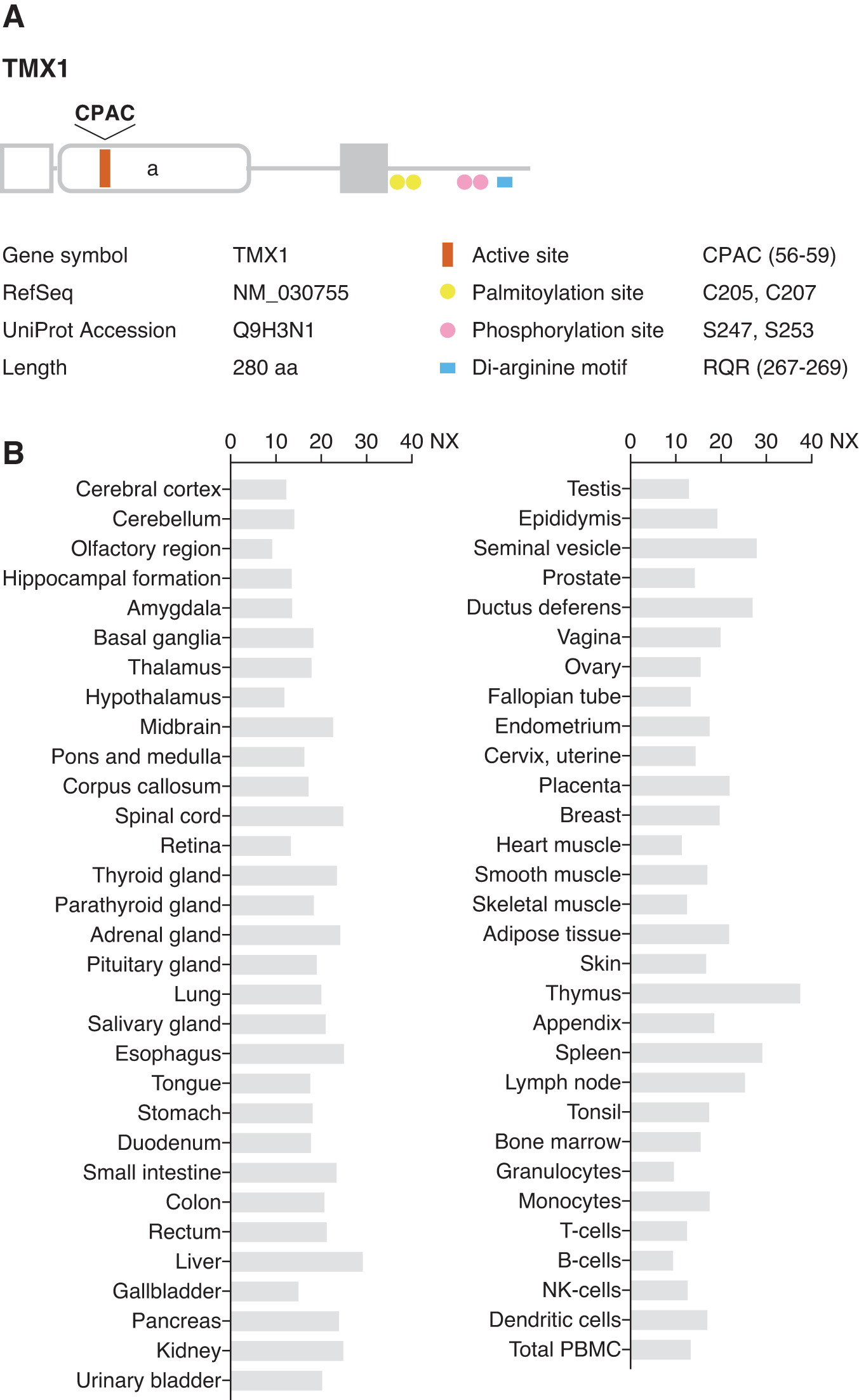
TMX1 messenger RNA (mRNA) expression is ubiquitous in human tissues (50) (Fig. 1B). Tmx1 knockout mice are born and develop normally (Mouse Genome Informatics, MGI [
Redox properties of TMX1
Environmental and pathological perturbations to ER function can cause accumulation of malfolded or denatured proteins in the ER, thereby activating the stress response pathway termed unfolded protein response (UPR) (58, 74). TMX1 expression is not induced by UPR (53). Disruption of ER proteostasis can alter the redox properties of ER-resident oxidoreductases. The active site cysteines in TMX1 are mainly reduced in the steady state, although a small protein fraction exists in the oxidized form (53). Under conditions of ER stress elicited by protein overload, TMX1 is readily converted to the oxidized state (51). This redox transition precedes the upregulation of ER stress markers downstream of the UPR pathway. Reactive oxygen species (ROS) produced under stress conditions do not seem to directly cause TMX1 oxidation. Considering that TMX1 preferentially binds to incompletely folded membrane proteins (26, 53), ER stress-induced TMX1 oxidation may occur as a consequence of thiol-disulfide exchange with accumulated substrates. In this scenario, TMX1 may catalyze the resolution of disulfide bonds formed within aberrantly folded proteins to cope with protein overload. Supporting this, oxidized TMX1 reverted to the basal reduced state after removal of ER stressors from the culture medium (51). Thus, protein accumulation in the ER disturbs the cellular redox balance, leading to the reversible oxidation of TMX1. These findings also suggest that cells are equipped with a reductive pathway to help restore ER homeostasis during post-stress recovery. However, the cellular component responsible for maintaining TMX1 in the reduced state has yet to be identified. Reduced glutathione has been proposed to provide reducing equivalents for the reduction of disulfides formed between the active site cysteines. Alternatively, an enzyme-catalyzed pathway may regulate the TMX1 redox state, enabling continuous rounds of thiol/disulfide exchange with substrate proteins.
Interaction of TMX1 with membrane-associated proteins
Several lines of evidence suggest that TMX1 favors membrane-bound substrates. The search for endogenous substrates for TMX1 has identified a series of cysteine-containing membrane proteins (10, 53, 69). TMX1 preferentially interacts with model proteins with transmembrane domains but not with soluble variants (26, 69), confirming the specificity of TMX1 for association with membrane-bound substrates.
The selective recognition of membrane proteins by TMX1 is partially mediated through its association with calnexin, a transmembrane chaperone in the ER (12, 76). This lectin-like molecular chaperone binds to N-glycosylated proteins and assists in their folding. Despite the lack of N-glycans in TMX1, the transmembrane domain is responsible for the binding of TMX1 to calnexin (53). The catalytic luminal portion of TMX1 does not contain a peptide-binding domain, such as the b-type thioredoxin-like domain found in PDI (30, 39), suggesting that TMX1 requires calnexin as a cofactor to recruit client proteins on the ER membrane. In fact, castanospermine, which blocks the glycan-dependent interactions between glycoproteins and calnexin, suppresses the disulfide-linked TMX1–substrate interaction (53) and destabilizes the TMX1–calnexin complex (69). These results suggest that TMX1 and calnexin form a functional complex that cooperatively acts on their clients during protein folding in the ER.
The role of thioredoxin-like ER oxidoreductases has been proposed in the reactivation of vitamin K epoxide reductase (VKOR) (10, 81, 87, 96). VKOR is an ER-resident transmembrane protein that catalyzes the conversion of vitamin K epoxide to vitamin K hydroquinone, an essential cofactor for activating blood coagulation factors (65). On catalyzing the reduction of vitamin K epoxide, VKOR is converted to the oxidized state and needs to be reverted to the reduced form through the vitamin K recycling system. TMX1 has been identified as a candidate redox partner that delivers reducing equivalents to VKOR (81). Considering that the proline residue in the CPAC motif in the TMX1 active site is required for stable interaction with VKOR, this residue may be a critical factor that determines substrate specificity and preference of TMX1 in the redox reaction. VKOR also interacts with TMX4 to a lesser extent, suggesting its preference for membrane-bound oxidoreductases. In contrast, VKOR does not form mixed disulfides with soluble ER oxidoreductases, with the exception of weak binding to ERp18 (81). No evidence has been reported on the involvement of TMX1 in thromboembolic events.
Disulfide reduction by TMX1
The active site cysteines of TMX1 are predominantly maintained in a reduced state, possibly reflecting the physiological role of TMX1 as a reductase. TMX1 exhibits reductase activity in vitro and cleaves interchain disulfide bonds in insulin (50). Moreover, it may putatively catalyze disulfide reduction; TMX1 is involved in the reductive activation of cytotoxins, such as ricin and abrin, belonging to the type 2 ribosome-inactivating protein (RIP) family (68). Type 2 RIPs are composed of two polypeptide chains linked by disulfide bonds (67). These proteins are incorporated into cells by endocytosis and translocated to the ER, where the interchain disulfide bond is cleaved to release a toxic A chain. On reduction in the ER, the catalytic A chain translocates to the cytosol and exerts cytotoxic effects by targeting eukaryotic ribosomes. In previous studies, PDI-catalyzed disulfide reduction has been implicated in the activation mechanism of these toxins (5, 85). TMX1 can efficiently reduce type 2 RIPs in the presence of reduced glutathione, and the cytotoxicity of the toxins was enhanced in TMX1-overexpressing cells. Conversely, TMX1 knockdown confers protection against type 2 RIP-induced cytotoxicity. This effect of TMX1 is specific for type 2 RIPs, and it does not increase the susceptibility of cells to other toxins with reduction-independent activities. These results represent general roles of TMX1 in the ER-to-cytosol retro-translocation of client proteins, which may be relevant for the ER-associated degradation pathway (26, 56).
Regulation of calcium homeostasis by TMX1
The pivotal effects of ER–mitochondria interplay and crosstalk have been implicated in various cellular functions, such as calcium homeostasis, lipid metabolism, autophagy, and inflammation (18). TMX1 is enriched on the mitochondria-associated membranes (MAM), specialized subdomains of the ER. Targeting TMX1 to these mitochondria–ER contact sites requires palmitoylation of two cytosolic cysteines (Cys205 and Cys207) adjacent to the transmembrane region of TMX1 (49). Palmitoylation also serves as an MAM-targeting signal for calnexin. The recruitment of palmitoylated TMX1 to MAM allows it to interact with sarco-ER Ca2+ transport ATPase (SERCA) 2b. SERCA2b facilitates Ca2+ uptake from the cytosol to the ER, thereby regulating cellular calcium flux. Interestingly, TMX1 overexpression decreases the ER Ca2+ content, whereas lack of TMX1 expression leads to increased Ca2+ import into the ER, indicating that TMX1 is a negative regulator of SERCA2b (71). TMX1 is targeted to the MAM more efficiently under oxidizing conditions, resulting in an enhanced association with SERCA2b, whereas reducing conditions produce the opposite effects. Further, an intact CPAC active site in TMX1 is essential for binding to SERCA2b; a catalytically inactive TMX1 in which two active site cysteines are mutated fails to interact with SERCA2b. Thus, TMX1 regulates Ca2+ flux through its interaction with SERCA2b in a redox-dependent fashion. However, the precise mechanism of TMX1 action in the control of cellular Ca2+ flux remains to be elucidated.
Reduced TMX1 expression indirectly impacts ER–mitochondrial Ca2+ flux by activating SERCA2b. Increased ER Ca2+ content in TMX1-deficient cells impairs Ca2+ transfer to the mitochondria. This can be attributed to the reduced ER–mitochondria contacts observed in cells with low TMX1 expression (71). Nevertheless, it remains unclear as to how TMX1 controls the physical association between these two organelles. Although speculative, TMX1 may be linked to the ER–mitochondria tethering complex, where it could catalyze redox-dependent conformational changes in tethering molecules. Alternatively, loss of tight ER–mitochondria contacts may be a result of a compensatory response to protect cells from mitochondrial Ca2+ overload, which triggers apoptosis (73).
From the perspective of mitochondrial bioenergetics, TMX1 deficiency reduces mitochondrial respiration, leading to a metabolic shift toward glycolysis. This may contribute to metabolic reprogramming (the Warburg effect) that occurs in cancer cells (94).
TMX2 (TXNDC14, PDIA12)
The complementary DNA (cDNA) clone of TMX2 was isolated from a human fetal cDNA library (55). Human TMX2 is located on chromosome 11. This gene encodes a 296 amino acid protein with an N-terminal signal sequence (Fig. 2A). The C-terminal half of the protein contains a single thioredoxin-like domain with an atypical SNDC sequence. A topological study of TMX2 predicted it to be a multi-spanning membrane protein with both the N- and C-termini on the cytosolic side (63). Thus, in contrast to other TMX family members, the thioredoxin-like domain of TMX2 is oriented toward the cytosol. The long C-terminal tail of TMX2 possesses a di-lysine ER retention signal (KKDK) (36).
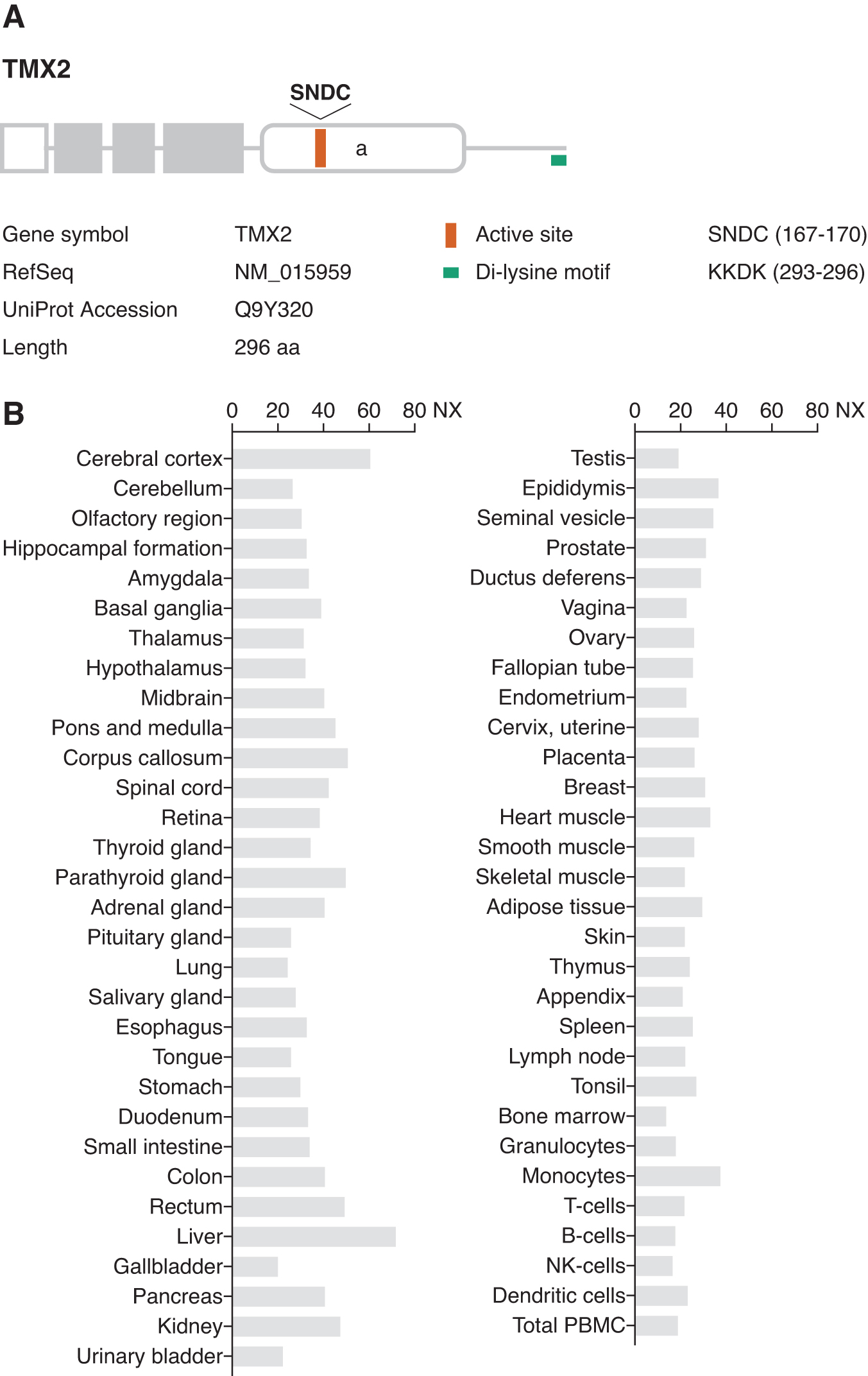
TMX2 mRNA is ubiquitously expressed in human tissues (55) (Fig. 2B). Tmx2 deficiency in mice results in embryonic lethality before implantation (MGI ID: 1914208), implying that TMX2 is essential in the early developmental stage.
Functional properties of TMX2
Endogenous TMX2 is predominantly localized in the ER, but a significant fraction of the protein is distributed in the outer membrane of the nuclear envelope (63). In a co-immunoprecipitation assay using TMX2 as bait, several nuclear membrane-associated proteins, including importin-β, were identified as candidate TMX2-interacting partners (63). Importin-β is an adaptor protein that mediates the nuclear transport of cargo molecules via the nuclear localization signal (91). TMX2 itself is not targeted for nuclear import through its interaction with importin-β. The nucleocytoplasmic transport of an importin–cargo complex is controlled by the Ras-related nuclear protein (Ran) GTPase cycle. It has been suggested that TMX2 regulates the compartmentalization of GTP- and GDP-bound Ran, generating a Ran gradient across the nuclear envelope. In support of this, small interfering RNA (siRNA)-mediated knockdown of TMX2 decreases the nuclear import of a model cargo protein expressed in cultured cells (63). Ran possesses a redox-sensitive cysteine (Cys112) (90), and the oxidized state of Ran can affect its subcellular distribution. TMX2 may control redox properties of Ran, thereby contributing to the formation of the Ran gradient that is essential for nucleocytoplasmic protein transport.
Notably, TMX2 is also targeted to MAM, where it can interact with calnexin and calcium pump SERCA2 (49, 95). It has been suggested that the interaction of TMX2 with MAM-associated proteins is crucial for regulating neuronal proliferation and migration (24, 42), which is discussed later in this article.
TMX3 (TXNDC10, PDIA13)
TMX3 was identified in a database search with a query sequence corresponding to the consensus thioredoxin-like domain (32). Human TMX3 is localized to chromosome 18 and encodes a type-I transmembrane protein comprising 454 amino acid residues with a predicted N-terminal signal sequence (Fig. 3A). TMX3 transcripts are found in a great variety of tissues (32) (Fig. 3B). TMX3 has two consensus sites for N-glycosylation in the luminal N-terminal portion of the protein; the presence of N-glycans was experimentally verified by sensitivity to endoglycosidase H treatment (32). TMX3 contains a classical KKXX-type consensus motif (KKKD) at the C-terminus for ER retention and, when exogenously expressed in cultured cells, TMX3 predominantly localizes to the ER. The luminal region is composed of one redox-active (a-type) and two non-catalytic (b- and b’-type) thioredoxin domains (33). The a-type domain contains a CGHC motif, a canonical active site sequence shared by most PDI family members in the ER lumen. Although the active site cysteines in TMX3 are predominantly reduced, a significant amount of the oxidized protein is present in living cells (32). Considering the structural similarity with the luminal ER oxidoreductases such as PDI and its closest homolog ERp57 (31), the enzymatically inactive b-type domains of TMX3 may be involved in substrate binding and co-factor recruitment (39). The b-type domains are also suggested to mediate structural stabilization of the redox-active a-type domain for efficient catalytic activity (33).
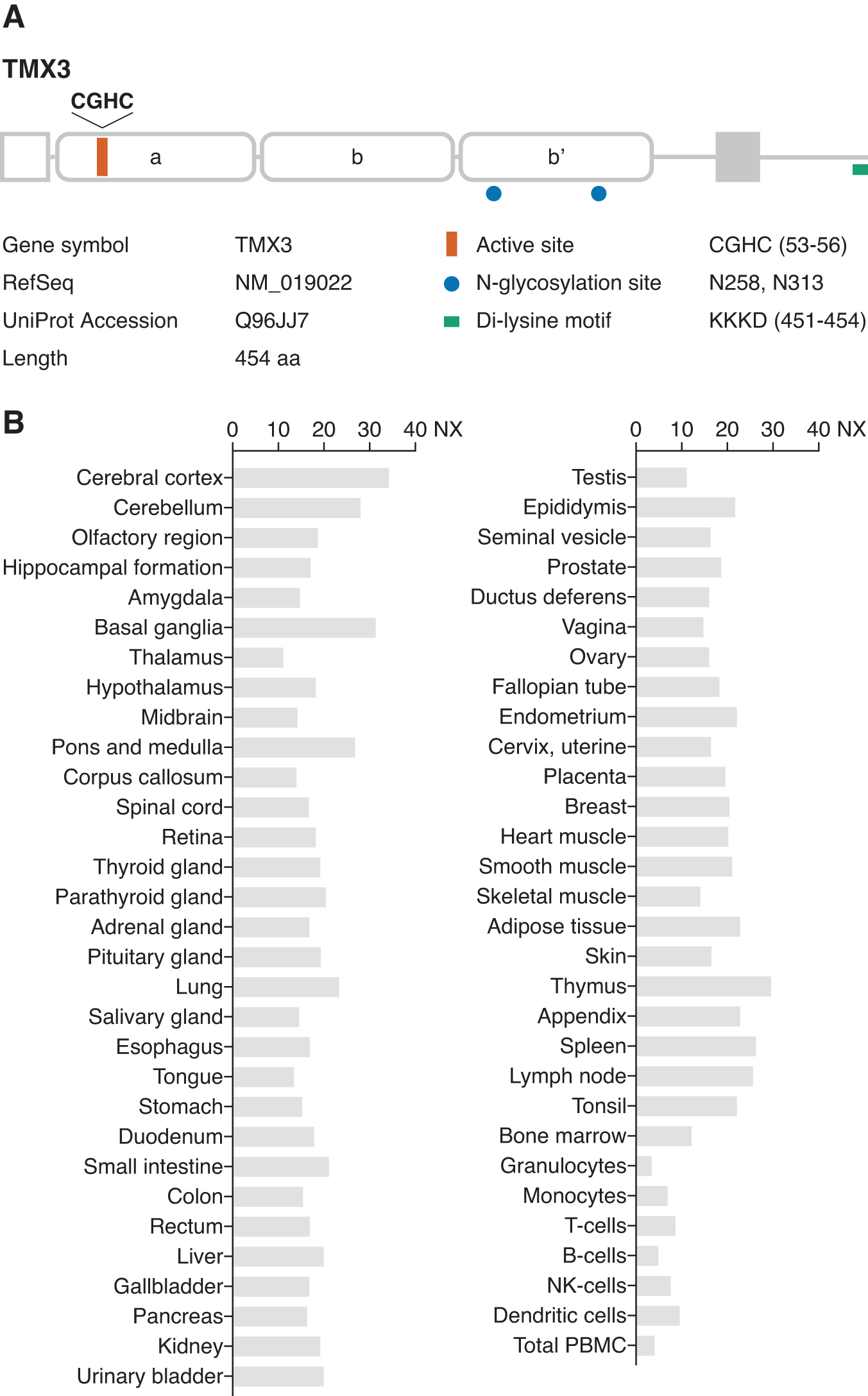
Functional properties of TMX3
The role for TMX3 as a foldase acting on membrane proteins has been demonstrated; TMX3 has been identified as a key cofactor for functional expression of insect nicotinic acetylcholine receptors (AChRs) (35). Co-expression of TMX3 can facilitate proper folding and assembly of the receptor subunits in the Xenopus oocyte system, resulting in the robust expression of functional AChRs. AChRs belong to the cysteine-loop ligand-gated ion channel superfamily that is characterized by a highly conserved cysteine loop motif consisting of two disulfide bond-forming cysteines (25, 61). Thus, TMX3 may catalyze the formation of disulfide bonds that are critical for the maturation of heteromeric AChRs.
TMX4 (TXNDC13, PDIA14)
Human TMX4 was identified in a database search for proteins with thioredoxin-like catalytic domains (75, 89). The gene is located on chromosome 20 and encodes a 349 amino acid protein with a cleavable signal sequence at the N-terminus and a single membrane-spanning region (Fig. 4A). TMX4 mRNA is ubiquitously expressed in human tissues (75, 89) (Fig. 4B). The membrane topology of TMX4 was established by determining sensitivity to proteinase treatment, revealing it to be a type I transmembrane protein with an N-terminal luminal region containing a thioredoxin-like catalytic domain (75, 89). TMX4 has a consensus N-glycosylation site in the luminal region, and the protein is sensitive to endoglycosidase H. TMX4 contains two phosphorylation sites (Sr251 and Ser259) within the C-terminal cytosolic tail (66). Exogenously expressed TMX4 is localized to the ER membrane, despite lacking a classical di-lysine motif that is required for ER localization. TMX4 possesses an RQR sequence that is located close to its C-terminus, which is conserved among TMX4 orthologs in different species. This di-arginine motif may be involved in the ER targeting of TMX4 (75).
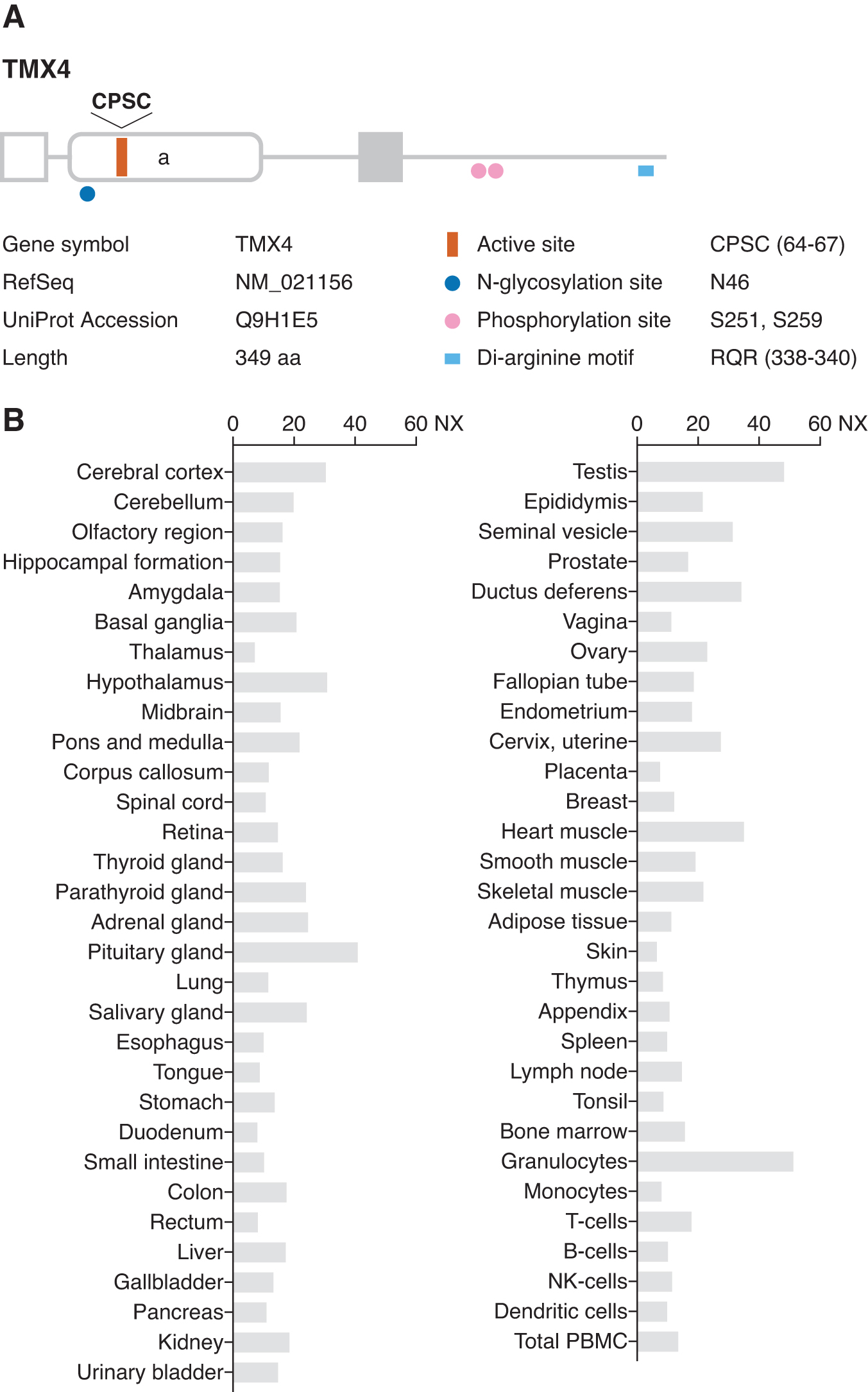
Functional properties of TMX4
The thioredoxin-like domain of TMX4 has a non-canonical CPSC active site sequence and shows the highest similarity with TMX1 among the five family members (75). The presence of a proline residue at the second position of the CXXC active site motif is a unique feature shared by TMX1 and TMX4. Both TMX1 and TMX4 form a mixed-disulfide intermediate with VKOR (81). The fact that VKOR exhibits a preference for these membrane-anchored thioredoxin family members suggests that the thiol-mediated ER quality control mechanisms are differentially regulated between luminal soluble proteins and membrane-associated molecules.
Phylogenetic analysis shows that TMX4 is a close paralog of TMX1 (Fig. 5A). However, the sequence similarities between TMX1 and TMX4 are confined to their thioredoxin-like domains, and TMX4 has some structural features that are different from TMX1, such as N-glycosylation and a longer cytoplasmic tail. In the steady state, TMX1 exists mainly in the reduced form (53), whereas TMX4 is primarily oxidized (75, 89). These properties suggest a distinct role for TMX4 in redox regulation on cell membranes. Moreover, molecular dynamics simulations in a comparative in silico analysis revealed that sequence-dependent structural and dynamical features of TMX4 are distinguishable from TMX1 and other ER oxidoreductases (87). Focusing on the CXXC active site motif, the folding state and the geometry (i.e., a distance between the cysteine residues and a dihedral angle) of the motif are predicted to be noticeably different among the oxidoreductases of the thioredoxin family. The CPAC motif of TMX1 constitutes a part of the well-folded α-helix, with the thiol groups in a restrained cis-configuration. In TMX4, the CPSC active site is located on a coiled linker connecting the β-strand and the α-helix. The coiled structure allows a highly divergent configuration of the TMX4 CPSC motif, and the thiol groups can adopt varying orientations from the synperiplanar to antiperiplanar conformation. These conformational differences may influence their specificity in target recognition during the thiol-disulfide reactions.
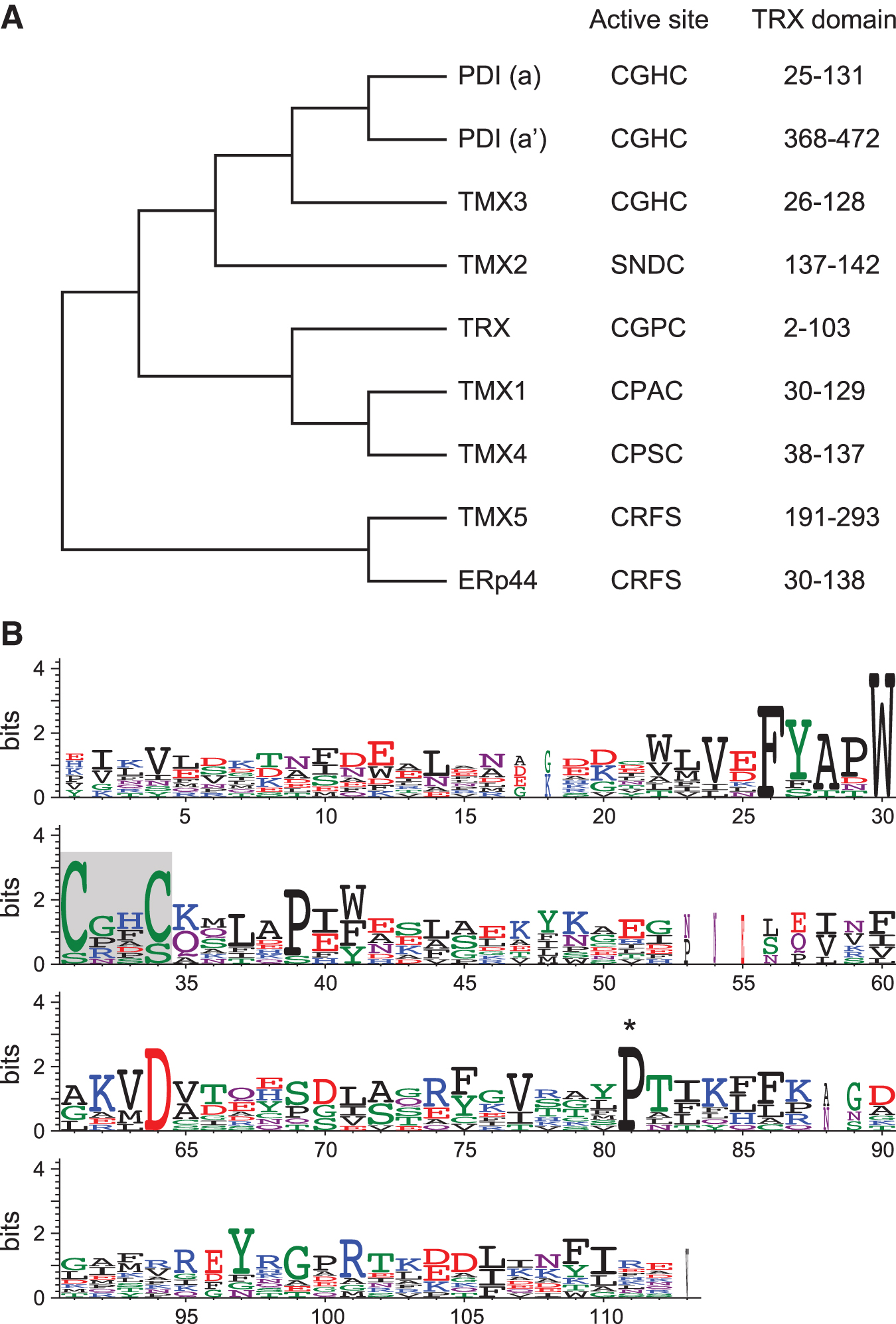
Recently, TMX4 was shown to localize on the inner membrane of the nuclear envelope in mesenchyme-derived cells (13). Although there are no reports on the physiological function of TMX4 on the nuclear membrane, this suggests that TMX4 may participate in redox-based regulation of the nuclear envelope structure.
TMX5 (TXNDC15)
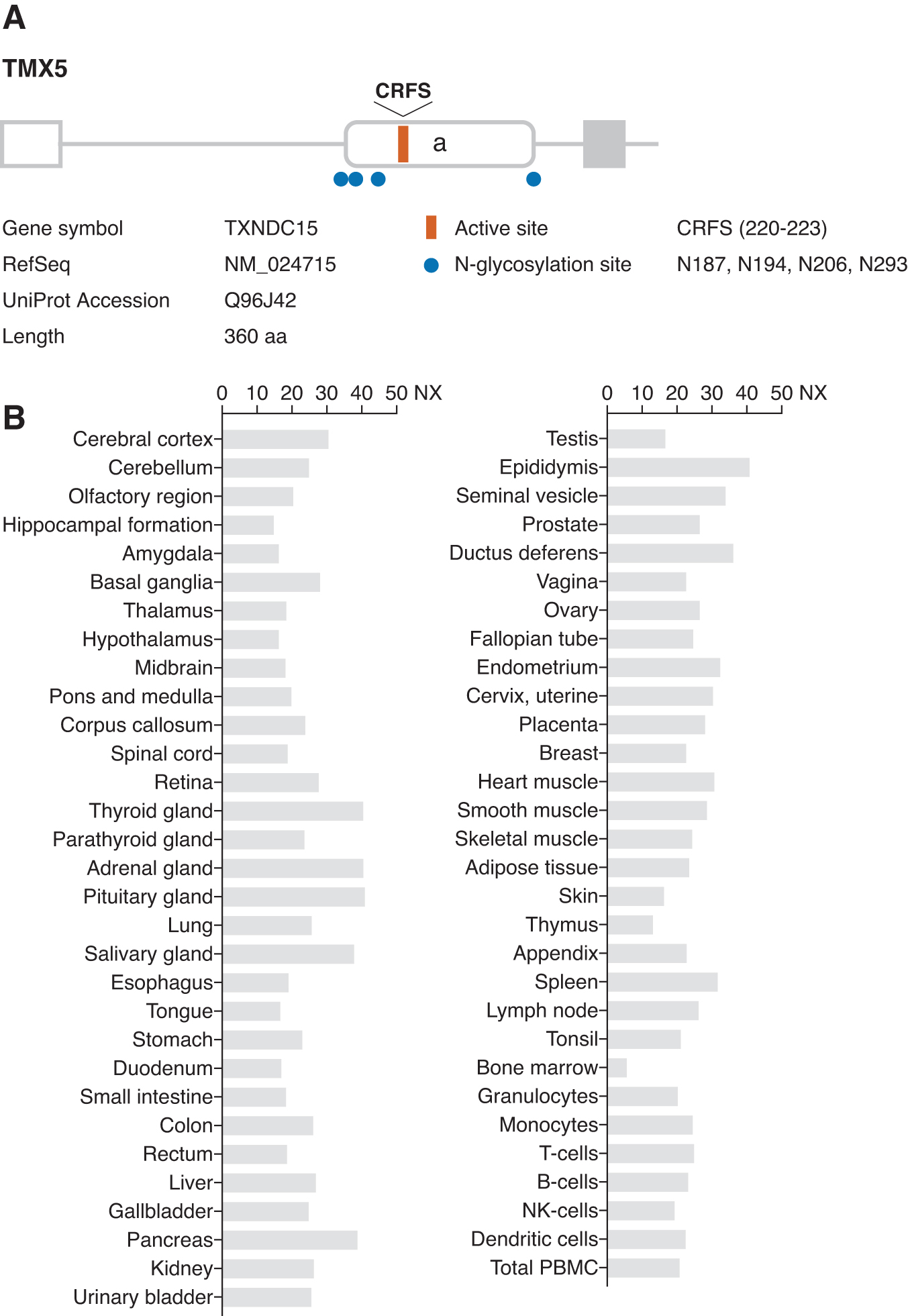
Human TMX5 (TXNDC15) was cloned during a large-scale screening for novel secreted and transmembrane proteins (14). The gene is localized to chromosome 5 and encodes a putative type I transmembrane protein comprising 360 amino acids with an N-terminal signal peptide (Fig. 6A). TMX5 transcripts are widely expressed in human tissues (Fig. 6B). Motif analysis indicates that the N-terminal region contains a single thioredoxin-like domain with a non-canonical CRFS active site motif and four predicted N-glycosylation sites. The protein lacks ER retention motifs, and its subcellular localization has not been precisely determined. According to the Human Protein Atlas (92), TMX5 was detected in the Golgi apparatus (
Functional properties of TMX5
Our understanding of TMX5 is very limited, and its physiological function has not been well characterized thus far. The thioredoxin-like domain of TMX5 is closer in structure to that of ERp44 (3), a soluble oxidoreductase of the PDI family in the ER lumen (Fig. 5A). ERp44 has been shown to form intermolecular disulfide bonds with client proteins, and the long-lived nature of this interaction allows for efficient retention of its substrates within the ER (2). The absence of the C-terminal active site cysteine in TMX5 suggests its ability to form stabilized intermolecular disulfide bonds with the client proteins. TMX5 may function in a manner similar to ERp44, regulating thiol-mediated protein quality control.
Identification of TMX Substrates
To determine whether these redox proteins share overlapping biological activities or whether they have separate and distinct functions, it is necessary to identify binding partners and substrate proteins for each oxidoreductase. The identification of physiological substrates of the thiol-disulfide isomerases has been challenging, which is in part due to the instability of the enzyme–substrate interaction during the thiol-disulfide exchange reaction. In this section, we describe the application of mechanism-based approaches to identify target proteins that engage in redox reactions with thiol-disulfide oxidoreductases. These methods should be applicable to a wide range of studies analyzing thiol-disulfide exchange reactions occurring inside or outside the cells.
Thiol-disulfide exchange reactions
The activity of thiol-disulfide oxidoreductases depends on a pair of cysteine residues in a conserved CXXC active site motif that shuttles between the oxidized (disulfide) and reduced (dithiol) states during the catalytic cycle (Fig. 7). The reduced form of the enzyme transfers reducing equivalents to disulfides in a substrate protein and catalyzes their reduction. Thereafter, a reduced substrate protein is released, and the oxidoreductase is converted to its oxidized form. When the active site cysteine residues are in the oxidized form, the oxidoreductase can receive electrons from a substrate and introduce a disulfide bond in the substrate protein. In this oxidative pathway, the reaction breaks the disulfide residue in the enzyme active site motif. Sequential reduction and oxidation result in the isomerization or rearrangement of a disulfide bond in a substrate protein.
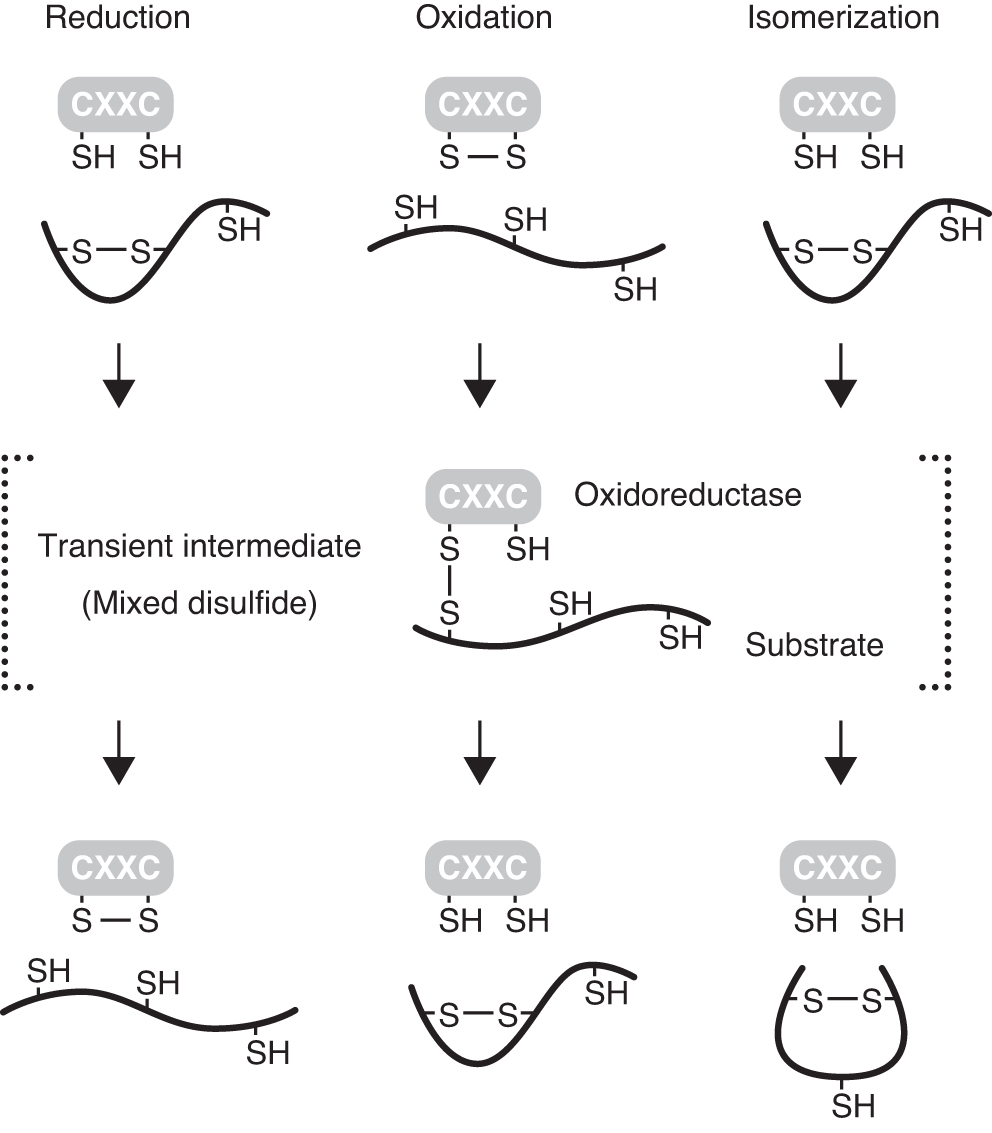
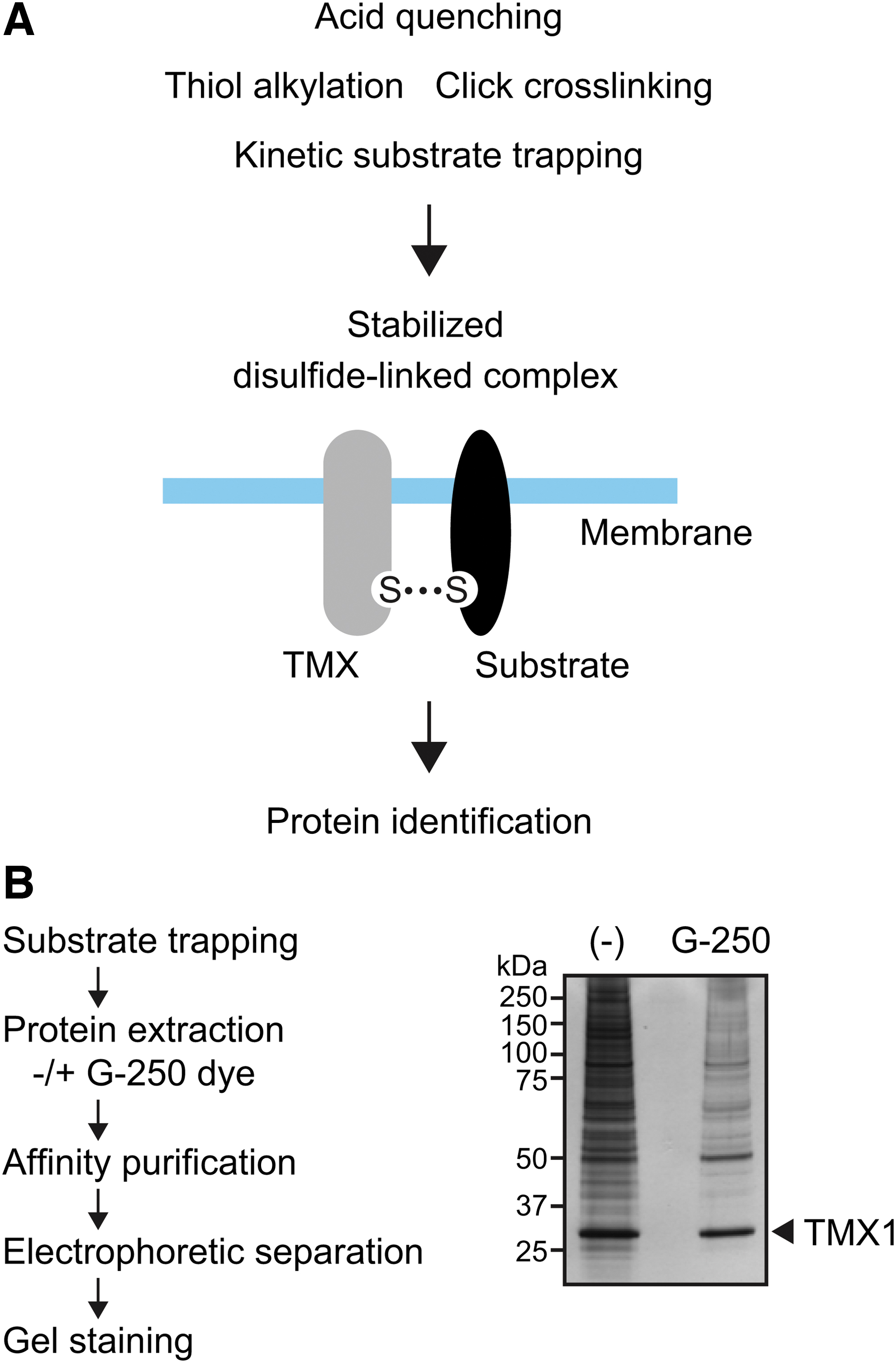
During the thiol-disulfide exchange reactions, a transient disulfide-linked intermediate (mixed disulfide) is formed between an oxidoreductase and its substrate (60). Because such transient mixed disulfides tend to be unstable, the identification of substrates for oxidoreductases has proven to be technically difficult. Several experimental approaches have been developed to stabilize reaction intermediates (4, 17, 21, 22, 28, 83), and these strategies can facilitate the identification of specific substrates of thioredoxin superfamily members (Fig. 8A).
Trapping substrates using a single-cysteine mutant
Considering the instability of the intermediates, experimental approaches have been used to trap substrates by mutating the C-terminal cysteine in the CXXC active sites of thiol-disulfide oxidoreductases. Thiol-disulfide oxidoreductase variants (trapping mutants), in which the C-terminal cysteine in the active site is replaced by Ala (CXXA) or Ser (CXXS), have been utilized to trap and identify substrate proteins (21). As the N-terminal cysteine is intact, the mutant retains the ability to attack the target disulfide in the substrate, leading to the formation of a mixed disulfide conjugate. Substitution of the C-terminal cysteine in the active site, however, retards resolution of the mixed-disulfide complex, trapping the substrate as a conjugate with the enzyme. The stabilized enzyme–substrate complex enables efficient recovery and isolation of the substrate protein for downstream analysis. Exogenous expression of oxidoreductase-trapping mutants causes accumulation of enzyme–substrate complexes linked by an intermolecular disulfide bond, leading to the successful identification of potential targets of various thiol-disulfide oxidoreductases, including TMX1 and TMX4 (53, 81, 89).
As an alternative approach, an oxidoreductase-trapping mutant may be prepared as a recombinant protein produced by bacteria or mammalian cells and administered to living cells to isolate the substrate on the cell surface. For example, a human thioredoxin-trapping mutant, in which the resolving cysteine at position 35 was replaced with serine, was allowed to react with the cell surface of lymphocytes, leading to the identification of CD30, a member of the tumor necrosis factor receptor superfamily, as the principal target of thioredoxin (82). Thioredoxin-mediated thiol-disulfide exchange causes a conformational change in the extracellular domain of CD30 and interferes with ligand–receptor binding. The recombinant trapping mutant was also used to capture substrates in tissue lysates and biological fluids. This method would be applicable to the catalytic domains of TMXs expressed as soluble proteins without the transmembrane regions. Identification of extracellular substrates for TMX family proteins will provide insights for elucidating the mechanism of action of TMXs on the cell surface.
Alternative approaches to stabilize mixed-disulfides
A drawback of the approach employing CXXA or CXXS trapping variants is that the method can only be used to identify substrates undergoing reduction or isomerization. To capture a substrate for oxidase, both cysteines in the active site must be intact and form disulfide bonds. Therefore, another strategy for substrate identification is required. To overcome the limitation of substrate trapping methods using CXXA or CXXS oxidoreductase mutants, researchers have generated several thiol-disulfide oxidoreductase trapping variants by substituting conserved amino acid residues other than cystine in the active site motif.
Conversion of the cis-proline 151 residue of DsbA, an Escherichia coli oxidase in the periplasm, results in the accumulation of disulfide-linked DsbA–substrate complexes (38). The crystal structure of the DsbA-substrate complex revealed that proline 151 is positioned close to the CXXC active site, suggesting its importance for substrate recognition by DsbA. Substitution of proline 151 with threonine causes a defect in the release of substrate from the enzyme. Thus, P151T substitution may affect the positioning of the mixed disulfide formed between DsbA and its substrate, leading to retarded resolution of the reaction intermediates. The corresponding proline residue is also conserved among mammalian thioredoxin-like proteins, including TMX family members (Fig. 5B). The TMX1 mutant harboring an analogous P101T substitution is capable of capturing disulfide-linked complexes (53). Thus, cis-proline mutants are useful for stabilizing mixed disulfides formed during thiol-disulfide exchange, leading to the identification of the substrates of TMX1. Based on these observations, it is suggested that the mutation of the corresponding proline residues may be useful in mammalian systems for the detection of mixed disulfide intermediates.
Another approach employing trapping variants involves changing the intervening residues between the cysteines in the CXXC motif. Screening for PDI variants that exhibit slower reaction kinetics against substrates revealed that substitution of the intervening histidine in the CGHC active site to either proline (CGPC) or arginine (CGRC) prolongs the enzyme–substrate interaction (88). The mechanism of action of these histidine trapping mutants has been attributed to a slower transition of the enzyme between the oxidized and reduced forms during the thiol-disulfide exchange catalytic cycle. It should be noted that the histidine-substituted trapping mutant retains both the active site cysteine residues and is enzymatically redox active. The histidine mutant of PDI with the reduced active site can trap substrates with disulfide bonds targeted for reduction by PDI. On the other hand, when the active site exists in the oxidized state, the histidine-trapping mutant is able to capture the substrate undergoing PDI-catalyzed oxidation. Thus, PDI histidine mutants allow the identification of protein substrates in both reductive and oxidative pathways. In fact, recombinant PDI variants with histidine substitution could capture candidate substrate proteins undergoing either reduction or oxidation by PDI during platelet activation. Considering the presence of CGHC active site motif in TMX3, this method would be beneficial to identify its physiological substrates.
Analysis of TMX–substrate complexes
In general, stabilized enzyme–substrate conjugates are purified by using specific antibodies or affinity chromatography resins, followed by mass spectrometry-based analysis. Considering that high background is a major problem in proteomic analysis, especially for integral membrane proteins with hydrophobic stretches (such as TMXs), it is necessary to minimize non-specific binding of contaminants to affinity matrices. In our approach to identify molecules interacting with TMX1, we included anionic Coomassie Brilliant Blue G-250 dye in the cell lysis buffer and successfully decreased the amount of co-purified contaminants (Fig. 8B). Coomassie G-250 dye has been used for blue-native polyacrylamide gel electrophoresis (97). It binds nonspecifically to proteins and confers a negative charge. The dye potentially masks hydrophobic sites, and negatively charged protein surfaces repel each other. Thus, the membrane proteins lose their hydrophobic properties on binding to the dye, and the G-250-treated molecules are expected to be less prone to aggregation (78). This approach is promising and would be useful for isolating specific interacting partners and physiological substrates for membrane-bound TMX family proteins.
Clinical Implications of TMX
A growing number of studies have provided evidence that dysregulation of TMX function is associated with the pathogenesis of a wide range of human diseases (Table 1).
Human Diseases Associated with Dysregulation of Thioredoxin-Related Transmembrane Protein Function
MIM, entry number in the Mendelian Inheritance in Man database.
Developmental disorders
Pathogenic variants of TMX2 have been identified in patients with neuronal migration disorders, such as microcephaly, lissencephaly, and polymicrogyria (Online Mendelian Inheritance in Man [OMIM;
Array-based comparative genomic hybridization analysis in patients with microphthalmia, a developmental defect of the eye, identified a deletion in chromosome 18, and TMX3 was included in the deleted region (99). Moreover, two missense mutations in TMX3 have been identified in patients with microphthalmia or anophthalmia (OMIM Phenotype MIM number: 300345) (11). These nucleotide changes result in substitution of amino acids (R39Q and D108N) in the catalytic thioredoxin-like domain of TMX3, possibly disrupting protein function. In a zebrafish model, morpholino-mediated gene knockdown of a TMX3 ortholog resulted in a small eye phenotype and abnormal retinal formation (11). In these knockdown larvae, co-injection of human TMX3 mRNA significantly reduced the frequency of ocular abnormalities; in contrast, co-injecting an mRNA encoding TMX3 mutant (R39Q) did not rescue the small eye phenotype, supporting the impaired functionality of the patient-derived mutant. Thus, despite its broad tissue distribution, deletion or haploinsufficiency of TMX3 causes more severe developmental defects in the eye than in other organs. Further studies are warranted to clarify the precise mechanism of TMX3 action in vertebrate eye development. The identification of physiological substrates of TMX3 in the developing eye would be of particular interest, as it may help further our understanding of the pathogenesis of human microphthalmia.
Genomic analysis of a large ciliopathy cohort identified loss-of-function mutations in TMX5 (84). TMX5 was found to be independently mutated in families with features of Meckel–Gruber syndrome (MKS), a lethal autosomal recessive ciliopathy caused by defective primary cilium formation. Lack of functional TMX5 causes mislocalization of the TMEM67 ciliary receptor to the transition zone, resulting in the perturbation of ciliary signaling and aberrant ciliogenesis. Several clinical studies have also reported pathogenic variants of TMX5 in fetuses with MKS (70, 72). In addition, an experimental approach based on genome-wide CRISPR-mediated gene disruption identified TMX5 as a candidate ciliopathy-associated gene (8). TMX5-knockout cells harbored a defect in Hedgehog signaling, which is indispensable for the normal development of embryo, and exhibited abnormal morphology, supporting its causative role in MKS. These reports suggest that TMX5 is a novel MKS-associated gene. However, the molecular basis for TMX5 action in ciliary signaling pathways remains poorly understood.
Neurodegenerative diseases
Mutations in the C9ORF72 gene are the most common causes of amyotrophic lateral sclerosis (ALS). The pathogenic mutation is a hexanucleotide-repeat expansion that results in the massive production of toxic dipeptide-repeat (DPR) proteins (16). DPR proteins are prone to aggregation and contribute to neurodegeneration. In CRISPR–Cas9 genome-wide genetic deletion screens, TMX2 was identified as a potent genetic modifier of DRP toxicity (42). TMX2 knockout suppressed DPR-induced toxicity in primary neurons. The DPR proteins induce ER stress that is believed to be involved in DPR toxicity and ALS pathogenesis. It has been suggested that TMX2 deficiency confers protection against DRP toxicity by modulating the ER stress response induced by DRP proteins. In view of disease prevention mechanisms, suppression of TMX2 would be beneficial for protecting neurons from DRP-induced cytotoxicity and may serve as a therapeutic target for ALS pathogenesis.
Accumulation of N-terminal fragments of mutant huntingtin protein (mHTT) has been implicated in the progression of Huntington's disease (HD) (77). It is suggested that the mHTT fragments containing expanded polyglutamine repeats are oxidized to form stable aggregates, thereby exerting their toxicity (19). In an experimental model of HD, TMX3 decreased the levels of exogenously expressed mHTT (48). Since mHTT accumulates in the cytosol, it is improbable that TMX3 directly regulates the redox properties of mHTT. The authors of that study also showed that overexpression of cytosolic thioredoxin attenuates the toxic effects of mHTT in the HD models (48). These results indicate that these oxidoreductases act to reduce the burden of protein aggregation by controlling protein redox states, thereby preventing mHTT-induced cell death. Although the implications of thiol-mediated protective mechanisms against neurodegenerative diseases can be postulated, the precise roles of oxidoreductases in the control of disease states have not yet been explored.
Cardiovascular diseases
The ER oxidoreductases, such as PDI, ERp57, and ERp5, are translocated to the cell surface where they regulate platelet function extracellularly (80). A recent study revealed that TMX1 is localized on the surface of human platelets and that its expression is increased on platelet activation (101). Moreover, the inhibition of cell surface TMX1 resulted in increased platelet aggregation. Tmx1-deficiency in mice potentiates platelet function with elevated platelet incorporation into the thrombus and shortened tail bleeding times, supporting the in vitro studies. These results indicate that TMX1 is a negative regulator of platelet aggregation and thrombosis. It has been proposed that TMX1 oxidizes integrin αIIbβ3, maintaining the integrin in an inactive or quiescent state and inhibiting its signal. Consistently, platelets from Tmx1-deficient mice showed increased thiols in the β3 subunit of αIIbβ3.
Whole-exome sequencing identified a novel pathogenic indel mutation in TMX3 in a Chinese cohort with a family history of coronary artery disease (46). A cell surface biotinylation assay revealed the presence of TMX3 on the platelet surface (34). Although speculative at present, TMX3 may act on certain extracellular substrates to control platelet function in a manner similar to TMX1 (98). Given that platelet aggregation and thrombosis are the major causes of coronary artery disease, it may be possible that TMX3 is trafficked from the ER to the platelet surface, where it regulates thrombus formation.
Cancer
The role of TMX1 in tumorigenesis remains controversial. Decreased TMX1 expression has been reported in metastatic melanoma (71). Low TMX1 expression in cancer cell lines decreases Ca2+ transport to the mitochondria, dampens mitochondrial respiration, and produces higher levels of ROS. Despite the growth suppressive phenotype of TMX1-knockdown cells in vitro, reduced TMX1 expression led to increased tumor growth in a mouse xenograft model, suggesting tumor suppressive properties of TMX1. These opposing effects of TMX1 on cell proliferation should be further explored. Further, lower expression levels of TMX1 were also reported in aggressive breast cancer cells (44) and asbestos-exposed lung cells (62).
Conversely, elevated expression of TMX1, as well as TMX3, has been reported in melanoma cells and patient-derived samples (100). Moreover, TMX1 expression levels positively correlate with the disease stage of aggressive melanoma, and patients with increased TMX1 expression have a lower survival expectancy. From a mechanistic point of view, TMX1 regulates nuclear factor of activated T cells 1 (NFAT1) signaling that is implicated in melanoma growth (59). TMX1 downregulation suppresses the nuclear translocation of NFAT1 in an ROS-dependent manner, thereby inhibiting the proliferation and invasive potential of melanoma cells. These results suggest that the TMX1–NFAT1 axis promotes cancer progression, and elevated TMX1 expression is indicative of cancer aggressiveness and poor survival outcome.
TP53-regulated inhibitor of apoptosis 1 (TRIAP1), an oncogenic protein highly expressed in lung adenocarcinoma, has been shown to mediate the upregulation of some anti-oxidative proteins, including TMX1 and TMX2 (29). The expression levels of TMX1 and TMX2 were inversely correlated with the overall survival of the patients. Further, their expression was enhanced in lung cancer cell lines on exposure to radiation. The results may imply that the pro-oncogenic property of TRIAP1 involves upregulation of the antioxidative capacity of cancer cells, thereby contributing to tumor resistance against radiation-induced oxidative damage.
Conclusion
Since the discovery of TMX1 in 2001, four additional members have been added to the human TMX family. They share the common property of being anchored to cellular membranes. The collective body of evidence indicates their preference for interacting with membrane-associated molecules. Most TMX members are localized mainly to the ER membrane, suggesting their primary roles in assisting the folding and assembly of membrane proteins in the secretory pathway. Further, recent studies have revealed that they have diverse functions outside of the ER. However, the precise function of individual TMX family members in human physiology and the underlying mechanism of action remain to be elucidated. The cellular membrane serves as a platform for signal transduction and cell–cell interaction, and membrane proteins are prime targets for drug discovery in several human diseases. Expanding our knowledge of the physiological roles of these membrane-bound oxidoreductases of the TMX family and determining the mechanistic details of their action will be of the utmost importance for developing diagnostics, potent molecular targets, and therapeutic approaches for the treatment of human disorders taken together with the elucidation of functional roles against other PDI family proteins in the cells, although extensive research is still required to obtain a complete picture.
Footnotes
Acknowledgments
The author thanks Kiichi Hirota (Kansai Medical University) for critical reading of the article. The author also thanks Editage (
Author Disclosure Statement
The author declares no competing interests.
Funding Information
This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant number JP19K09339 to Y.M.).